
Abstract
A computational protocol based on the complete and restricted active space self-consistent field (CASSCF/RASSCF) methods and their second-order perturbation theory extensions (CASPT2/RASPT2) is employed to benchmark the highly excited-state manifold of the DNA/RNA canonical purine nucleobase guanine in vacuo. Several RASPT2 schemes are tested, displaying a steady convergence of electronic transition energies and dipole moments upon active space enlargement toward the reference values. The outcome allows calibrating and optimizing computational efforts by considering cheaper and more approximate RAS schemes that could enable the characterization of the excited-state manifolds of multi-chromophoric systems, such as DNA/RNA nucleobase dimers or multimers. Simulations of two-dimensional electronic spectra show similar trends to those observed on the other purine nucleobase adenine, deviating from this and other pyrimidine nucleobases in featuring its main excited-state absorption signal, embodied by sizable double HOMO to LUMO excitation contributions, in the UV probing window.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Two-dimensional electronic spectroscopy (2DES) is [1,2,3,4,5,6,7,8,9,10] steadily becoming a favorable tool to track photoinduced phenomena in complex systems [1,2,3,4,5,6,7,8,9,10,11]. This is due to its enhanced spatial resolution as compared to its one-dimensional pump-probe (1D-PP) counterpart, stemming from the use of an extra laser pulse in the buildup of the third-order nonlinear response signal compared to the latter: A typical experiment is performed by scanning time delays between the first and second laser pulses (t1) for a given time delay between the second and third pulses (t2), which can be considered as the analogue of the waiting time between pump and probe pulses in PP spectroscopy, and between a third pulse and a local oscillator (t3), which heterodynes the field emitted by the sample in response to the perturbation by the three incident pulses. By Fourier transforming the signal along t1 and t3, two-dimensional frequency maps with signals S(Ω1, t2, Ω3) can be obtained at different waiting times t2, where all electronic transitions involved in the three field–matter interactions are recorded [12,13,14]. The enhanced spatial resolution of 2DES along the pump frequency (Ω1) is particularly important for studying systems containing several absorbing chromophores in neighboring spectral regions that produce congested signals in PP setups, as is the case in DNA/RNA single- and double-strand multimers, where the different nucleobases contained in the sequence absorb at similar wavelengths and their signals overlap making their disentanglement in specific decay channels unfeasible and thus requiring more complex techniques for their adequate and unequivocal characterization.
The interpretation of two-dimensional electronic spectra strongly relies on theoretical modeling working as a road map for their understanding. These range from those based on model Hamiltonians [15,16,17,18,19], which employ pre-computed ab initio parameters from the different (isolated) monomers contained in the macromolecular system to mixed quantum mechanics/molecular mechanics (QM/MM) approaches that explicitly account for the description of the solvent combined with supermolecular schemes (involving dimeric or multimeric species), thus accounting explicitly and more accurately for electronic coupling [20,21,22]. Such advanced modeling involves electronic structure theory evaluations of the high-energy excited-state manifolds probed by 1D-PP and 2DES experiments, which can add up to 9 eV from the ground state in the case of UV-active DNA/RNA nucleobases, i.e., featuring a couple dozen electronic states in monomers [23, 24] and over a hundred excited states in dimeric species [25,26,27]. Thus, robust theoretical approaches able to describe in principle all types of excitations (localized, singly and doubly excited and/or charge transfer) on an even footing are required. Suitable candidates are multiconfigurational complete active space self-consistent field (CASSCF) [28] and its second-order perturbation theory extension (CASPT2) [29, 30], which are able to accurately compute high-lying states despite featuring different known caveats such as those arising from valence-Rydberg mixing [31] and a potential unbalanced treatment of ionic and covalent states at certain points of the potential energy surface upon addition of the dynamic correlation [32,33,34].
In this work, we evaluate and benchmark the capabilities of CASPT2 and restricted active space RASPT2 methods to simulate the electronic excited states (and their associated transition dipole moments) for the DNA/RNA nucleobase guanine, yielding the most accurate theoretical estimates to date for its high-lying excited-state manifold. Given the lack of experimental data in the high-energy windows, we resort to a systematic protocol where an incremental active space is employed until convergence, yielding the best estimates computationally affordable. Due to the large computational expense associated with these techniques and their dependence on the size of the active space employed, we focus on less-costly restricted active space (RASSCF/RASPT2) approaches, which are thoroughly benchmarked to allow considering larger dimeric and multimeric sequences in future studies. Moreover, the outcome completes our previous efforts on pyrimidine and adenine (purine) bases [23, 24] and it can eventually be straight on used for building Frenkel excitonic Hamiltonian-type models to simulate nonlinear spectra on DNA/RNA hetero-multimers.
2 Computational details
2.1 Electronic structure theory computations
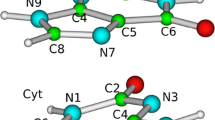
Complete/restricted active space self-consistent field (CASSCF/RASSCF) [28, 35, 36] and their second-order perturbation theory extensions (CASPT2/RASPT2) [30, 37, 38] have been employed as implemented in the MOLCAS 8 package [39]. The reference computations of guanine, used to calibrate different reduced active space schemes, were obtained with a procedure previously introduced by Roos and co-workers [40] and applied by us to adenine [24] and all canonical pyrimidine nucleobases [23]: A set of additional diffuse and uncontracted basis functions is added to the center of charge of the molecule, followed by a systematic removal of π* Rydberg-like orbitals (21 in this particular case) to minimize the expected CASPT2 overestabilization due to the presence of Rydberg-type orbitals in the secondary space, which are not properly represented within the ANO basis set employed, and to avoid the appearance of spurious quasi-Rydberg states [40]. A more detailed description is given in [32]. A full π complete active space reference comprising all π bonding and π* antibonding orbitals (see Fig. 1) is then employed for all energy evaluations and geometry optimizations enforcing Cs symmetry. The following RAS nomenclature is adopted: RAS (maximal number of holes in RAS1, number of RAS1 orbitals/number of electrons in RAS2, number of RAS2 orbitals/maximal number of electrons in RAS3, number of RAS3 orbitals). A systematic increase in the RAS3 subspace allowing up to two electrons leads to RAS(0,0|14,11|2,4), RAS(0,0|14,11|2,6), and RAS(0,0|14,11|2,8) schemes, which are analyzed to evaluate the energy convergence upon active space enlargement. The exclusion of the n lone pair orbitals and the corresponding nπ* transitions is based on the relatively small dependence these states have on the dynamic correlation (being of covalent nature within valence bond theory) here benchmarked at the CASPT2/RASPT2 levels of theory [33], the present schemes being able to properly describe them upon their inclusion given their lesser dependence on the active space, as shown by their already accurate estimates at the CASSCF level. Moreover, their mediating role in the photoinduced events taking place in guanine [41] heavily depends on the environment surrounding the system [42, 43], as the nπ* state lays relatively low in energy and is thus accessible in vacuo from both bright La and Lb states, being blue-shifted in polar solvents for this and all other nucleobases and thus reducing their probability of non-adiabatic population [42, 44]. An imaginary level shift [45] of 0.2 a.u. was employed in the perturbation treatment to minimize the appearance of intruder states except when explicitly stated as in Sect. 3.2, while the IPEA shift was set to zero for all cases [46, 47]. The required transition dipole moments were obtained making use of the restricted/complete active space state interaction (RASSI/CASSI) method [48, 49]. Additional RASPT2 computations with less demanding RAS1/RAS3 schemes were considered within the RAS(4,7|0,0|4,4) by artificially shifting the excitation energies with the imaginary level shift (IMAG) as previously done in adenine [24], and by systematically increasing the number of holes and electrons in the RAS1/RAS3 subspace leading to RAS(5,7|0,0|5,4), RAS(6,7|0,0|6,4), RAS(7,7|0,0|7,4), and RAS(8,7|0,0|8,4). Cholesky decomposition was employed throughout to speed up the two-electron integrals [50,51,52].

Guanine molecular orbitals contained in the CASSCF simulations featuring all π bonding and antibonding orbitals
2.2 Nonlinear spectroscopy
The resulting energy levels and transition dipole moments have then been employed to simulate the two-dimensional electron spectra making use of the Spectron 2.7 program [53], which can compute quasi-absorptive 2DES maps via the sum-over-states approach [54] within the dipole approximation [20]. Further details on the working equations have been given elsewhere [24, 53]. Spectral line shapes are simulated assuming that the dephasing is caused by pure dephasing in the Markovian approximation. Thus, the signals were homogeneously broadened with a constant line broadening of 500 cm−1. The waiting time t2 between the pump pulse pair and the probe pulse is set to zero; excited-state dynamics is thus neglected. Within this static approximation, only computations at the Franck–Condon (FC) region are required to simulate the spectra. Protocols for computing 2DES considering spectral diffusion and non-adiabatic effects on an ultrashort timescale are well established [55,56,57,58]. However, our approximation is sufficient for our aim of benchmarking the effect of various computational parameters on the shape (position and intensity of the peaks) of the two-dimensional maps and to qualitatively separate the main signals characterizing the spectra of guanine at the FC region. Future studies could tackle the fine line shape given by the ultrashort vibrational dynamics as well as by inhomogeneities present in the sample, once the best affordable computational protocol is firmly established. All signals reported use the all-parallel xxxx pulse polarization configurations and are plotted on a linear scale. Both ground state bleaching (GSB) and stimulated emission (SE) contributions appear as negative (blue) peaks, whereas excited-state absorptions (ESAs) appear as positive (red) peaks in the two-dimensional spectra.
3 Results and discussion
3.1 Reference spectrum
Table 1 displays the reference excited-state computations of guanine based on a SA-30-RAS (0,0|14,11|2,8)/SS-RASPT2 computation. This level is achieved by performing the protocol described in Sect. 2, providing converged results with respect to the systematic increase in the RAS3 subspace [23, 24]. The specific configuration state functions (CSFs) defining the nature of the electronic transitions are reported in Table 1. As can be seen, low-energy lying states feature contributions of one or two CSFs with large weights, whereas those placed in the high-energy window display strong multiconfigurational character, by featuring multiple CSFs with rather small weights contributing to the total excited-state wave functions.
A systematic enlargement of the RAS3 subspace has been carried out starting from the CAS(14,11) level, or CAS(0,0|14,11|0,0), its effects on the electronic excited-state energies and dipole moments being directly displayed along the NUV-pump/Vis-probe (Fig. 2, in the 10,000–30,000 cm−1 range) and the NUV-pump/NUV-probe (Fig. 3, in the 30,000–46,000 cm−1 range) 2D spectra. Evaluating the different energetic trends upon active space enlargement through 2DES maps is particularly appealing, given it provides a visual aid for its interpretation, the position of the peaks and their relative intensities being proportional to the computed vertical excitation energies and transition dipole moments, respectively. As can be seen, the NUV-pump/Vis-probe (Fig. 2) and NUV-pump/NUV-probe (Fig. 3) spectra show relatively small changes by going from RAS(0,0|14,11|2,6) to RAS(0,0|14,11|2,8), providing evidence of convergence already upon addition of six additional RAS3 orbitals, while considering the permutation of up to two electrons.

Two-dimensional NUV-pump/Vis-probe spectra of guanine in gas phase for different active space sizes: a CAS(14,11); b RAS(0,0|14,11|2,4); c RAS(0,0|14,11|2,6); d RAS(0,0|14,11|2,8). Peak numbering follows the excited-state assignment displayed in Table 1, red labels (left) denote ESAs along the 1La trace, and blue labels (right) refer to ESAs along the 1Lb trace

2D NUV-pump/NUV-probe spectra of guanine in gas phase for different active space sizes: a CAS(14,11); b RAS(0,0|14,11|2,4); c RAS(0,0|14,11|2,6); d RAS(0,0|14,11|2,8). Peak numbering follows the excited-state assignment displayed in Table 1, red labels (left) denoting ESAs along the 1La trace, blue labels (right) refer to ESAs along the 1Lb trace and black labels denote GSB signals
A closer look at the NUV-pump/Vis-probe spectra shows a negligible dependence of the different ESA signals among Ω3 upon active space enlargement, both for energetic position and associated relative intensities, whereas more pronounced differences are registered among Ω1 in terms of a systematic energy shift toward higher energies as has been previously reported for the other nucleobases [23, 24]. This energy shift totals ~ 1000 cm−1 along both 1La (centered at 37,000–38,000 cm−1) and 1Lb (centered at 41,000–42,000 cm−1) by going from CAS(0,0|14,11|0,0) to RAS(0,0|14,11|2,8). These differences are smaller than those observed for the other nucleobases, mainly due to the shared CFSs within both 1La and 1Lb, which remain stable upon active space enlargement. The reported excitation energy values for 1La (4.68 eV) and 1Lb (5.20 eV) are in agreement with the cross section of guanine, showing a band with two shoulders peaking at ~ 275 nm (4.51 eV) and ~ 250 (4.96 eV) [59], thus being within 0.2 eV of the theoretical estimates here reported. These are in agreement with equation of motion excitation energy coupled cluster (EOMEE)-CCSD(T)/aug-cc-pVTZ estimates reported by Szalay et al. [60] of 4.86 and 5.37 eV for the 1La and 1Lb states, respectively, and with previous CASPT2 computations of Roos and co-workers [61] yielding 4.51 and 5.25 eV, respectively. Unfortunately, only theoretical values up to state 4 (ππ*) are available at the (EOMEE)-CCSD(T) level [60], making its comparison for the high-energy window unfeasible, yet presenting an excitation energy of 6.26 eV, also in agreement with our estimate of 6.00 eV (see Table 1). The oscillator strength estimates for the 1La and 1Lb transitions are 0.145 and 0.269, respectively, which are lower than those obtained in the literature at the EOMEE-CCSD/aug-cc-pVTZ level of 0.16 and 0.37 [43]. Unfortunately, no estimates for the oscillator strength are available at the higher (EOMEE)-CCSD(T)/aug-cc-pVTZ level, which has a more accurate description of the excitation energies, for a better comparison. The active space enlargement effect along Ω1 is also observed in the low-lying excited-state absorptions, mainly referring to states 6, 7, and 9 along the 1La trace and state 7 along the 1Lb trace, which consequently causes the preservation of the energy gap among them and roughly retains the peak position along Ω3. This is associated with the similar nature of the excited ππ* states, highly dependent on dynamic correlation [33], which react analogously toward its systematic increase. Higher energy signals in the Vis-probe window show a more pronounced dependence on the dynamic correlation included in the model (Fig. 2): Peak 11 along the 1La trace oscillates slightly upon active space enlargement, its associated intensity being overestimated in the less-correlated CAS(0,0|14,11|0,0) with respect to the RAS(0,0|14,11|2,8) reference, a feature also observed for peaks 5 and 7 along the 1La trace, while peak 11 along the 1Lb displays a similar behavior and provides, together with state 7, the most intense and characteristic signals of guanine in the Vis window. Peak 14 along the 1Lb trace appears to lose intensity upon active space enlargement and is pushed beyond the Vis window and completely obscured in the UV range in the reference. It is worth noting that, as opposed to what has been found so far for the remaining nucleobases [23, 24], the leading CSFs of the signals featured in this window feature predominantly singly excited character (see Table 1), which may facilitate its description with computationally cheaper approaches such as those related to linear or quadratic response theories [62, 63].
Moving onto NUV-pump/NUV-probe spectra (see Fig. 3), larger differences can be discerned: As previously observed [23, 24], higher-lying electronic excited states seem to oscillate more pronouncedly compared to those found in the lower-lying Vis window. Besides those arising from GSB (peaks 2 and 3 for 1La and 1Lb, respectively), two signals feature in this window with sizable intensity, i.e., the off-diagonal GSB negative (blue) signals that are expected to appear when two or more bright states fall under the envelope of the utilized pump pulse. The off-diagonal bleach is thus a characteristic fingerprint of the 2D-NUV-probe spectrum of purine nucleobases [24], and it can be considered to individuate guanine traces along Ω1 in 2DUV spectra of multimeric systems. Along the 1La trace, signal 19 featuring pronounced doubly excited character is placed ~ 38,000 cm−1 in CAS(0,0|14,11|0,0) and it is systematically blue-shifted to ~ 40,000 cm−1 in RAS(0,0|14,11|2,8), RAS(0,0|14,11|2,6) displaying a strong overestimation, placing the signal at 43,000 cm−1. This large overestimation appears due to an artificial mixing with a close-lying CAS state, which strongly affects its energetic position upon the subsequent perturbation theory treatment; a more in-depth explanation may be found in [34]. Along the 1Lb trace, signal 17 is placed at ~ 31,000 cm−1 at CAS(0,0|14,11|0,0), moving up to ~ 34,000 cm−1 for the reference RAS(0,0|14,11|2,8). It is worth noting that this signal features a pronounced doubly excited HOMO to LUMO component, being suggested as a monomer-specific fingerprint for other nucleobases in the Vis probing window, while appearing in the UV in the case of guanine. As it was previously discussed for the pump-UV/probe-Vis spectra, CAS(0,0|14,11|0,0) overestimates the intensity of some signals, as can be seen for peaks 18 and 19 along the 1Lb trace (Fig. 3), which vanish upon active space enlargement.
3.2 Cost-effective approaches to 2DES
While reference RAS(0,0|14,11|2,8) results provide the highest accuracy available for the high-lying excited-state manifold, its routine use in applications is limited due to its exceedingly elevated cost, featuring well over 5 million CSFs and 12 million Slater determinants in its configuration interaction (CI) expansion. This elevated cost, despite being affordable within a single gas-phase molecule exploiting Cs symmetry, becomes unaffordable for (more interesting) dimeric and multimeric systems. To reduce costs, two different approaches have been followed: the use of artificial level shifts and of larger restricted active space schemes.
In a series of benchmarks on aromatic systems [24, 64, 65], we have documented that the RASPT2 protocol using schemes with small active spaces or featuring a moderate number of CSFs can dramatically underestimate transition energies as it overestimates the dynamic correlation of ionic (in valence bond terms) states. In the search for a cost-effective protocol to counteract (or at least damp) this effect, we observe that real and imaginary shift parameters (originally introduced to solve intruder state problem) invoke a non-uniform decrease of the correlation contribution, much more pronounced for ionic than for covalent states. This makes the use of the imaginary level shift well suited for improving performances of minimal RAS schemes toward characterization of highly excited states, even if we need to resort to larger shift values than suggested in the literature (a detailed argumentation can be found in Ref. [65]). We emphasize that the application of the large shift parameters makes sense only in the framework of semiempirical parameterization against a reliable reference data set and its use is otherwise discouraged.
Figure 4 shows the NUV-pump/Vis-probe spectra (lower panels) for the reduced RAS(4,7|0,0|4,4) approach with different imaginary level shifts. The less-expensive RAS(4,7|0,0|4,3), featuring one less π* orbital in the active space, displays much larger deviations with respect to the reference as opposed to what was observed in adenine [24]. As can be seen in Fig. 4, upon increase in the imaginary level shift, the energies along Ω1 are progressively blue-shifted, 1La and 1Lb traces approaching the RAS(0,0|14,11|2,8) reference for IMAG values of 0.5 (Fig. 4d), and being slightly blue-shifted with respect to the reference for the 0.6 value (Fig. 4e). The same trend can be observed along Ω3 in the Vis window (lower panels) with a couple exceptions, the splitting between signals 10 and 11 along the 1La trace, which is not reproduced at the RAS(4,7|0,0|4,4) approach regardless of the imaginary level shift employed, and the position of peak 7 along the 1Lb trace, which is shown to be extremely red-shifted within this cost-effective approach. The red-shifted signal 14 along 1Lb trace is pushed to the NUV window along Ω3 for larger IMAG (Fig. 4d, e), in agreement with reference values. As it has also been documented for other canonical nucleobases [23, 24], the deviations from reference computations in cheaper RAS approaches increase in the NUV-probe window (upper panels in Fig. 4), particularly for signal 17 along the 1Lb trace, which is the main fingerprint for this state in the NUV range and that is shown to still be within ~ 2000 cm−1 of the reference, while signals 18 and 19 appear in the right energetic position but displaying an artificially enhanced intensity in the reduced RAS approaches.

Two-dimensional NUV-pump/Vis-probe (lower panels) and NUV-pump/NUV-probe (upper panels) spectra of guanine in gas phase for a RAS(4,7|0,0|4,4) approach with different imaginary level shifts: a 0.2, b 0.3, c 0.4, d 0.5, e 0.6, and f reference RAS(0,0|14,11|2,8) values. Dashed lines represent positions of the 1La and 1Lb traces along Ω1 in RAS(0,0|14,11|2,8) reference computations for the sake of visual aid
Whereas all previous estimates shown above rely on a RAS(4,7|0,0|4,4) approach, an arguably more robust and correct way to increase the correlation in the model would be to extend its flexibility in terms of the holes and electrons accessible to the RAS1 and RAS3 subspaces, respectively. A systematic increase in the number of holes and electrons accessible within the RAS1/RAS3 subspaces was thus considered, incrementing symmetrically the number of holes and electrons one by one, up to eight, while retaining the imaginary level shift to a standard value of 0.2. As can be seen in Fig. 5 (lower panels), negligible gains along Ω3 can be observed in the NUV-pump/Vis-probe by going from four to five electrons/holes, from there onward being fully converged and showing no appreciable differences among the different RAS schemes considered besides an increase in the computational time, thus supporting the use of the RAS(4,7|0,0|4,4) for large systems. All estimates appear red-shifted with respect to the reference along Ω1, and remaining red-shifted regardless of the number of holes/electrons included in the active space, showing the strong active space dependence as discussed in the previous section. Moving to the higher energy NUV-pump/NUV-probe window (Fig. 5 upper panels), we observe lesser differences as those displayed in Fig. 4: The overall spectra seem analogous except for the intensity displayed by peak 19 along the 1La trace, which is obscured upon active space enlargement and in agreement with the reference computations, thus showing an overestimation on the transition dipole moments for this state in the smaller RAS approaches. Overall, we observe very little gains in moving to RAS approaches containing a larger number of electrons/holes, and also displaying an elevated cost with respect to the RAS(4,7|0,0|4,4) initially considered, which is here shown to be good enough for a qualitative description of the spectra and to be used for dimeric or multimeric species.

To-dimensional NUV-pump/Vis-probe (lower panels) and NUV-pump/NUV-probe (upper panels) spectra of guanine in gas phase for a RAS(4,7|0,0|4,4), b RAS(5,7|0,0|5,4), c RAS(6,7|0,0|6,4), d RAS(7,7|0,0|7,4), e RAS(8,7|0,0|8,4) schemes, and f RAS(0,0|14,11|2,8) reference values. Dashed lines represent positions of the 1La and 1Lb traces along Ω1 in RAS(0,0|14,11|2,8) reference computations for the sake of visual aid
4 Conclusions
We report a thorough assessment of the electronic high-lying singlet excited-state manifold of canonical purine nucleobase guanine in vacuo at the multiconfigurational wave function RASSCF/RASPT2 level. This approach allows us to obtain accurate estimates for transition energies and their associated dipole moments, which are then employed to simulate the two-dimensional NUV-pump/Vis-probe and NUV-pump/NUV-probe spectra of guanine for waiting times t2 = 0. The nonlinear electronic spectra show several ESA signals that may represent the particular fingerprint for this DNA/RNA nucleobase. As has been shown for other nucleobases [23, 24], the NUV-pump/Vis-probe spectral window features the clearest ESA fingerprints to be employed in order to discern among the different bases, for clearly tracking their monomer-specific decays and separating their contributions to those arising due to intermolecular interactions in complex dimeric/multimeric species. As expected, guanine displays similar signals to the other purine nucleobase, adenine [24], being slightly displaced along both pump and probe frequencies and displaying an analogous dependence on the dynamic correlation added, which blue-shifts both 1La and 1Lb signals along Ω1 upon its increase, while showing its main signal of partial doubly excited HOMO to LUMO character in the UV range along Ω3 at difference with the other nucleobases previously considered. Several reduced RAS schemes featuring all active orbitals in the RAS1/RAS3 subspaces to reduce costs were benchmarked, showing negligible gains over the inclusion of more than four holes/electrons in the active space, while a judicious use of the imaginary level shift is shown to artificially blue-shift the excitation energies moving them closer to the RAS(0,0|14,11|2,8) reference and thus making this approach applicable to larger dimeric and oligomeric systems.
References
Mukamel S (2000) Multidimensional femtosecond correlation spectroscopies of electronic and vibrational excitations. Annu Rev Phys Chem 51(1):691–729
Mukamel S (1995) Principles of nonlinear optical spectroscopy. O.U.P, New York
Scholes GD, Fleming GR, Chen LX, Aspuru-Guzik A, Buchleitner A, Coker DF, Engel GS, van Grondelle R, Ishizaki A, Jonas DM, Lundeen JS, McCusker JK, Mukamel S, Ogilvie JP, Olaya-Castro A, Ratner MA, Spano FC, Whaley KB, Zhu X (2017) Using coherence to enhance function in chemical and biophysical systems. Nature 543(7647):647–656
Scholes GD (2014) Extreme cross-peak 2d spectroscopy. Proc Natl Acad Sci 111(28):10031–10032
Prokhorenko VI, Picchiotti A, Pola M, Dijkstra AG, Miller RJD (2016) New insights into the photophysics of DNA nucleobases. J Phys Chem Lett 7(22):4445–4450
Halpin A, JohnsonPhilip JM, Tempelaar R, Murphy RS, Knoester J, JansenThomas LC, Miller RJD (2014) Two-dimensional spectroscopy of a molecular dimer unveils the effects of vibronic coupling on exciton coherences. Nat Chem 6(3):196–201
Oliver TAA, Lewis NHC, Fleming GR (2014) Correlating the motion of electrons and nuclei with two-dimensional electronic–vibrational spectroscopy. Proc Natl Acad Sci 111(28):10061–10066
Bakulin AA, Morgan SE, Kehoe TB, Wilson MWB, Chin AW, Zigmantas D, Egorova D, Rao A (2016) Real-time observation of multiexcitonic states in ultrafast singlet fission using coherent 2d electronic spectroscopy. Nat Chem 8(1):16–23
Jonas DM (2003) Two-dimensional femtosecond spectroscopy. Annu Rev Phys Chem 54(1):425–463
Polli D, Rivalta I, Nenov A, Weingart O, Garavelli M, Cerullo G (2015) Tracking the primary photoconversion events in rhodopsins by ultrafast optical spectroscopy. Photochem Photobiol Sci 14:213–228
Rivalta I, Nenov A, Garavelli M (2014) Modelling retinal chromophores photoisomerization: from minimal models in vacuo to ultimate bidimensional spectroscopy in rhodopsins. Phys Chem Chem Phys 16(32):16865–16879
C-h Tseng, Sándor P, Kotur M, Weinacht T, Matsika S (2012) Two-dimensional fourier transform spectroscopy of adenine and uracil using shaped ultrafast laser pulses in the deep uv. J Phys Chem A 116:2654–2661
West BA, Moran AM (2012) Two-dimensional electronic spectroscopy in the ultraviolet wavelength range. J Phys Chem Lett 3(18):2575–2581
Brańczyk AM, Turner DB, Scholes GD (2014) Crossing disciplines—a view on two-dimensional optical spectroscopy. Ann Phys 526(1–2):31–49
Egorova D (2008) Detection of electronic and vibrational coherences in molecular systems by 2d electronic photon echo spectroscopy. Chem Phys 347(1–3):166–176
Egorova D, Gelin MF, Domcke W (2007) Analysis of cross peaks in two-dimensional electronic photon-echo spectroscopy for simple models with vibrations and dissipation. J Chem Phys 126(7):074314
Yuen-Zhou J, Aspuru-Guzik A (2011) Quantum process tomography of excitonic dimers from two-dimensional electronic spectroscopy. I. General theory and application to homodimers. J Chem Phys 134(13):134505
Jiang J, Mukamel S (2011) Two-dimensional near-ultraviolet spectroscopy of aromatic residues in amyloid fibrils: a first principles study. Phys Chem Chem Phys 13(6):2394–2400
Jiang J, Abramavicius D, Bulheller BM, Hirst JD, Mukamel S (2010) Ultraviolet spectroscopy of protein backbone transitions in aqueous solution: combined qm and mm simulations. J Phys Chem B 114(24):8270–8277
Rivalta I, Nenov A, Cerullo G, Mukamel S, Garavelli M (2014) Ab initio simulations of two-dimensional electronic spectra: the sos//qm/mm approach. Int J Quantum Chem 114(2):85–93
Nenov A, Beccara S, Rivalta I, Cerullo G, Mukamel S, Garavelli M (2014) Tracking conformational dynamics of polypeptides by nonlinear electronic spectroscopy of aromatic residues: a first-principles simulation study. ChemPhysChem 15(15):3282–3290
Nenov A, Rivalta I, Cerullo G, Mukamel S, Garavelli M (2014) Disentangling peptide configurations via two-dimensional electronic spectroscopy: ab initio simulations beyond the Frenkel exciton hamiltonian. J Phys Chem Lett 5:767–771
Giussani A, Segarra-Martí J, Nenov A, Rivalta I, Tolomelli A, Mukamel S, Garavelli M (2016) Spectroscopic fingerprints of DNA/rna pyrimidine nucleobases in third-order nonlinear electronic spectra. Theor Chem Acc 135(5):1–18
Nenov A, Giussani A, Segarra-Martí J, Jaiswal VK, Rivalta I, Cerullo G, Mukamel S, Garavelli M (2015) Modeling the high-energy electronic state manifold of adenine: calibration for nonlinear electronic spectroscopy. J Chem Phys 142(21):212443
Li Q, Giussani A, Segarra-Martí J, Nenov A, Rivalta I, Voityuk AA, Mukamel S, Roca-Sanjuán D, Garavelli M, Blancafort L (2016) Multiple decay mechanisms and 2d-uv spectroscopic fingerprints of singlet excited solvated adenine-uracil monophosphate. Chem Eur J 22(22):7497–7507
Nenov A, Segarra-Martí J, Giussani A, Conti I, Rivalta I, Dumont E, Jaiswal VK, Altavilla SF, Mukamel S, Garavelli M (2015) Fd 177: probing deactivation pathways of DNA nucleobases by two-dimensional electronic spectroscopy: first principles simulations. Faraday Discuss 177:345–362
Segarra-Martí J, Jaiswal VK, Pepino AJ, Giussani A, Nenov A, Mukamel S, Garavelli M, Rivalta I (2017) Two-dimensional electronic spectroscopy as a tool for tracking molecular conformations in DNA/rna aggregates. Faraday DiscussDOI. https://doi.org/10.1039/C1037FD00201G
Roos BO (1987) The complete active space self-consistent field method and its applications in electronic structure calculations. In: Advances in chemical physics. Wiley, pp 399–445. https://doi.org/10.1002/9780470142943.ch7
Andersson K, Malmqvist PA, Roos BO (1992) 2nd-order perturbation-theory with a complete active space self-consistent field reference function. J Chem Phys 96(2):1218–1226
Roca-Sanjuán D, Aquilante F, Lindh R (2012) Multiconfiguration second-order perturbation theory approach to strong electron correlation in chemistry and photochemistry. Wiley Interdiscip Rev Comput Mol Sci 2(4):585–603
Rubio M, Serrano-Andrés L, Merchán M (2008) Excited states of the water molecule: analysis of the valence and Rydberg character. J Chem Phys 128:104305
Nenov A, Rivalta I, Mukamel S, Garavelli M (2014) Bidimensional electronic spectroscopy on indole in gas phase and in water from first principles. Comput Theor Chem 1040–1041:295–303
Segarra-Martí J, Garavelli M, Aquilante F (2015) Multiconfigurational second-order perturbation theory with frozen natural orbitals extended to the treatment of photochemical problems. J Chem Theory Comput 11(8):3772–3784
Serrano-Andrés L, Merchán M, Lindh R (2005) Computation of conical intersections by using perturbation techniques. J Chem Phys 122:104107
Roos BO, Lindh R, Malmqvist PA, Veryazov V, Widmark PO (2016) Multiconfigurational quantum chemistry. Wiley, Hoboken
Malmqvist PA, Rendell A, Roos BO (1990) The restricted active space self-consistent-field method, implemented with a split graph unitary-group approach. J Phys Chem 94(14):5477–5482
Andersson K, Malmqvist P-Å, Roos BO (1992) 2nd-order perturbation-theory with a complete active space self-consistent field reference function. J Chem Phys 96:1218–1226
Roos BO, Andersson K, Fulscher MP, Malmqvist PA, Serrano-Andrés L, Pierloot K, Merchán M (1996) Multiconfigurational perturbation theory: applications in electronic spectroscopy. Adv Chem Phys 93:219–331
Aquilante F, Autschbach J, Carlson R, Chibotaru L, Delcey MG, De Vico L, Fernández Galvan I, Ferré N, Frutos LM, Gagliardi L, Garavelli M, Giussani A, Hoyer C, Li Manni G, Lischka H, Ma D, Malmqvist PA, Müller T, Nenov A, Olivucci M, Pedersen TB, Peng D, Plasser F, Pritchard B, Reiher M, Rivalta I, Schapiro I, Segarra-Martí J, Stenrup M, Truhlar DG, Ungur L, Valentini A, Vancoillie S, Veryazov V, Vysotskiy V, Weingart O, Zapata F, Lindh R (2016) Molcas 8: new capabilities for multiconfigurational quantum chemical calculations across the periodic table. J Comput Chem 37(5):506–541
Lorentzon J, Malmqvist P-Å, Fülscher M, Roos BO (1995) A caspt2 study of the valence and lowest Rydberg electronic states of benzene and phenol. Theor Chim Acta 91(1):91–108
Giussani A, Segarra-Martí J, Roca-Sanjuán D, Merchán M (2015) Excitation of nucleobases from a computational perspective I: reaction paths. In: Barbatti M, Borin AC, Ullrich S (eds) Photoinduced phenomena in nucleic acids I, vol 355. Topics in current chemistry. Springer, New York, pp 57–97. https://doi.org/10.1007/128_2013_501
Altavilla SF, Segarra-martí J, Nenov A, Conti I, Rivalta I, Garavelli M (2015) Deciphering the photochemical mechanisms describing the uv-induced processes occurring in solvated guanine monophosphate deciphering the photochemical mechanisms describing the uv-induced processes occurring in solvated guanine monophosphate. Front Chem 3:29
Improta R, Santoro F, Blancafort L (2016) Quantum mechanical studies on the photophysics and the photochemistry of nucleic acids and nucleobases. Chem Rev 116(6):3540–3593
Pepino AJ, Segarra-Martí J, Nenov A, Improta R, Garavelli M (2017) Resolving ultrafast photoinduced deactivations in water-solvated pyrimidine nucleosides. J Phys Chem Lett 8(8):1777–1783
Forsberg N, Malmqvist PA (1997) Multiconfiguration perturbation theory with imaginary level shift. Chem Phys Lett 274(1–3):196–204
Zobel JP, Nogueira JJ, Gonzalez L (2017) The IPEA dilemma in CASPT2. Chem Sci 8:1482–1499
Ghigo G, Roos BO, Malmqvist P-Å (2004) A modified definition of the zeroth-order hamiltonian in multiconfigurational perturbation theory (caspt2). Chem Phys Lett 396(1–3):142–149
Malmqvist P-Å, Roos BO (1989) The casscf state interaction method. Chem Phys Lett 155(2):189–194
Malmqvist PÅ, Roos BO, Schimmelpfennig B (2002) The restricted active space (ras) state interaction approach with spin–orbit coupling. Chem Phys Lett 357(3–4):230–240
Aquilante F, Lindh R, Bondo Pedersen T (2007) Unbiased auxiliary basis sets for accurate two-electron integral approximations. J Chem Phys 127(11):114107
Aquilante F, Pedersen TB, Lindh R (2007) Low-cost evaluation of the exchange Fock matrix from Cholesky and density fitting representations of the electron repulsion integrals. J Chem Phys 126(19):194106
Bostrom J, Delcey MG, Aquilante F, Serrano-Andres L, Pedersen TB, Lindh R (2010) Calibration of Cholesky auxiliary basis sets for multiconfigurational perturbation theory calculations of excitation energies. J Chem Theory Comput 6(3):747–754
Abramavicius D, Palmieri B, Voronine DV, Šanda F, Mukamel S (2009) Coherent multidimensional optical spectroscopy of excitons in molecular aggregates; quasiparticle versus supermolecule perspectives. Chem Rev 109(6):2350–2408
Mukamel S, Oszwaldowski R, Abramavicius D (2007) Sum-over-states versus quasiparticle pictures of coherent correlation spectroscopy of excitons in semiconductors: femtosecond analogs of multidimensional nmr. Phys Rev B 75(24):245305
Tempelaar R, van der Vegte CP, Knoester J, Jansen TLC (2013) Surface hopping modeling of two-dimensional spectra. J Chem Phys 138(16):164106
Nenov A, Giussani A, Fingerhut BP, Rivalta I, Dumont E, Mukamel S, Garavelli M (2015) Spectral lineshape in nonlinear electronic spectroscopy †. Phys Chem Chem Phys 17:30925–30936
Richter M, Fingerhut BP (2016) Simulation of multi-dimensional signals in the optical domain: quantum-classical feedback in nonlinear exciton propagation. J Chem Theory Comput 12(7):3284–3294
van der Vegte CP, Dijkstra AG, Knoester J, Jansen TLC (2013) Calculating two-dimensional spectra with the mixed quantum-classical ehrenfest method. J Phys Chem A 117(29):5970–5980
NIST Chemistry WebBook (2017). http://webbook.nist.gov/cgi/inchi/InChI%3D1S/C5H5N5O/c6-5-9-3-2(4(11)10-5)7-1-8-3/h1H%2C(H4%2C6%2C7%2C8%2C9%2C10%2C11). Accessed 20 July 2017
Szalay PG, Watson T, Perera A, Lotrich VF, Bartlett RJ (2012) Benchmark studies on the building blocks of DNA. 1. Superiority of coupled cluster methods in describing the excited states of nucleobases in the Franck–Condon region. J Phys Chem A 116(25):6702–6710
Fülscher MP, Serrano-Andrés L, Roos BO (1997) A theoretical study of the electronic spectra of adenine and guanine. J Am Chem Soc 119(26):6168–6176
Elliott P, Goldson S, Canahui C, Maitra NT (2011) Perspectives on double-excitations in tddft. Chem Phys 391(1):110–119
Gritsenko OV, Jan Baerends E (2009) Double excitation effect in non-adiabatic time-dependent density functional theory with an analytic construction of the exchange-correlation kernel in the common energy denominator approximation. Phys Chem Chem Phys 11(22):4640–4646
Nenov A, Mukamel S, Garavelli M, Rivalta I (2015) Two-dimensional electronic spectroscopy of benzene, phenol, and their dimer: an efficient first-principles simulation protocol. J Chem Theory Comput 11(8):3755–3771
Giussani A, Marcheselli J, Mukamel S, Garavelli M, Nenov A (2017) On the simulation of two-dimensional electronic spectroscopy of indole-containing peptides. Photochem Photobiol 93:1368–1380
Acknowledgments
MG acknowledges support from the European Research Council Advanced Grant STRATUS (ERC-2011-AdG No. 291198). IR acknowledges support from the French Agence National de la Recherche (FEMTO-2DNA, ANR-15-CE29-0010). SM gratefully acknowledges the support from the National Science Foundation (Grant CHE-1663822), and of the Chemical Sciences, Geosciences, and Biosciences division, Office of Basic Energy Sciences, Office of Science, US Department of Energy, through Award No. DE-FG02-04ER15571.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Published as part of the special collection of articles “Festschrift in honour of A. Rizzo.”
Rights and permissions
About this article

Cite this article
Segarra-Martí, J., Pepino, A.J., Nenov, A. et al. The highly excited-state manifold of guanine: calibration for nonlinear electronic spectroscopy simulations. Theor Chem Acc 137, 47 (2018). https://doi.org/10.1007/s00214-018-2225-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-018-2225-0