
Abstract
In order to assess the potential role of a single water molecule as a catalyst, the gas-phase oxidation reaction of hydroxyl radical with glyoxylic acid has been investigated by the means of quantum mechanical computations using CCSD(T), MP2 and DFT methods. The pre-reaction complexes along the oxidation pathways are systematically explored through a global reaction route mapping method. The computations reveal that a single water molecule stabilizes the pre-reaction complexes as well as respective transition states, resulting in the lowering of the energy barrier, however, the transition state theory computed rate constants for the water-catalysed pathways are found to be an order of magnitude lower than that for the water-free pathways. Notably, the abstraction of formyl hydrogen is observed to be most favourable both in the presence and absence of a single water molecule. Besides these, a couple of triple-proton exchange pathways involving simultaneous proton transfer between the glyoxylic acid, OH radical and a single water molecule are also explored. All the pathways are observed to proceed through conventional free radical mechanism when analysed using BHandHLYP exchange–correlation (XC) functional of DFT. However, DFT method using other XC functionals revealed that one of the relevant acidic H-abstraction pathways proceeds through a proton-coupled electron-transfer mechanism and also results in the dissociation of glyoxylic acid. The pathways for trans form of glyoxylic acid has also been compared with those explored for the cis form. The standard Gibbs free-energy profiles for the reactions studied indicate that the hydrogen abstraction, particularly in trans-glyoxylic acid, may be more feasible than the dissociation, particularly at lower temperatures. This study may assist future investigation of similar atmospheric reactions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Glyoxylic acid is a natural component of plants [1], and is also a metabolite in the biochemical pathways in mammals [2]. It is commonly used as a cleaning agent, a chemical and biodegradable copolymer feedstock in industries [3] and as a constituent in cosmetics [4], thereby releasing to the environment through various waste streams. Glyoxylic acid is also a secondary organic compound formed in clouds by photochemical oxidation reactions of isoprene, alkene and aromatic compounds, where it is further oxidized by the OH radical, thereby participating in cloud processing [5, 6]. The reaction of OH radical with polar molecules has also been a topic of considerable interest owing to the impact of hydrogen-bonded molecular complexes on the kinetics and dynamics of gas-phase free radical reactions [7–10]. The pre-reactive intermediates in these reactions significantly affect the kinetics, structural and reactive behaviour [11–24].
Recently [11–21], various theoretical and experimental studies had demonstrated the catalytic effect of a single water molecule on the reactions involving OH radical. For example, gas-phase single water catalysis is proposed to promote the oxidation reaction of acetaldehyde by hydroxyl radical [11]. It should be noted that water not only influences the reaction mechanism but may also decreases the rate constant which has been observed in the hydrogen abstraction reactions of the CH4 [17] and of HNO3 with the OH radical [18]. We had recently explored the water catalysis in the oxidation reaction of thioformic acid (TFA) [19] and dithioformic acid (DTFA) with OH radical [20]. The water-catalysed reaction pathways were predicted through a systematic and automated exploration of pre-reaction complexes, transition states (TSs) and product structures, using a global reaction route mapping (GRRM) method [15, 25–31]. It was found that the water-catalysed oxidation was thermodynamically as well as kinetically more feasible than the non-catalytic oxidation. Similar results were also observed in our previous works exploring the pathways for water-migration in the water complexes of TFA [32] and l-proline [33], leading to isomerization in the latter. However, in our recent work [34], exploring the detailed mechanism and kinetics of hydrogen abstraction pathways in the gas-phase reaction of glycolaldehyde with OH radical, it was found that a single water molecule does not accelerate the reaction under tropospheric conditions, though it facilitates cis–trans interconversion in the glycolaldehyde during the reaction pathways which otherwise is infeasible in the absence of water.
In the present work, to the best of our knowledge, a detailed investigation on the mechanism of hydrogen abstraction in the gas-phase reaction of glyoxylic acid with hydroxyl radical, with and without a single water molecule, is being investigated for the first time. The hydrogen abstraction pathways are systematically explored through the GRRM method, which employs an anharmonic downward distortion (ADD) following approach to map the reaction routes. GRRM method is highly efficient in searching the pre-reaction complexes of water-catalysed reactions. The paper is organized as follows: The next section describes the computational details employed for exploring the oxidation pathways of glyoxylic acid in the absence and presence of a single water molecule, which is followed by the results and discussion, presenting a detailed mechanism for the hydrogen abstraction reaction. Finally, the last section makes a few concluding remarks.
2 Computational details
For the reaction of OH radical with glyoxylic acid in the absence as well as in the presence of a single water molecule, the binary and ternary pre-reaction complexes, depicted, respectively, as Bs and Cs, in Figs. 1, 2 and 3, are explored through GRRM using a large-ADD-following method at the DFT/BHandHLYP/6-311++G(d,p) level of the theory using Becke-half-and-half-Lee–Yang–Parr (BHandHLYP) [35–38] exchange–correlation (XC) functional of the density functional theory (DFT) which is known to exhibit reliable performance and accuracy for the hydrogen-bonded complexes [39]. The lower regions of potential energy surfaces of the reaction systems were searched employing five largest ADDs around 10 random structures in the case of binary pre-reaction complexes for the reaction in the absence of water and 40 random structures in the case of ternary pre-reaction complexes for the reaction in the presence of a single water molecule. It should be noted that two of the pre-reaction complexes were found intuitively followed by a minimization through the GRRM. The binary complexes, [glyoxylic acid···(H2O)] and [(H2O)···(OH)], as depicted in Figs. 1, 2 and 3, were optimized by removing the OH and glyoxylic acid, respectively, from the ternary pre-reaction complexes. All the transition state (TS) structures were obtained intuitively and by applying saddle point optimization [31] through the GRRM. Note that GRRM using ADDs has been used only to explore the pre-reaction complexes (PRCs). The transition states are intuitively guessed from the PRCs explored, which were further optimized using saddle option followed by intrinsic reaction coordinate (IRC) [40, 41] calculations through the GRRM program. Further, in order to test if the optimized geometry obtained is a minimum or a transition state, and to obtain the zero-point energy (ZPE) correction, harmonic vibrational frequency analysis was performed at the same level of the theory. The connectivity between the reactants and products was further checked using the IRC computations on the obtained transition states. The single-point energies were further refined employing coupled cluster theory [42] at the single-point level of CCSD(T)/6-311++G(d,p)//BHandHLYP/6-311++G(d,p).

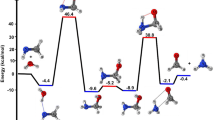
Non-catalytic (water-free) oxidation pathways I–II for the formyl hydrogen abstraction, and III for the acidic hydrogen abstraction in trans-glyoxylic acid. The values, depicted in bold below the structures, refers to the ZPE and BSSE corrected relative energies ΔE (in kcal/mol) at 0 K with respect to the isolated reactants A1, computed at the CCSD(T)/6-311++G(d,p)//BHandHLYP/6-311++G(d,p) level of the theory (for cis-glyoxylic acid, see supporting information Figure S3)


Same as Fig. 1, but for the oxidation pathways involving water-catalysed hydrogen abstraction of: a formyl hydrogen, b acidic hydrogen, starting from the binary pre-reaction complexes of trans-glyoxylic acid with a single water molecule. The relative energies depicted in bold are with respect to the isolated reactants A2 (for cis-glyoxylic acid, see supporting information Figure S4)


Same as Fig. 2, but for the oxidation pathways involving water-catalysed hydrogen abstraction of: a formyl hydrogen, b acidic hydrogen in trans-glyoxylic acid, starting from the binary pre-reaction complexes of hydroxyl radical with a single water molecule (for cis-glyoxylic acid, see supporting information Figure S5)
The geometries of relevant TSs obtained at the DFT/BHandHLYP/6-311++G(d,p) level of the theory were further optimized, and the IRC paths were obtained employing frozen-core (FC) Møller-Plesset (MP) perturbation theory [43] at the MP2(FC)/6-311++G(d,p) level of the theory, and single-point energies were refined at the CCSD(T)/6-311++G(2d,2p)//MP2(FC)/6-311++G(2d,2p) level of the theory for a comparative study. The energies have also been corrected for the basis set superposition error (BSSE) using the counterpoise (CP) method of Boys and Bernardi [44]. In order to check the effect of basis set in the CCSD(T) computations, the single-point energy values were also calculated using cc-pVTZ basis set at the CCSD(T)/cc-pVTZ//BHandHLYP/6-311++G(d,p) level of the theory for two pathways explored in the present work. Further, in order to confirm the reaction pathways traced at the level of DFT/BHandHLYP/6-311++G(d,p), the relevant transition states, along the important pathways discussed later, were optimized and IRC computations were also carried using hybrid B3LYP [37, 38] and dispersion-corrected ω-B97XD [45] XC functionals of the DFT with basis set 6-311++G(d,p). In the present work, all the required computations for the GRRM program are performed along with Gaussian 03 and 09 quantum chemistry software [35, 36]. Note that two different versions of Gaussian software were deployed only due to their availability though any one may suffice for the computations presented in the present work. Further, to check the dependability of the theoretical approach applied with respect to a multi-reference character of the wave function at the stationary points, T1 diagnostics [46] were performed at the CCSD/6-311++G(d,p)//BHandHLYP/6-311++G(d,p) level of the theory. As evident in Table 1, the T1 diagnostic values were found to be less than 0.02, depicting the reliability of a single-reference method employed for the present investigation.
2.1 Reaction kinetics
For the non-catalytic oxidation (in the absence of water), the mechanism of H-abstraction process in glyoxylic acid can be described as,

where k 1 and k −1 are the rate constants for forward and backward reactions in first step, and k 2 is rate constant for the step involving formation of products. The pre-reaction binary complex formed can be assumed to be in pseudo-equilibrium with the reactants [13, 22–24], and applying the steady state approximation gives the total rate constant as [47],
where
Note that while arriving at Eq. (2) \(k_{ - 1}\) is assumed to be much greater than k 2 because entropy change in k −1 is much larger than that for the product formation due to a loose transition state involved along the pathway [14–22, 47].
For the catalytic oxidation (in the presence of water), the mechanism can be depicted as,

The rate constant for the water-catalysed reaction can then be given as,
where the equilibrium constants K weq0 and K weq1 correspond to the first two steps. For the present work, the rate constants are calculated using conventional transition state theory (TST) [48–50] taking account of reaction path degeneracy (σ). The equilibrium constant for the purpose is estimated as,
while k 2, the unimolecular rate constant for last step leading to the formation of products, is determined as,
where G PRC, G reactants, G TS are Gibbs free energies for pre-reaction complexes, reactants and transition states, respectively, whereas k B and h are the Boltzmann and Planck constants, T is the temperature (K). In the present work, the reaction path degeneracy is taken to be 1 since there is only one site for both acidic and formyl hydrogen abstraction.
3 Results and discussion
Our present work is mainly focused on the hydrogen abstraction reactions of trans-glyoxylic acid since it is found to be relatively more stable than the cis form, at the different levels of the theories employed, as had also been predicted in a previous study [51]. For example, trans-glyoxylic acid is 0.9 kcal/mol more stable than the cis form (in terms of relative energy ∆E including ZPE at 0 K) at the CCSD(T)/6-311++G(d,p)//BHandHLYP/6-311++G(d,p) level of the theory. However, the pathways involving cis-glyoxylic acid were also explored in the present work but are reported in supporting information (SI) Figures S3–S5, though these have been compared in the discussions below.
Various oxidative reaction pathways, identified for the reaction of trans-glyoxylic acid with OH radical, involve an abstraction of formyl hydrogen (C–H) or acidic hydrogen (O–H) as shown in Fig. 1 for the reaction in the absence of water and in Figs. 2 and 3 for that in the presence of a single water molecule. The schematic enthalpy change at 298.15 K, for the hydrogen abstraction pathways at the CCSD(T)/6-311++G(d,p)//BHandHLYP/6-311++G(d,p) level of the theory, is reported in Fig. 4, while Fig. 5 depicts the corresponding standard Gibbs free-energy profile (in terms of ΔG). Table 1 contains enthalpy change and Gibbs free-energy change computed relative to the sum of separated reactants, and T1 diagnostic values of various reaction species at the CCSD(T)//BHandHLYP level of the theory, while Table 3 gives enthalpy change at 298.15 K of a few relevant reaction species at CCSD(T)//MP2(FC), B3LYP and ω-B97XD levels of the theories. The spin density distribution determined from the natural bond orbital (NBO) [52] analysis of transition states is further provided in SI Table S2, whereas Table 2 lists the ΔG values relative to the isolated species at different temperatures for the dissociation reaction of pre-reaction complexes into the glyoxylic acid, OH and/or H2O. The rate constants calculated at 298.15 K, along various pathways of reaction of glyoxylic acid with hydroxyl radical, are depicted in Table 4 while SI Table S3 compares rate constants in temperature range of 273–400 K. Besides these, one reaction pathway (depicted in SI Figure S2) is explored in which the OH radical adds to the carbonyl carbon. The effect of basis set was also analysed by using cc-pVTZ basis set at the level of CCSD(T)/cc-pVTZ//BHandHLY/6-311++G(d,p) in Table 5. The pathways explored for trans-glyoxylic acid was also compared with that for cis-glyoxylic acid (as depicted in SI Figures S3–S7). A total of 9 pathways were explored for cis-glyoxylic acid in comparison with 12 pathways observed for trans form as analysed below.

Enthalpy change (ΔH at 298.15 K), in kcal/mol, including the corrections for ZPE and BSSE, with respect to separated reactants A1 and A2, for various reaction pathways depicted in Figs. 1, 2 and 3 for: a formyl H-abstraction, and b acidic H-abstraction, in trans-glyoxylic acid, at the CCSD(T)/6-311++(d,p)//BHandHLYP/6-311++G(d,p) level of the theory, in the presence and absence of a single water molecule (for cis-glyoxylic acid, see supporting information Figure S6)

Schematic standard Gibbs free energy (ΔG at 298.15 K), in kcal/mol for various reaction pathways: a formyl H-abstraction, and b acidic H-abstraction, in trans-glyoxylic acid, at the CCSD(T)/6-311++G(d,p)//BHandHLYP/6-311++G(d,p) level of the theory, in the presence and absence of a single water molecule (for cis-glyoxylic acid, see supporting information Figure S7)
3.1 Non-catalytic oxidation of trans-glyoxylic acid
In the oxidation reaction of trans-glyoxylic acid with OH radical, in water-free environment, three pathways were traced: path I and II involve formyl hydrogen abstraction, whereas path III involves acidic hydrogen-abstraction. Each reaction path explored begins with the formation of hydrogen-bonded pre-reaction complex before the transition state. Three minima and three TSs connecting them are obtained in the pre-reaction hydrogen-bonded complex region which finally leads to the production of a single water molecule along with (HOOC)CO or OOC(CHO) fragments. Furthermore, as evident in Fig. 1, the relative stability (in terms of ∆E including ZPE at 0 K in kcal/mol) of these three reaction complexes follows the order as: B3 (−3.9) > B2 (−1.8) > B1 (−1.6) at CCSD(T)/6-311++G(d,p)//BHandHLYP/6-311++G(d,p) level of theory. This can be attributed to the fact that B3 and B2 form a more stable six-membered planar ring as compared to five-membered ring in B1. Moreover, hydrogen bonding seems to be stronger in B3 than in B2. However, the relative energy (∆E including ZPE in kcal/mol) of governing TSs with respect to the sum of separated reactants follows as: TS2 (+3.7) < TS1 (+5.2) < TS3 (+12.0). Moreover, in TS 2, the oxygen atom of hydroxyl radical approaches towards the formyl hydrogen atom from top of the glyoxylic acid molecule. The energetics in Table 1 and Figs. 4 and 5 suggest that though the pre-reaction complex B2 is more stable, but the formyl hydrogen abstraction along path II is more favourable since enthalpy change and standard Gibbs free-energy change at 298.15 K of the governing TS and products are lowest with a barrier height of only 1.2 and 6.59 kcal/mol, respectively, at CCSD(T)/6-311++G(d,p)//BHandHLYP/6-311++G(d,p) level of the theory. Moreover, the rate constant for path II was found to be highest among water-free pathways. All the three pathways undergo hydrogen atom abstraction via conventional radical mechanism since OH radical in the transition states is oriented in the plane of glyoxylic acid, as also can be seen from the spin density analysis in Table S2 which demonstrates that the unpaired electron is shared between atoms C1 and O4 along path I and II, and between O1 and O4 atoms along path III forming 3c–3e bonds which causes triplet repulsions. Moreover, in our previous study [19] on the reaction of thiol form of thioformic acid (TFA) with OH radical, formyl hydrogen abstraction was observed to be kinetically more favourable, while acidic hydrogen abstraction is observed to be thermodynamically more feasible. In our other study [20] involving the reaction of dithioformic acid (DTFA) with hydroxyl radical, inverse results were observed demonstrating acidic H-abstraction to be more favourable and the pathways followed a proton-coupled electron-transfer (PCET) mechanism [55].
3.2 Comparison with cis-glyoxylic acid
In the case of cis-glyoxylic acid, for water-free oxidation, only two pathways could be traced as displayed in Figure S2. Path Ia and IIa of cis-glyoxylic acid resembles the aforementioned path II and III of trans-glyoxylic acid for formyl and acidic H-abstraction, respectively. For cis-isomer also, the formyl H-abstraction is observed to be more favourable since barrier height for the formyl abstraction is quite low in terms of both ∆H and ∆G at 298.15 K, as depicted in SI Figure S5 and S6. Notably, in the case of cis-isomer, the acidic H-abstraction leads to not only abstraction of hydrogen in glyoxylic acid but also to its dissociation resulting in HC=O, CO2 and H2O, as observed along path IIa.
3.3 Catalytic oxidation of trans-glyoxylic acid
The catalytic effect of a single water molecule on the title oxidation reaction was studied by taking into account that (a) glyoxylic acid can first form stable complex with water and then react with OH radical, (b) glyoxylic acid can form stable complex with the complex of OH and water: [H2O···HO] or [HOH···OH]. All these binary complexes including the [glyoxylic acid···(H2O)] complex and [(H2O)···(OH)] complex were found to lie lower in energy than the sum of separated reactants by 1.32–6.15 kcal/mol, at CCSD(T)/6-311++G(d,p)//BHandHLYP/6-311++G(d,p) level of the theory. Among all the binary complexes, B4 is the most stable due to the presence of six-membered ring between trans-glyoxylic acid and OH, while B5 and B6 forms five-membered and seven membered rings, respectively, causing ring strain. It should be noted that two of the [(H2O)···(OH)] complexes, B8 and B9, traced had also been reported previously [18, 28].
As depicted in Fig. 2, pre-reaction binary complex B4 reacts with OH radical to form ternary pre-reaction complexes: C1 along path IV and C2 along path V involving formyl hydrogen abstraction. Moreover, complexes B5, B7 and B6 interact with OH radical to form ternary complex C3 along path VI, C4 along path VII and C5 along path VIII, respectively, leading to acidic hydrogen abstraction. As evident in Fig. 2a, C2 is more stable than C1 due to formation of a bicyclic six-membered ring separated by O2–C2 bond forming a butterfly-like structure, while in C1, two rings are formed: one six-membered ring and other five-membered ring lying on the opposite side. It should be noted that the transition state TS5 along path V, which lays 5.3 kcal/mol higher in energy than the reaction complex C2, is the lowest-lying TS among all the explored TSs along water-catalysed reaction channels. Also, it lays 1.6 kcal/mol lower in energy than the second lowest-lying transition state TS4 along path IV involving formyl H-abstraction. It should be noted that the TSs along the various pathways explored do not directly lead to the products as evident from the IRC paths depicted in the SI Figure S7(a, b). From the paths, it is clear that the transition states TS4 and TS5 lead to the respective product structures, however, before passing to the latter, a few intermediates complexes, though with insignificant barriers, are also encountered.
Among the pre-reaction complexes C3–C5 along pathways VI–VIII, the stabilization effect of a single water molecule was found to be slightly more in C5 (along path VIII) than in C3 and C4 due to similar reason as explained above for the formyl H-abstraction. Moreover, the transition state TS7 along path VII lies lower than TS6 and TS8 by 0.9 and 1.9 kcal/mol, respectively, leading to acidic H-abstraction as depicted in Fig. 2b. Similar trends were seen for the standard Gibbs free energy. It is demonstrated that path VII is more feasible among pathways VI–VIII, since the barrier height to reach TS7 is lowest among TS6 and TS8.
Further, the interaction of complex B9 with trans-glyoxylic acid, leads to ternary pre-reaction complexes: C7 (along path X involving formyl H-abstraction), C8 and C9 (along path XI and XII involving acidic H-abstraction), however, complex B8 forms only one three-body pre-reaction complex, C6 along path IX involving formyl H- abstraction in trans-glyoxylic acid. Since water only interacts with the OH radical in C6, this ternary complex can also be considered to be formed by interaction of B2, [glyoxylic acid···(OH)] complex, with a single water molecule. As evident in Fig. 3a, complex C6 is only 0.1 kcal/mol less stable in energy than C7 while stability becomes vice versa in case of their governing transition states. Noticeably, C8 along path XI is the most stable complex among all the explored three-body pre-reaction complexes due to formation of strong H-bonded eight-membered ring, while C9 along path XII is a six-membered complex and the second lowest-lying, which leads to acidic H-abstraction. Interestingly, C9 type of complex has never been reported in the literature for similar reactions. Besides these, the acidic H-abstraction pathway having the lowest energy barrier occurs through TS11 which has eight-membered planar ring structure, as evident in Fig. 3b.
All the aforementioned results in terms of relative energies and ∆H suggest that the formyl H-abstraction via transition state TS5 along path V is the most favourable with the lowest barrier (relative energy of only 3.0 kcal/mol) among all the pathways traced in the present work. Moreover, the standard Gibbs free-energy profile, as depicted in Fig. 5, demonstrates that the pre-reaction complexes for acidic H-abstraction have similar stability as compared to products, but their governing TSs are high-lying, indicating that they are not likely to undergo acidic H-abstraction to form products. However, in the case of formyl H-abstraction, TSs are low-lying than that in the case of acidic H-abstraction, and the pre-reaction complexes are more likely to undergo H-abstraction. Also, as depicted in Table 4, the calculated rate constant (at 298.15 K) for path V was found to be highest among all water-catalysed pathways. All these pathways involve free radical mechanism, as evident from the spin density analysis (in Table S2) where the unpaired electron is observed to be shared by C and O atoms along the formyl H-abstraction pathway, whereas along the acidic H-abstraction pathway, it is shared between O-atom of trans-glyoxylic acid and O-atom of OH radical. It should be noted that in our previous study [19] on TFA(thiol), the formyl and acidic hydrogen abstractions in the presence of a single water molecule were found to be kinetically and thermodynamically more feasible, respectively, and follow free radical mechanism. However, in our study [20] on DTFA, the catalytic acidic H-abstraction is found to be more feasible and proposed to follow proton-coupled electron-transfer (PCET) mechanism.
From the GRRM search for pre-reaction complexes, besides the above-mentioned ternary pre-reaction complexes, a few other three-body complexes were also traced for the trans-glyoxylic acid. However, these complexes do not undergo direct hydrogen abstraction but are likely to undergo double- or triple-proton exchange involving hydrogen abstraction. For double-proton exchange, various attempts to locate the transition states were failed, however, as depicted in Figure S1, two pathways involving triple H-transfer could be traced where all the three reactants (OH, H2O and glyoxylic acid) are actively involved as a hydrogen donor or acceptor, leading to acidic hydrogen abstraction. The reaction crop up via a simultaneous hydrogen abstraction from the O–H group in trans-glyoxylic acid by the OH radical, while the hydrogen of the OH radical is transferred to the oxygen atom of water whose hydrogen atom gets attached to the oxygen atom of C=O or OH group in glyoxylic acid, as evident from transition states TS13 and TS14, respectively, in Figure S1. The products formed in these two transformations are geometric isomers of glyoxylic acid. These transformations are least feasible since governing TSs are high-lying. Moreover, as depicted in SI Figure S2, a pathway involving nucleophilic addition of hydroxyl radical to carbonyl carbon leading to dissociation into formic acid and COOH radical was also explored at the BHandHLYP/6-311++G(d,p) level of the theory. As can be seen from the relative energy values, this pathway is also highly probable.
On comparing the non-catalytic oxidation with water-catalysed oxidation of trans-glyoxylic acid by hydroxyl radical, it was noted that the catalytic oxidation seems to be more feasible whether analysis is performed in terms of relative energies and ∆H, because formation of hydrogen-bonded complexes with water leads to lowering of the potential energy surface. Similar observations were made in the previous studies on TFA and DTFA [19, 20]. For example, ∆H of lowest-lying transition state TS2 along water-free pathway is 5.1 kcal/mol more than the transition state TS5 along the water-catalysed pathway involving formyl H-abstraction. The products, (HOOC)CO or OOC(CHO), formed in the water-catalysed oxidation are also more stabilized due to hydrogen bonding by two water molecules than that in the case of water-free pathway where one water molecule is present. Contrary to this, the standard Gibbs free-energy profile at CCSD(T)/6-311++G(d,p)//BHandHLYP/6-311++G(d,p), as depicted in Fig. 5, demonstrates water-free pathways to be more feasible than water-catalysed pathways. This is further reflected in the higher rate constants observed for the water-free pathways when compared with that for the water-catalysed pathways. As evident from Fig. 5, the standard Gibbs free energy of activation (at 298.15 K) to reach the transition state (TS2) along the water-free pathway is relatively lower than that required for TS5 along the water-catalysed pathway, due to considerable lowering of entropy in water complexes. It should also be noted that there is strong preference for making the radical at the carbon atom in glyoxylic acid rather than over the oxygen atom, which may be due to lesser bond dissociation energy of C–H bond (337.2 kJ/mol) over O–H bond (428.0 kJ/mol), making C–H bond weaker. Also, the radical product [(HOOC)CO] formed due to formyl H-abstraction is more stable than [OOC(CHO)] resulting due to acidic H-abstraction. Notably, paths IV, V and IX lead to the same product (P4, P5 or P9) passing through various intermediates as depicted in IRC paths of Figure S7(a–c).
Further, to check the strength of pre-reaction complexes relative to respective dissociation fragments, the Gibbs free-energy change at different temperatures was analysed as provided in Table 2. The dissociation of ternary complexes (C1–C11) and binary complexes (B1–B9) of glyoxylic acid, OH radical and H2O molecule, into their respective separated reactants is found to increase with the increase in temperature, as evident from the increasingly negative standard free-energy change (∆G) and positive entropy factor (T∆S). Interestingly, all the complexes seem to be stable at low temperature particularly below 150 K. It should be noted that the strength of ternary complexes is more than binary complexes at lower temperatures due to increase in the number of hydrogen bonds following the insertion of a single water molecule.
A further aspect to be noted in this study is that the most stable binary complex (B4) predicted is H2O···organic(glyoxylic acid) which is followed (in order of stability) by OH···organic complex B3, and OH···H2O complex B9. The latter is ca. 3.3 kcal/mol less stable than B4 at CCSD//BHandHLYP level of the theory. An OH···H2O complex with one hydrogen bond (as in complex B9) is likely to be more stable than the one with the two hydrogen bonds. The reason is simple that an OH···H2O complex with two hydrogen bonds has a highly strained four-membered ring-like framework. The second hydrogen bond is unable to compensate for the strain energy. Therefore, it is relatively unstable not only with respect to the single hydrogen-bonded OH···H2O complex B9, but also relative to the H2O···organic complex B4, and OH···organic complex B3, both of which have a more stable six-membered ring-like hydrogen-bonded framework with two hydrogen bonds.
Finally, to check the reliability of the applied theoretical methods for the present work, additional computations were also performed for four most relevant pathways (II, III, V, XI) at the CCSD(T)/6-311++G(2d,2p)//MP2(FC)/6-311++G(d,p) level of the theory. Similar to the case of BHandHLYP computed pathways, the formyl H-abstraction pathway (V) was also found to be most feasible through the MP2(FC) method. However, the product formed along path V differs in the orientation of water molecule. The aforementioned relevant pathways traced at the level of DFT/BHandHLYP/6-311++G(d,p) were further verified using different exchange–correlation functionals, namely the hybrid B3LYP, and a dispersion-corrected ω-B97XD along with 6-311++G(d,p) basis set. Surprisingly, while employing these functionals of the DFT, all the acidic H-abstraction pathways lead to both H-abstraction and the dissociation of complex to HC=O, CO2 and two molecules of H2O. Moreover, the water-free acidic hydrogen abstraction along path III is observed to follow the PCET mechanism rather than conventional free radical mechanism. However, at the B3LYP/6-311++G(d,p) level of the theory, pathway II could not be confirmed since TS2 could not be optimized at this level of the theory. Notably, while employing ω-B97XD and B3LYP levels of theory, the catalytic formyl H-abstraction along path V was found to be most feasible. It should be noted that at ω-B97XD/6-311++G(d,p) level of theory, the product formed along path V was similar to that formed at MP2(FC) level of the theory. However, note that as evident in Table 3, the frozen-core MP2(FC) absolute values for energy of the reaction species differ entirely from other methods employed though CCSDT//MP2(FC) results correlates quite well. Besides these, for a few pathways, the effect of basis set was also analysed by using cc-pVTZ basis set at the level of CCSD(T)/cc-pVTZ//BHandHLY/6-311++G(d,p). As evident in Table 5, the species explored along path II were observed to relatively more stabilized at this level compared to that using 6-311++G(d,p) basis set for CCSD(T) computations, whereas for species along path III, a destabilizing effect was observed. Such different results using cc-pVTZ basis sets are expected because it does not include diffuse functions as in aug-cc-pVTZ. However, note that, though there is a significant change in energies using cc-pVTZ basis set, the relative order of different species along the potential energy profile remains the same as evident in Table 5.
3.4 Comparison with cis-glyoxylic acid
In the case of catalytic oxidation of cis form of glyoxylic acid in the presence of a single water molecule, the pathways depicted in supporting information Figures S3 and S4 were successfully traced out. The formyl H-abstraction pathways: IIIa, IVa, VIIa, and acidic H-abstraction pathways: Va, VIa resemble path IV, V, IX and VI, VIII, respectively, observed for the trans form. It was further noted that except path VIa, acidic H-abstraction pathways in cis-glyoxylic acid lead to abstraction as well as dissociation to HC=O, CO2 and two molecules of H2O. Moreover, formyl H-abstraction along Path IVa is observed to be most feasible since the barrier height is lowest as evident in Figure S5. Though in the case of cis-glyoxylic acid, the pathways similar to path VII, X and XII, observed for the catalytic oxidation of trans form by a single water molecule, could not be located, however, as depicted in Figure S4, a new pathway (path VIIIa) was successfully traced out which was not observed for the trans form.
4 Conclusions
In the present work, the reaction pathways explored for the gas-phase oxidation reaction of glyoxylic acid by the hydroxyl radical, in the absence and presence of a single water molecule, are analysed to observe any catalytic effect water. The pathways were traced through the pre-reaction complexes explored via a systematic and automated search performed using the global reaction route mapping employing the DFT, CCSD(T) and MP2 quantum mechanical methods. Importantly, though the single water molecule seems to lower the potential energy surface of the studied reaction system mainly due to formation of hydrogen-bonded complexes with water, however, it does not accelerate the reaction because of significant loss of entropy during the complexation. Besides this, the formyl hydrogen abstraction in both cis- and trans-glyoxylic acid was found to be most probable during water-catalysed as well as water-free non-catalytic oxidation. All the pathways explored seem to follow a conventional free radical mechanism as observed from the computations employing BHandHLYP exchange–correlation functional of the DFT. However, using other DFT functionals such as ω-B97XD and B3LYP, one of the relevant water-free acidic H-abstraction pathways was observed to follow a proton-coupled electron-transfer mechanism, which also leads to dissociation of glyoxylic acid. These reaction pathways may provide significant insights into the similar reactions occurring in the atmosphere.
References
Budavari S (1996) The merck index, 12th edn. Merck & Co., Inc, Whitehouse Station, p 648
Mattioda G, Christidis Y (1989) Ullmann’s Encyclopedia of Industrial Chemistry, vol 12, 5th edn. VCH Publ VA, New York, pp 405–407
Datta R (1995) Kirk-Othmer encyclopedia of chemical technology, vol 13, 4th edn. Wiley, New York, p 1055
Boga C, Taddei P, Micheletti G, Ascari F, Ballarin B, Morigi M, Galli S (2014) Int J Cosmet Sci 36:459
Xiao M, Wu F (2014) J Environ Sci 26:935–954
Lim H-J, Carlton AG, Turpin BJ (2005) Environ Sci Technol 39:4441–4446
Iuga C, Olea R, Vivier-Bunge A (2008) J Mex Chem Soc 52:36–46
Anglada JM, Olivella S, Sole A (2006) J Phys Chem A 110:1982–1990
Izet N, Ali Drea AA (2012) Basrah J Sci 30(1C):92–104
Shannon RJ, Caravan RL, Blitz MA, Heard DE (2014) Phys Chem Chem Phys 16:3466–3478
Vohringer-Martinez E, Tellbach E, Liessmann M, Abel B (2010) J Phys Chem A 114:9720–9724
Vohringer-Martinez E, Hansmann B, Hernandez H, Francisco JS, Troe J, Abel B (2007) Science 315:497–501
Iuga C, Alvarez-Idaboy JR, Vivier-Bunge A (2010) Chem Phys Lett 50:11–15
Aloisio S, Francisco JS (2000) Acc Chem Res 33:825–830
Luo Y, Maeda S, Ohno K (2009) Chem Phys Lett 469:57–61
Treo J (1994) J Chem Soc, Faraday Trans 90:2303–2317
Allodi MA, Dunn ME, Livada J, Kirschner KN, Shields GC (2006) J Phys Chem A 110:13283–13289
Gonzalez J, Anglada JM (2010) J Phys Chem A 114:9151–9162
Kaur G, Vikas (2014) J Phys Chem A 118:4019–4029
Kaur G, Vikas (2015) RSC Adv 5:50989–50998
Long B, Zhang W, Tan X, Long Z, Wang Y, Ren D (2011) J Phys Chem A 115:1350–1357
Cordova-Gomez M, Iuga C, Alvarez-Idaboy JR (2012) Int J Quantum Chem 112:3508–3515
Prasanthkumar KP, Alvarez-Idaboy J (2014) RSC Adv 4:14157–14164
Galano A, Alvarez-Idaboy JR (2014) J Comp Chem 35:2019–2026
Ohno K, Maeda S (2004) Chem Phys Lett 384:277–282
Maeda S, Ohno K (2006) J Phys Chem A 110:8933–8941
Maeda S, Ohno K (2005) J Phys Chem A 109:5742–5753
Maeda S, Ohno K (2005) Chem Phys Lett 404:95–99
Maeda S, Ohno K, Morokuma K (2013) Phys Chem Chem Phys 15:3683–3701
Maeda S, Taketsugu T, Morokuma K, Ohno K (2014) Bull Chem Soc Jpn 87:1315–1334
Maeda S, Osada Y, Morokuma K, Ohno K (2011) GRRM 11 user manual. http://grrm.chem.tohoku.ac.jp/GRRM/
Kaur G, Vikas (2014) Phys Chem Chem Phys 16:24401–24416
Kaur G, Vikas (2015) RSC Adv 5:82587–82604
Kaur R, Vikas K (2016) RSC Adv 6:29080–29098
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven T Jr, Kudin KN, Burant JC et al (2004) Gaussian 03, revision E.01. Gaussian Inc, Wallingford
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA et al (2013) Gaussian 09, revision D.01. Gaussian Inc, Wallingford
Becke AD (1993) J Chem Phys 98:1372–1377
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785–789
Zhao Y, Truhlar DG (2005) J Chem Theory Comput 1:415–432
Gonzalez C, Schlegel HB (1989) J Chem Phys 90:2154–2161
Gonzalez C, Schlegel HB (1990) J Chem Phys 94:5523–5527
Raghavachari K, Trucks GW, Pople JA, Head-Gordon M (1989) Chem Phys Lett 157:479–483
Head-Gordon M, Pople JA, Frisch MJ (1988) Chem Phys Lett 153:503–506
Boys SF, Bernardi F (1970) Mol Phys 19:553–566
Chai D, Head-Gordon M (2008) Phys Chem Chem Phys 10:6615–6620
Lee TJ, Taylor PR (1989) Int J Quantum Chem 36:199–207
Singleton DL, Cvetanovic RJ (1976) J Am Chem Soc 98:6812–6819
Eyring H (1935) J Chem Phys 3:107–115
Evans MG, Polanyi M (1935) Trans Faraday Soc 31:875–894
Truhlar DG, Hase WL, Hynes JT (1983) J Phys Chem 87:2664–2682
George P, Bock CW, Trachtman M (1982) J Mol Struct 87:1–18
Weinhold F (2012) J Comput Chem 33:2363–2379
Wagnera W, Pruß A (2002) J Phys Chem Ref Data 31:387–535
Murphy DM, Koop T (2005) Q J R Meteorol Soc 131:1539–1565
Huynh MHV, Meyer T (2007) J Chem Rev 107:5004–5064
Acknowledgements
One of the authors, GK, thanks University Grants Commission (UGC), India, for providing financial support in the form of UGC-SRF (NET) fellowship. The authors are also grateful to Prof. Koichi Ohno for providing GRRM program, and to Dr. Neetu Goel and the Department of Chemistry, Panjab University, Chandigarh, for providing other computational software and resources.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kaur, G., Vikas Global reaction route mapping of water-catalysed gas phase oxidation of glyoxylic acid with hydroxyl radical. Theor Chem Acc 135, 259 (2016). https://doi.org/10.1007/s00214-016-2019-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-016-2019-1