
Abstract
A non-empirical global hybrid exchange–correlation energy functional which leads to an exchange potential with correct asymptotic behavior is presented. The exchange functional combines one-fourth of exact exchange with three-fourths of the correct asymptotic potential (CAP) generalized gradient approximation functional. It is combined with the Perdew–Burke–Ernzerhof correlation energy with a slightly modified parameterization so as to cancel the gradient terms of CAP exchange with that of correlation, in the limit of slowly varying density. The resulting global hybrid functional, called CAP0, gives heats of formation, ionization potentials, electron affinities, proton affinities, binding energies of weakly interacting systems, barrier heights for hydrogen and non-hydrogen transfer reactions, bond distances, and harmonic frequencies on standard test sets that are competitive with results from other long-range-corrected, Coulomb-attenuated, or global hybrid functionals. In fact, they are generally superior to or competitive with CAM-PBE0 and, except for heats of formation, with CAM-B3LYP as well. Advantageously, the Rydberg excitation energies from CAP0 are superior to those of other global hybrids and of the long-range-corrected hybrids. They are similar to those from CAM-B3LYP and modestly inferior to the CAM-PBE0 errors. For the valence excitations, we did not find substantial differences for all the hybrid functionals considered, while the oscillator strengths from CAP0 are better to those of other global hybrids and comparable to those obtained with long-range-corrected and Coulomb-attenuated hybrids.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The conceptual and practical advantages of density functional theory (DFT) [1], especially in its Kohn–Sham (KS) [2, 3] form, are so well known that rehearsal of those merits is un-needed. The result is wide-spread use to investigate diverse systems [3–13]. Within KS, the primary ingredient which theory must supply is an approximation to the exchange–correlation (XC) energy functional, \( E_{\text{XC}} [\rho ] \) of useful accuracy and generality, where \( \rho ({\mathbf{r}}) \) is the electron density. The distinguishing feature of the KS equations is the XC potential,
which is a consequence of the variational property of the Hohenberg–Kohn–Levy–Lieb functional. Immediately, it is evident that the accuracy of KS calculations depends both upon the quality of the XC energy functional and upon the quality of its functional derivative. This is particularly the case for expressions in which the KS eigenvalues and orbitals are used explicitly, as for example, in computing a response property.
Time-dependent density functional theory (TDDFT) within the KS formulation [1, 2, 14–24] also has been extensively applied to the study of dynamical phenomena. At least formally, one defines an action XC functional, \( A_{\text{XC}} [\rho ] \), that plays a role similar to that of the XC energy functional in the time-independent case. Most commonly the adiabatic approximation is made. In it, the explicitly time-dependent XC potential is approximated by the time-independent XC potential evaluated with the time-dependent density. Introducing the notation \( \rho_{t} \) to denote \( \rho \) evaluated at the time \( t \) [24],
For the calculation of excitation energies, it is common to use linear response theory [24–27] with TDDFT (though there are exceptions [28–33]). The outcome is an eigenvalue equation for which the eigenvalues are the square of the excitation energies and the eigenvectors contain information about the molecular orbitals involved in each excitation and hence can be used to determine the oscillator strengths. Within this approach the so-called XC kernel is determined from the adiabatic XC potential [24],
From Eqs. (2) and (3), one can see that the accuracy of excitation energies in adiabatic TDKS calculations is determined by the quality of the first and second functional derivatives of the XC energy functional.
Thus, to build an approximate XC energy functional that could give a satisfactory description of total energies, energy differences (e.g., atomization energies or activation barriers), and response properties (e.g., static and dynamic polarizabilities and hyperpolarizabilities) or excitation energies, one needs to consider both the exact properties of \( E_{\text{XC}} [\rho ] \), and the exact properties of its first and second functional derivatives. The first functional derivative, which is the XC potential, has a direct effect upon the occupied and unoccupied KS orbitals and eigenvalues. For the total energies and energy differences, the correct description of the orbitals in the physically important regions is crucial, while the asymptotic behavior (far from the nuclei) is of little consequence. However, for response properties and excitation energies the asymptotic behavior of the orbitals becomes crucial. This situation implies that the XC potential needs to be correctly described in both the region important for binding and in the asymptotic region [34–36]. In the latter, for a neutral system, the exact exchange (X) potential adopts the form
This behavior has important consequences in the occupied and unoccupied KS orbitals and eigenvalues, particularly the highest occupied and lowest unoccupied molecular orbital (HOMO, LUMO, respectively). This is relevant in time-dependent systems because the electrons can explore regions far from the nuclei. Another significant feature that is closely related with the behavior of the KS orbitals and eigenvalues is the discontinuity of the XC potential with respect to electron number N [37–41]. Incorporation of this behavior in approximate XC functional is a difficult task that we do not undertake here.
Current expressions for the XC functional, like the local density approximations (LDA) [42, 43] or generalized gradient approximations (GGA) such as PBE [44] with its different parameterizations [45–48], RPBE [49], BLYP [50, 51], OLYP [51, 52], VMT [53], VT{8,4} [54], PBE-LS [55], SOGGA11 [56], lsRPBE [57], and others, all yield an X potential that decays exponentially with distance from the nuclei. This asymptotic behavior limits the applicability of these functionals for the calculation of excited states. In particular, Casida [58] has shown that the LDA does not give a good description of excitation energies for high-lying excited states. Those excitation energies tend to be severely underestimated with respect to experimental values, a failure traceable to the incorrect asymptotic behavior of the LDA X potential. The GGA results are similar for the same reason. The underestimation can be alleviated by introducing corrections directly to the X potential. Examples include GGA-type potentials asymptotically corrected [59], model potentials with both correct asymptotic behavior and derivative discontinuity [37–39, 60], exact local potentials [61, 62], and non-local potentials with the correct asymptotic behavior [63–70]. But because these all are direct modifications of the X potential, the corresponding X functional \( E_{\text{X}} [\rho ] \) in general is unknown. Though there have been efforts [71–75] to establish a procedure to obtain the X functional (or correlation functional, C) that corresponds to a model potential, the resulting total energies must be interpreted with caution, because the reconstructed density functional is unique only if its KS potential is a functional derivative. Conversely, it has been shown [76] that the quality of calculated excitation energies is severely limited in the case that \( v_{\text{XC}} [\rho ]({\mathbf{r}}) \) is not the functional derivative of \( E_{\text{XC}} [\rho ] \).
There have been efforts, beyond the level of the GGA, to construct an X functional with a functional derivative that describes the binding region well and that has correct asymptotic behavior. Some incorporate the Laplacian of the density [77–79], others the Fermi-Amaldi term [80]. In contrast, we recently proposed [81] a non-empirical GGA X functional, CAP, whose functional derivative leads to the correct asymptotic potential. In combination with the PBE correlation energy, CAP leads to a good description of energy differences, when compared to other GGA functionals and gives improved description of response properties like static and dynamic polarizabilities and hyperpolarizabilities through the use of time-dependent auxiliary density perturbation theory (TDADPT) [82–86].
However, it is known that global, long-range-corrected and Coulomb-attenuated hybrid exchange energy functionals, which combine exact X with a GGA X, provide, in general, better descriptions of energy differences and excitation energies than GGA X functionals alone, although the computational effort is increased. Thus, the object of the present work is to study the non-empirical global hybrid built from CAP for the GGA component, combined with one-fourth of exact exchange [87], to assess the capability to predict the properties just mentioned.
2 Global hybrid exchange energy functional with correct asymptotic potential
Global hybrid functionals arose via analysis of the adiabatic connection between the non-interacting system and the fully interacting real system [88–92]. The basic concept is that XC energy functionals could be composed of a fraction of exact X, \( E_{\text{X}}^{\text{exact}} [\rho ] \), combined with GGA XC. Originally, Becke [93] suggested a three-parameter formula involving the B88 X functional and the Perdew–Wang C functional [94, 95] correlation. That proposal gave rise to the well-known empirical XC energy functional B3LYP [96, 97], in which the PW91 C energy functional was replaced by the LYP C energy functional. Later, Becke [98] produced a simplified one-parameter expression
Subsequently, Perdew et al. [87] used DF perturbation theory [99, 100] to rationalize a non-empirical value of \( a_{0} \approx 1/4. \) When the GGA used in Eq. (5) is the PBE XC functional one is led to the also well-known, non-empirical, hybrid functional PBE0 [101–103], where the zero indicates that there are no adjustable parameters.
Both hybrids, B3LYP (empirical) and PBE0 (non-empirical), lead to a rather reasonable description of energy differences and excitation energies [104–107], among other properties. In general, it is found that global hybrids perform better, over a wide range of properties, than their GGA counterparts, indicating that the fraction of exact exchange included plays an important role. However, in both cases, the X potential determined from the functional derivative of the GGA X energy functional component of the hybrid does not lead to correct asymptotic behavior of the X potential. Moreover, the fraction of exact exchange, according to Eq. (5), leads asymptotically to \( - a_{0}/r \), which underestimates the \( - 1/r \) behavior, because \( a_{0} < 1 \). Thus one is motivated to construct a global hybrid with the correct asymptotic potential, i.e., one in which the GGA contribution yields an asymptotically correct potential. The CAP X functional fits that specification. Observe that, unless the optimized effective potential procedure is used [108], hybrid functionals do not give simple multiplicative potentials, and hence, it is customary to implement hybrids via the generalized Kohn–Sham procedure. We do so here.
Conventionally, a GGA X energy functional is written in terms of an enhancement factor with respect to local X, \( F_{\text{X}} (s) \), as
where \( e_{\text{X}}^{\text{LDA}} [\rho ]({\mathbf{r}}) = A_{\text{X}} \rho ({\mathbf{r}})^{1/3} \), with \( A_{\text{X}} = - 3(3\pi^{2} )^{1/3} /4\pi \), and \( s({\mathbf{r}}) = \left| {\nabla \rho ({\mathbf{r}})} \right|/2k_{F} ({\mathbf{r}})\rho ({\mathbf{r}}) \), is the reduced density gradient, with \( k_{F} ({\mathbf{r}}) = (3\pi^{2} \rho ({\mathbf{r}}))^{1/3} \).
The CAP X energy enhancement factor is [81]
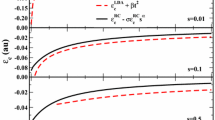
where \( \alpha \) and \( c \) are determined from constraint satisfaction. That is, by requiring that
one finds that the functional derivative of \( E_{\text{X}}^{\text{CAP}} [\rho ] \) gives an asymptotically correct X potential. This implies that \( c = \alpha /(3\pi^{2} )^{1/3} \). On the other hand, since for small values of \( s \) one has that,
where the constant \( \mu \) may be fixed through several non-empirical procedures, one finds that \( \alpha = - A_{x} \,\mu \). Thus, a given value of \( \mu \) fixes \( \alpha \), which in turn fixes \( c \). We have found that by using the value \( \mu = 0.2195 \) (as in the PBE X functional [44]), CAP X combined with PBE C gives a rather good description of the heats of formation of the G3 test set [109]. We note that in PBE the value of \( \mu \), which is associated with the second-order term of the X gradient expansion [Eq. (9)], was fixed to cancel the second-order term of the C gradient expansion so as to recover the benefits of LSDA linear response. The C gradient is dependent on a parameter \( \beta \). The cancelation implies that
The value of \( \mu = 0.2195 \) was obtained by using the value [110] \( \beta_{\text{PBE}} = 0.066725 \). However, it has become common practice to use Eq. (10) instead to fix \( \beta \) from a given \( \mu \). Here we propose a new global hybrid XC energy functional based in Eq. (5), with \( a_{0} = 1/4 \), where the GGA X functional is CAP [Eqs. (6), (7)], and the C functional is PBE C with \( \beta = (3/4)\beta_{\text{PBE}} \), in order to achieve second-order gradient cancelation. This new hybrid is named CAP0, because as in the case of PBE0 it has no adjustable parameters. We note that the CAP potential has somewhat unusual properties, including a pragmatically valuable but anomalous localization of the LUMO. Details are in Ref. [80]. The focus here is not on those properties but upon the extent to which CAP X yields a broadly accurate global hybrid functional.
A relevant aspect of the assessment of CAP0 is its performance compared with long-range-corrected or Coulomb-attenuated hybrid XC functionals [111–116]. In those, the two-electron operator \( 1/r_{12} \) is separated into short-range and long-range parts via the standard error function,
where \( a \), \( b \) and \( \eta \) are parameters, that must satisfy the relations \( 0 \le a + b \le 1 \), \( 0 \le a \le 1 \) and \( 0 \le b \le 1 \). To obtain a functional with the correct asymptotic potential, the long-range part (the second term in the right hand side of Eq. (11) is associated with exact exchange, whereas the short-range effects are obtained with conventional-type XC functionals adapted to the range-separated interaction. For \( a = 0 \) and \( b = 1 \) this kind of functional is commonly called long-range-corrected (LC), while other combinations lead to the so-called Coulomb attenuating method (CAM) [117, 118].
3 Results and discussion
The performance of CAP0 is assessed on the basis of results for energy differences, bond distances, and excitation energies for molecules in standard data sets designed for each property. All calculations were done with a developmental version of the program NWChem-6.0 [119].
For various energy differences the properties studied for the dataset indicated in parenthesis were heats of formation (G3/99) [109, 120], ionization potentials (IP13/3) [121], electron affinities (EA13/3) [122], proton affinities (PA8) [120, 123–126], binding energies of weakly interacting systems (HB6/04 [127], CT7/04 [127], D16/04 [127], W17/05 [128], and PPS5/05 [128]), and barrier heights for forward and backward transition states of hydrogen and non-hydrogen transfer reactions (HTBH38/04 and NHTBH38/04 [128–131]). In the case of geometries, the properties were bond distances (T-96R [132, 133]) and harmonic frequencies (T-82F [132–135]). The detailed protocols used in all these cases can be found in Ref. [48].
We treated the new hybrid functional CAP0, the global hybrids PBE0, B3LYP, and M06-2X [136], the long-range-corrected hybrids LC-BLYP, LC-PBE, and the Coulomb-attenuated CAM-B3LYP and CAM-PBE0. All but PBE0 and CAP0 rely upon empirically adjusted parameters. The functional M06-2X provides a reference to the improvement achieved by the other functionals, since the 32 adjustable parameters in M06-2X were determined to minimize the deviations for several of the test sets considered here. The parameters of Eq. (11) are for LC-BLYP [137] \( a = 0 \), \( b = 1.0 \) and \( \eta = 0.33 \), for LC-PBE [118] \( a = 0 \), \( b = 1.0 \) and \( \eta = 0.30 \), for CAM-B3LYP [117] \( a = 0.19 \), \( b = 0.46 \) and \( \eta = 0.33 \), and finally, for CAM-PBE0 [118] \( a = 0.25 \), \( b = 0.75 \) and \( \eta = 0.30 \).
Table 1 displays the mean absolute deviation (MAD) for all these properties, for the specific datasets mentioned. One sees that for heats of formation PBE0 and B3LYP perform slightly better than CAP0, whereas CAM-B3LYP improves upon them, but LC-BLYP, LC-PBE, and CAM-PBE0 show very large errors. For ionization potentials, CAP0 has the lowest MAD. In the case of electron affinities all functionals give similar errors. For the proton affinities, the global hybrid functionals show similar results, while long-range-corrected and Coulomb-attenuated hybrid functionals lead to slightly worst results. For binding energies of weakly interacting systems CAP0 shows a relatively larger deviation from experiment than the other functionals. In the case of the barrier heights for forward and backward transition states of hydrogen transfer reactions, CAP0 improves upon all the other functionals. For the non-hydrogen transfer reaction barrier heights, CAM-B3LYP and CAM-PBE0 provide improvements over the other functionals. However, both CAP0 and the LC functionals provide a better description than B3LYP and PBE0. For bond distances CAP0, PBE0, and B3LYP lead to similar results, whereas the LC and CAM approximations tend to give larger deviations from the experimental values. In frequencies B3LYP is better than PBE0 and CAP0. Nonetheless, the LC and CAM approximations give larger deviations than B3LYP, PBE0 and CAP0. One also can see in Table 1 that the functional M06-2X provides the best description for all the properties considered, except for bond distances and frequencies. However, as already mentioned, M06-2X is heavily parametrized to minimize the deviations for several of the test sets utilized here. Hence M06-2X serves here simply as a reference to attainable accuracy. In the Supplementary Material, we present the individual deviations of the properties reported in Table 1.
To analyze the performance of CAP0 in the calculation of the X energy, Table 2 shows results for X-only calculations on the noble gas atoms. One can see that the MAD for CAP0 is slightly larger than for all the other functionals. This situation is similar to the one for CAP itself in comparison with other GGA functionals. It seems that the constraint given by Eq. (8) induces small changes in the binding region that lead to an underestimation of the X energy. That underestimation is partially canceled when calculating energy differences, as shown by properties reported in Table 1.
To test the reliability of the new X functional in the calculation of excited states, we employed TDDFT within the adiabatic approximation for calculating oscillator strengths and excitation energies. In particular we used the TDDFT module of NWChem-6.0, which calculates single vertical excitations through the linear response of TDDFT. Valence and Rydberg excited states were determined for a set of four molecules [58], N2, CO, CH2O, and C2H4 at experimental geometries [134, 138]. These excited states were calculated with the aug-cc-pVDZ+ basis set [60], which has an additional set of diffuse functions with exponents set at 1/3 of the most diffuse function exponents of the original aug-cc-pVDZ basis sets [139, 140]. Such diffuse functions are mandatory for treating Rydberg excited states. Note that this combination of basis sets has already proven to be reliable [60]. In addition, we calculated some of the lowest-energy valence excitations that dominate the UV/visible absorption spectra of several aromatic molecules. For this purpose, we selected molecules for which accurate experimental excitation energies as well as gas phase oscillator strengths have been published. The structures of benzene, naphthalene, anthracene, phenol, aniline, and fluorobenzene were optimized at the LDA level of approximation, with the DZVP basis sets [141] and frequency analysis was performed. TDDFT calculations were done with the 6-311++G** basis sets [142], that has proven, in a recent work [143], to be a good choice.
In Table 3 we show the MAD with respect to the experimental values [134, 144–147] for the valence and Rydberg excitations, from TDDFT calculations on the N2, CO, CH2O and C2H4 for the LDA and several GGA functionals. Although the results are rather similar for the four functionals considered, CAP and LDA lead to somewhat better results than PBE and BLYP. It may also be noted that the description of valence excitations is better than the description of Rydberg excitations. In Table 4 we present the same information for the global, long-range-corrected and Coulomb-attenuated hybrid functionals. One can see from the analysis of the MAD that, in comparison with the other global hybrids, CAP0 gives a substantial improvement over PBE0 and B3LYP in the case of the Rydberg excitations, and gives a slightly better description in this case than the highly parameterized M06-2X functional. Note also that for the Rydberg excitations CAP0 provides a better description than the LC functionals, and it delivers also slightly better results than CAM-B3LYP, but is modestly inferior to CAM-PBE0 performance. The description of the valence excitations by CAP0 is similar to what is obtained with the long-range-corrected and Coulomb-attenuated functionals, is slightly better than PBE0 and B3LYP, but is worse than M06-2X. The Supplementary Material contains the excitation energies that lead to the values reported in Tables 3 and 4.
In Table 5 we present excitation energies and corresponding oscillator strengths for some low-energy valence excitations of benzene, naphthalene, anthracene, phenol, aniline, and fluorobenzene, calculated with LDA and GGA functionals, and compare them with experimental values. With respect to the excitation energies, one can see that in general these are underestimated by all the functionals, but CAP leads systematically to larger values that lie closer to the experimental ones. Notice that in the case of aniline, the functionals LDA, PBE, and BLYP show a splitting which is not present in the experimental results. However, for the oscillator strengths, they all lead to rather similar results (in the case of the split excitations, one needs to add all the oscillator strengths).
In Table 6 we present global, long-range-corrected and Coulomb-attenuated hybrid results for the same excitations as in Table 5 and compare them with both coupled-cluster singles and doubles (CCSD) results and with experimental values. Clearly the excitation energies obtained from CAP0, PBE0, LC, and CAM-B3LYP are very similar. All give small overestimations with respect to the experiment, while B3LYP gets closer to the experimental values, and CAM-PBE0, as well CCSD also overestimates them. One can also see that for aniline, all functionals except CAP0 and LC-PBE generate a splitting which is not present in the experimental results. In the case of the oscillator strengths, all the functionals show similar behavior, except for CAM-PBE0 and CCSD, which yield generally larger values.
4 Concluding remarks
The results reported in Sect. 3 show that the global hybrid of CAP with one-fourth exact exchange, CAP0, yields a description of properties depending on energy differences that is competitive in quality with other global functionals like PBE0 and B3LYP. This confirms again that for thermodynamic, kinetic, and structural properties the GGA contribution depends basically on the behavior of the enhancement factor in the physically important region of reduced gradients (\( 0 \le s \le 3 \)). For the Rydberg excitation energies, however, among the global hybrids, CAP0 leads to a much better description than PBE0 and B3LYP. In fact, CAP0 provides better results than the long-range-corrected hybrids, of a quality similar to those obtained from CAM-B3LYP, though not as good as CAM-PBE0. Both, CAP0 and the long-range-corrected and CAM-PBE0 have in common a fraction of exact exchange and the correct asymptotic behavior of the exchange potential, indicating that both contributions can be rather important in TDDFT. However, it is important to note that correct asymptotic behavior of CAP occurs at very large distances from the molecule, so that the fraction of CAP that enters into CAP0 also reaches the full asymptotic behavior far away. Thus, it would seem desirable to reach the full \( - 1/r \) behavior at smaller distances, in order to obtain a better description of the Rydberg excitations.
These conclusions can also be observed through the comparison of the results obtained for the GGAs, particularly CAP, which has the correct asymptotic behavior, with the global, long-range-corrected and Coulomb-attenuated hybrids. Tables 3 and 5 show that the GGAs tend to underestimate the excitation energies. Tables 4 and 6 show that global hybrids such as PBE0 (25 % of exact X) and B3LYP (20 % of exact X) increase the excitation energies because of the inclusion of exact exchange, as had already been observed [103, 143, 148, 149]. But Tables 4 and 6 also show that CAP0 (25 % of exact X) and the long-range-corrected and Coulomb-attenuated hybrids (which contain a fraction of exact X) increase the values even more, perhaps because of the correct asymptotic behavior. The global hybrid M06-2X does not have the correct asymptotic behavior, but since it incorporates 54 % of exact X, it gets closer to the \( - 1/r \) behavior required for the exchange potential than PBE0 and B3LYP.
Thus, one may conclude that the non-empirical CAP0 provides a good description of a wide range of properties, leading, in general, to better results than the non-empirical hybrid PBE0. Also, CAP0 provides a similar description to that of the empirical M06-2X hybrid except for the heats of formation and the barrier heights for the non-hydrogen transfer reactions. However, M06-2X depends upon a large number of fitted parameters and incorporates slightly more than twice the amount of exact exchange of CAP0.
References
Hohenberg P, Kohn W (1964) Phys Rev 136:B864–B871
Kohn W, Sham LJ (1965) Phys Rev 137:A1697–A1705
Engel E, Dreizler RM (2011) Density functional theory. Springer, Berlin
Parr RG, Yang WT (1989) Density functional theory of atoms and molecules. Oxford University Press, New York
Dreizler RM, Gross EKU (1990) Density functional theory. Springer, Berlin
Perdew JP, Kurth S (2003) In: Fiolhais C, Nogueira F, Marques MAL (eds) A primer in density functional theory. Springer, Berlin, pp 1–55
Perdew JP, Ruzsinszky A, Tao JM, Staroverov VN, Scuseria GE, Csonka GI (2005) J Chem Phys 123(6):062201
Scuseria GE, Staroverov VN (2005) In: Dykstra C, Frenking G, Kim KS, Scuseria GE (eds) Theory and applications of computational chemistry: the first forty years. Elsevier, Amsterdam, pp 669–724
Burke K (2012) J Chem Phys 136(15):150901
Cohen AJ, Mori-Sanchez P, Yang WT (2012) Chem Rev 112:289–320
Becke AD (2014) J Chem Phys 140:18A301
Calaminici P, Domínguez-Soria VD, Moreno RF, Gamboa GU, Geudtner G, Goursot A, Salahub DR, Köster AM (2011) In: Leszczynski J (ed) Handbook of computational chemistry. Springer, Berlin
Geudtner G, Calaminici P, Carmona-Espíndola J, del Campo JM, Domínguez-Soria VD, Moreno RF, Gamboa GU, Goursot A, Köster AM, Reveles JU, Mineva T, Vásquez-Pérez JM, Vela A, Zúñiga-Gutiérrez B, Salahub DR (2012) Wiley Interdiscip Rev Comput Mol Sci 2:548–555
Chakravarty S, Fogel MB, Kohn W (1979) Phys Rev Lett 43:775–778
Bartolotti LJ (1981) Phys Rev A 24:1661–1667
Bartolotti LJ (1982) Phys Rev A 26:2243–2244
Deb BM, Gosh SK (1982) J Chem Phys 77:342–348
Gosh SK, Deb BM (1982) Chem Phys 71:295–306
Runge E, Gross EKU (1984) Phys Rev Lett 52:997–1000
Mearns D, Kohn W (1987) Phys Rev A 35:4796–4799
Gross EKU, Kohn W (1990) Adv Quantum Chem 21:255–291
Marques MAL, Ullrich CA, Nogueira F, Rubio A, Burke K, Gross EKU (eds) (2006) Time dependent density functional theory, vol 706. Lecture notes in physics. Springer, Berlin
Marques MAL, Maitra NT, Nogueira FMS, Gross EKU, Rubio A (eds) (2012) Fundamentals of time-dependent density functional theory, vol 837. Lecture notes in physics. Springer, Berlin
Casida ME (1996) In: Seminario JM (ed) Recent developments and applications of modern density functional theory. Theoretical and computational chemistry, vol 4. Elsevier, Amsterdam, pp 391–439
Casida ME (1995) In: Chong DP (ed) Recent advances in density functional methods, vol I. World Scientific, Singapore
Jamorski C, Casida ME, Salahub DR (1996) J Chem Phys 104:5134–5147
Ipatov A, Fouqueau A, dell Valle CP, Córdova F, Casida ME, Köster AM, Vela A, Jamorski CJ (2006) J Mol Struct (THEOCHEM) 179:179–191
Yabana K, Bertsch GF (1999) Int J Quantum Chem 75:55–66
López X, Marques MA, Castro A, Rubio A (2005) J Am Chem Soc 127:12329–12337
Malloci G, Mulas G, Cappellini G, Joblin C (2007) Chem Phys 340:43–58
Koponen L, Puska MJ, Nieminen RM (2008) J Chem Phys 128:154307
Cappellini G, Malloci G, Mulas G (2009) Superlattices Microstruct 46:14–18
Kawashita Y, Yabana K, Noda M, Nobusada K, Nakatsukasa T (2009) J Mol Struct (THEOCHEM) 914:130–135
Sham LJ (1985) Phys Rev B 32(6):3876–3882
Almbladh CO, vonBarth U (1985) Phys Rev B 31(6):3231–3244
Harbola MK, Sahni V (1989) Phys Rev Lett 62(5):489–492
Tozer DJ, Handy NC (1998) J Chem Phys 109(23):10180–10189
Casida ME, Salahub DR (2000) J Chem Phys 113(20):8918–8935
Gruning M, Gritsenko OV, van Gisbergen SJA, Baerends EJ (2001) J Chem Phys 114(2):652–660
Gázquez JL, Garza J, Hinojosa FD, Vela A (2007) J Chem Phys 126(21):214105
Gázquez JL, Garza J, Vargas R, Vela A (2008) In: Diaz-Herrera E, Juaristi E (eds) Recent developments in physical chemistry, vol 979. AIP conference proceedings, pp 11–20
Dirac PAM (1930) Proc Camb Philos Soc 26:376–385
Vosko SH, Wilk L, Nusair M (1980) Can J Phys 58:1200–1211
Perdew JP, Burke K, Ernzerhof M (1996) Phys Rev Lett 77:3865–3868
Zhang YK, Yang WT (1998) Phys Rev Lett 80(4):890–893
Perdew JP, Ruzsinszky A, Csonka GI, Vydrov OA, Scuseria GE, Constantin LA, Zhou XL, Burke K (2008) Phys Rev Lett 100(13):136406
Constantin LA, Fabiano E, Laricchia S, Della Sala F (2011) Phys Rev Lett 106(18):186406
del Campo JM, Gázquez JL, Trickey SB, Vela A (2012) J Chem Phys 136(10):104108
Hammer B, Hansen LB, Norskov JK (1999) Phys Rev B 59(11):7413–7421
Becke AD (1988) Phys Rev A 38:3098–3100
Lee C, Yang WT, Parr RG (1988) Phys Rev B 37:785–789
Handy NC, Cohen AJ (2001) Mol Phys 99:403–412
Vela A, Medel V, Trickey SB (2009) J Chem Phys 130:244103
Vela A, Pacheco-Kato JC, Gázquez JL, del Campo JM, Trickey SB (2012) J Chem Phys 136:144115
Gázquez JL, del Campo JM, Trickey SB, Alvarez-Mendez RJ, Vela A (2013) In: Ghosh SK, Chattaraj PK (eds) Concepts and methods in modern theoretical chemistry. CRC Press, Boca Raton, pp 295–311
Peverati R, Zhao Y, Truhlar DG (2011) J Phys Chem Lett 2(16):1991–1997
Pacheco-Kato JC, del Campo MJ, Gázquez JL, Trickey SB, Vela A (2016) Chem Phys Lett (in press)
Casida ME, Jamorsky C, Casida KC, Salahub DR (1998) J Chem Phys 108:4439–4449
van Leuwen R, Baerends EJ (1994) Phys Rev A 49:2421–2431
Hirata S, Zhan CG, Apra E, Windus TL, Dixon DA (2003) J Phys Chem A 107:10154–10158
Görling A (1999) Phys Rev Lett 83:5459–5462
Ivanov S, Hirata S, Bartlett RJ (1999) Phys Rev Lett 83:5455–5458
Garza J, Nichols JA, Dixon DA (2000) J Chem Phys 112:7880–7890
Garza J, Nichols JA, Dixon DA (2000) J Chem Phys 112:1150–1157
Umezawa N (2006) Phys Rev A 74:032505
Gaiduk AP, Mizzi D, Staroverov VN (2012) Phys Rev A 86:052518
Gaiduk AP, Firaha DS, Staroverov VN (2012) Phys Rev Lett 108:253005
Gritsenko OV, Schipper PRT, Baerends EJ (1999) Chem Phys Lett 302:199–207
Gritsenko OV, Schipper PRT, Baerends EJ (2000) Int J Quantum Chem 76:407–419
Schipper PRT, Gritsenko OV, van Gisbergen SJA, Baerends EJ (2000) J Chem Phys 112:1344–1352
Gaiduk AP, Chulkov SK, Staroverov VN (2009) J Chem Theory Comput 5(4):699–707
Gaiduk AP, Staroverov VN (2009) J Chem Phys 131(4):044107
Gaiduk AP, Staroverov VN (2010) J Chem Phys 133(10):101104
Gaiduk AP, Staroverov VN (2011) Phys Rev A 83(1):012509
Elkind PD, Staroverov VN (2012) J Chem Phys 136(12):124115
Karolewski A, Armiento R, Kummel S (2013) Phys Rev A 88(5):052519
Engel E, Vosko SH (1994) Phys Rev B 50(15):10498–10505
Jemmer P, Knowles PJ (1995) Phys Rev A 51(5):3571–3575
Filatov M, Thiel W (1998) Phys Rev A 57(1):189–199
Gledhill JD, Tozer DJ (2015) J Chem Phys 143(2):024104
Carmona-Espindola J, Gázquez JL, Vela A, Trickey SB (2015) J Chem Phys 142(5):054105
Carmona-Espíndola J, Moreno RF, Köster AM (2010) J Chem Phys 133:084102
Shedge SV, Carmona-Espíndola J, Pal S, Köster AM (2010) J Phys Chem A 114:2357–2364
Carmona-Espíndola J, Moreno RF, Köster AM (2012) Int J Quantum Chem 112:3461–3471
Calaminici P, Carmona-Espíndola J, Geudtner G, Köster AM (2012) Int J Quantum Chem 112:3252–3255
Shedge SV, Pal S, Köster AM (2012) Chem Phys Lett 552:146–150
Perdew JP, Emzerhof M, Burke K (1996) J Chem Phys 105(22):9982–9985
Harris J, Jones RO (1974) J Phys F 4:1170–1186
Gunnarsson O, Lundqvist BI (1976) Phys Rev B 13:4274–4298
Langreth DC, Perdew JP (1977) Phys Rev B 15:2884–2901
Harris J (1984) Phys Rev A 29:1648–1659
Becke AD (1993) J Chem Phys 98:1372–1377
Becke AD (1993) J Chem Phys 98:5648–5652
Perdew JP (1991) In: Ziesche P, Eschrig H (eds) Electronic structure of solids ‘91. Akademie, Berlin, pp 11–20
Perdew JP, Chevary JA, Vosko SH, Jackson KA, Pederson MR, Singh DJ, Fiolhais C (1992) Phys Rev B 46:6671–6687
Kim K, Jordan KD (1994) J Phys Chem 98:10089–10094
Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ (1994) J Phys Chem 98:11623–11627
Becke AD (1996) J Chem Phys 104:1040–1046
Görling A, Levy M (1993) Phys Rev B 47:13105–13113
Görling A, Levy M (1994) Phys Rev A 50:196–204
Adamo C, Barone V (1999) J Chem Phys 110:6158–6170
Ernzerhof M, Scuseria GE (1999) J Chem Phys 110:5029–5036
Adamo C, Scuseria GE, Barone V (1999) J Chem Phys 111:2889–2899
Ando S, Fujigaya T, Ueda M (2002) Jpn J Appl Phys 41:L105–L108
Burke K, Werschnik J, Gross EKU (2005) J Chem Phys 123:062206
Jacquemin D, Perèpte EA, Scuseria GE, Ciofini I, Adamo C (2008) J Chem Theory Comput 4:123–135
Li YL, Han L, Mei Y, Zhang JZH (2009) Chem Phys Lett 482:217–222
Heßelmann A, Götz AW, Della Sala F, Görling A (2007) J Chem Phys 127(5):054102
Curtiss LA, Raghavachari K, Redfern PC, Pople JA (2000) J Chem Phys 112:7374–7383
Ma SK, Brueckner KA (1968) Phys Rev 165(1):18–31
Savin A, Flad HJ (1995) Int J Quantum Chem 56:327–332
Savin A (1996) In: Seminario JM (ed) Recent developments of modern density functional theory. Elsevier, Amsterdam
Savin A (1996) In: Chong DP (ed) Recent advances in density functional methods. World Scientific, Singapore
Leininger T, Stoll H, Werner HJ, Savin A (1997) Chem Phys Lett 275:151–160
Likura H, Tsuneda T, Yanai T, Hirao K (2001) J Chem Phys 115:3540–3544
Toulouse J, Colonna F, Savin A (2004) Phys Rev A 70:062505
Yanai T, Tew DP, Handy NC (2004) Chem Phys Lett 393:51–57
Rohrdanz MA, Herbert JM (2008) J Chem Phys 129:034107
Valiev M, Bylaska EJ, Govind N, Kowalski K, Straatsma TP, van Dam HJJ, Wang D, Nieplocha J, Apra E, Windus TL, de Jong WA (2010) Comput Phys Commun 181:1477–1489
Curtiss LA, Raghavachari K, Redfern PC, Pople JA (1997) J Chem Phys 106:1063–1079
Lynch BJ, Zhao Y, Truhlar DG (2003) J Phys Chem A 107:1384–1388
Zhao Y, Schultz NE, Truhlar DG (2006) J Chem Theory Comput 2:364–382
Parthiban S, Martin JML (2001) J Chem Phys 114:6014–6029
Zhao Y, Truhlar DG (2006) J Phys Chem A 110:10478–10486
Curtiss LA, Raghavachari K, Trucks GW, Pople JA (1991) J Chem Phys 94:7221–7230
Curtiss LA, Redfern PC, Raghavachari K, Pople JA (1998) J Chem Phys 109:42–55
Zhao Y, Truhlar DG (2005) J Chem Theory Comput 1:415–432
Zhao Y, Truhlar DG (2005) J Phys Chem A 109:5656–5667
Lynch BJ, Zhao Y, Truhlar DG. http://t1.chem.umn.edu/db/
Zhao Y, González-García N, Truhlar DG (2005) J Phys Chem A 109:2012–2018
Zhao Y, Lynch BJ, Truhlar DG (2005) Phys Chem Chem Phys 7:43–52
Staroverov VN, Scuseria GE, Tao JM, Perdew JP (2003) J Chem Phys 119:12129–12137
David R. Lide (2002) CRC handbook of chemistry and physics, 83rd edn. CRC Press, Boca Ratón
Huber K, Herzberg G (1979) Molecular spectra and molecular structure. IV Constants of diatomic molecules. Van Nostrand Reinhold, New York
Martin JML (1999) Chem Phys Lett 303:399–407
Zhao Y, Truhlar DG (2008) Theor Chem Acc 120(1–3):215–241
Tawada Y, Tsuneda T, Yanagisawa S, Yanai T, Hirao K (2004) J Chem Phys 120:8425–8433
Herzberg G (1966) Electronic spectra and electronic structure of polyatomic molecules. Van Nostrand Reinhold, New York
Dunning TH (1989) J Chem Phys 90:1007–1023
Kendall RA, Dunning TH, Harrison RJ (1992) J Chem Phys 96:6796–6806
Godbout N, Salahub DR, Andzelm J, Wimmer E (1992) Can J Chem 70:560–571
Krishnan R, Binkley JS, Seeger R, Pople JA (1980) J Chem Phys 72:650–654
Miura M, Aoki Y, Champagne B (2007) J Chem Phys 127:084103
Nielsen ES, Jrgensen P, Oddsershede J (1980) J Chem Phys 73:6238–6246
Clouthier DJ, Ramsay DA (1983) Ann Rev Phys Chem 34:31–58
Ben-Shlomo SB, Kaldor U (1990) J Chem Phys 92:3680–3682
Serrano-Andrés L, Merchán M, Nebot-Gil I, Lindh R, Roos BO (1993) J Chem Phys 98:3151–3162
Stratmann RE, Scuseria GE, Frisch MJ (1998) J Chem Phys 109:8218–8224
Matsuzawa NN, Ishitani A, Dixon DA, Uda T (2001) J Phys Chem A 105:4953–4962
de Castro EVR, Jorge FE (1998) J Chem Phys 108(1):5225–5229
Pantos E, Philis J, Bolovinos A (1978) J Mol Spec 72:36–43
Pickett LW, Muntz M, McPherson EM (1951) J Am Chem Soc 73:4862–4865
George GA, Morris GC (1968) J Mol Spectrosc 26:67–71
Koch EE, Otto A, Radler K (1973) Chem Phys Lett 21:501–504
Kimura K, Nagakura S (1965) Mol Phys 9:117–135
Gilbert R, Sandorfy C (1971) Chem Phys Lett 9:121–124
Philis J, Bolovinos A, Andritsopoulos G, Pantos E, Tsekeris P (1981) J Phys B 14:3621–3635
Acknowledgments
We thank the Laboratorio de Supercómputo y Visualización of Universidad Autónoma Metropolitana-Iztapalapa for the use of their facilities. J.C.E. was supported in part by Conacyt and by Universidad Autónoma Metropolitana through postdoctoral fellowships. J.L.G. thanks Conacyt for Grant 155698, and A.V. thanks Conacyt for Grant 128369. S.B.T. was supported in part by U.S. NSF Grant DMR-1515307.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Carmona-Espíndola, J., Gázquez, J.L., Vela, A. et al. Global hybrid exchange energy functional with correct asymptotic behavior of the corresponding potential. Theor Chem Acc 135, 120 (2016). https://doi.org/10.1007/s00214-016-1864-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-016-1864-2