
Abstract
In recent years, diagnosis of diseases worldwide has been of much interest to the scientific community. Among these diagnosis methods, fluorescence spectroscopy has shown promise. Naphthoquinone and their halogenated derivatives have fluorescent properties and the presence of such substituents promote changes in the spectroscopic properties of the compounds. These properties can be studied by time dependent density functional theory methods. Relativistic effects such as spin–orbit coupling, the Hamiltonian relativistic and the basis set including relativistic corrections are essential for the accurate calculation of spectroscopic properties. For the selection of which of these factors are important for the halogenated derivatives naphthoquinone (F, Cl, Br and I) were employed in a factorial design of the 33 Type, known as a Box–Benhken design. It was observed that the DKH2 Hamiltonian and the basis set TVZ_DKH were significant for studying spectroscopic properties of these compounds. Using these parameters, the ESIPT process was investigated for halogenated compounds of naphthoquinone. It was observed that compounds containing Cl, Br and I do not have the ESIPT process, while a compound containing F showed the process having energy values, 4.69 eV for absorption energy, −1.58 eV for the proton transfer energy and 1.87 eV for the emission energy. We believe that the current study can assist in understanding the ESIPT behavior of ANQ derivatives and why the relativistic effects affect this process.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
In the last decade, the interest in photophysics and photochemistry processes has dramatically grown [2]. To date, fluorescence techniques exhibit several industrial and biomedical applications, such as photocatalytic [2], photoinduced isomerization of cis–trans [3], white organic light emitted dye [4] and can be used as fluorescent probes for image diagnosis [5]. This outlook is currently also reinforced by the large number of fluorescent compounds with biological and technological interest. In this context, one important class of compounds that exhibit fluorescent properties are the naphthoquinone derivatives [6]. These compounds of natural origin can be considered antitumor, antifungal, and antimalarial drugs [7]. Naphthoquinone derivatives have also been tested by many research groups [6, 8] as fluorescent probes for breast and buccal cancer with promising results [6, 9].
It is well known that structural modifications, such as a halogen addition, of potential fluorescent probe candidates can provide dramatic changes in their spectroscopic properties, exhibiting new absorption and emission regions, which can be appreciated for the generation of more efficient fluorescent probes [10]. In this context, halogen derivatives of naphthoquinones were studied by fluorescent spectroscopy [11], Raman [12] and UV–VIS techniques [3] for evaluation of their absorption and emission parameters [11]. Recently, theoretical calculations [13] have also been employed for the study of the Homo–Lumo barrier and other electronic properties [11] of this class of compounds.
Nevertheless, one of the greatest challenges is to investigate the spectroscopic parameters of halogen derivatives due to several electronic and structural effects that can modulate the excited-state properties of those compounds [11]. Despite several efforts, surprisingly little detailed theoretical work has appeared on the analysis of the different fluorescence mechanisms for naphthoquinone derivatives as well as the development of computational strategies, including relativistic effects, able to accurately reproduce spectroscopic parameters of halogen derivatives.
Different theoretical methodologies such as the complete active space self-consistent field (CASSCF), complete active space with second-order perturbation theory (CASPT2), multireference methods and TDDFT techniques can be used for analysis of emission and absorption parameters [3, 14]. It should be kept in mind, however, that the use of time dependent density functional theory (TD-DFT) has provided a promising and accurate alternative for the study of spectroscopic properties, such as vertical energy values, fluorescent parameters or excitation properties of molecules in condensed phase [15–17].
In this context, the accurate description of electronic properties of each atom has great impact on the spectroscopic parameters of the molecule [18]. Among the various parameters and effects that can modulate the electronic properties of heavy atoms, relativistic effects play a crucial role on the spectroscopic properties [18]. However, these effects are complicated to model and generally neglected. In fact, various aspects of the emission and absorption process of organic compounds containing bromide and iodide atoms reveal a great influence of relativistic effects [19].
Currently, relativistic effects are in general employed for theoretical calculations of gold [19], platinum [20], and gadolinium [21] atoms as well as metallic complexes with heavy atoms. Even though this kind of effect in organic systems has been widely neglected spin–orbit coupling, mass/velocity and Darwin effects can strongly influence spectroscopic properties, such as emission and absorption parameters of organic molecules in condensed phase [19, 22].
One way of including the relativistic effects is to apply the relativistic Hamiltonian, which involves the resolution of Dirac’s equation [23, 24]. The Douglas–Kroll–Hess (DKH) approximation, which transforms the Dirac’s Hamiltonian of four components into two components, is one of the most used methods to access the relativistic effects [25]. Another method extensively cited in the literature is the zero-order of regular approximation (ZORA) [26], employing the Pauli’s approximation for the resolution of Dirac’s equation for chemical compounds [27, 28]. This method points out an alternative to solve the Dirac’s equation for the non-relativistic limit and ignores the contributions of the first order within the Pauli method [25, 29].
Besides modifying the Hamiltonian [30] of the system to take basis sets that include relativistic correction is another way of introducing the relativistic effects in the calculations [28]. The basic sets simulate the relativistic corrections implemented by the Dirac equation, by incorporating the relativistic correction for electronic properties. Many research groups have developed basis sets that have generated similar results to the ZORA [31] and DKH approximations [32]. For instance, the Ahlrichs TZV basis set [31–33] has some basis set with relativistic approximation, like TZV_DKH or TZV_ZORA [32]. Finally, relativistic effects can also be included into the system by the introduction of spin–orbit coupling. The spin–orbit coupling is related to the splitting of the energy levels for the heavy atoms. In many cases, this splitting can influence electronic properties of the compounds [34]. The spin–orbit coupling can be investigated using approaches, such as effective nuclear charge (SOC-ENC) or effective nuclear charge with mean-field approach (SOC-ENC-MFA) [32, 35, 36].
Since many theoretical strategies exist to incorporate the relativistic effects on the electronic structure calculation, to select one specific approach is a difficult task and a critical evaluation is necessary. This outlook is aggravated in the case of fluorescent probes that contain heavy atoms, because the impact of relativistic effects on spectroscopic properties of those organic molecules has been little explored in literature and can be significant.
Keeping this in view, the Hamiltonian used, the incorporation or not of spin–orbit coupling and relativistic corrections for basis sets are the three main parameters in relativistic calculations, which in turn can provide vital clues about the type of modifications that might be desirable to increase the accuracy.
Chemometric techniques such as the experimental design or factorial model can assist in the choosing the best conditions and parameter calculation evaluations [37]. Actually, the factorial model is a statistical approach involving the building of the factor combinations selecting those that are significant from the parameters under study [38, 39]. The factorial model can be modeled in different ways employing two or three levels for each parameter (2K, 3K…), and in each level employing two (22, 23,…), three (32, 33,…) or a larger number of parameters in order to obtain the correct interpretation of the results [40].
In the present work, the 33 factorial model, the Hamiltonian, the spin–orbit coupling and the basis sets including relativistic corrections have been applied for the evaluation of the relativistic methods effect on the absorption process of halogen derivatives of 2-amino-1,4-naphthoquinones. The energy barriers involved in the excited-state intramolecular proton transfer (ESIPT) process for these halogen derivatives employing the selected methodology were evaluated afterward.
2 Computational details
The theoretical calculations were performed in the ORCA program [41]. The calculations for the ground state (S 0) and eight excited states were carried out at DFT and TDDFT level of theory using the B3LYP functional [42], respectively. The basis set employed in the study is described in Table 1 as the parameter B. The halogen derivatives for the 2-amino-3-halo-1,4-naphthoquinone were evaluated for the four halogens, F, Cl, Br and I [43] according to Fig. 1. In order to take into account environment effect methanol was taken, as there are experimental results available for this solvent [43, 44]. The COSMO approximation [11, 28, 35] was used in the absorption and fluorescence properties calculations. The Tamm–Dancoff approximation (TDA) [45] method was employed for simulation of the relaxation effect of the solvent in the excited state within the TD-DFT method.

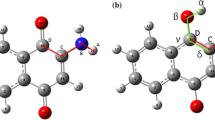
Chemical structures of 3-halogen derivatives of 2-amino-1,4-naphthoquinone: a AFNQ, b ACNQ, c ABNQ and d AINQ
The factorial model calculations were performed in the Statistica® program [46]. The Factorial model employed the 3K method called the Box–Behnken design [40]. The parameters and levels of the model are reported in Table 1. The first factorial model parameter was the presence of the spin–orbit coupling, in which we evaluate the influence of the spin–orbit coupling on the system, by means of the method spin–orbit coupling effective charge nuclear (SOC-ECN) [41] as well as employing the most complete method spin–orbit coupling effective charge nuclear with mean-field approach (SOC-ECN-MFA) [41] and finally removing the SOC effect. The second parameter in the factorial model was the basis set, and its levels were a non-relativistic basis set was employed (TVZ) [31, 32], the basis set with ZORA implementation (TVZ_ZORA) [41] was used as a second level of a basis set and the third level was a DKH basis set (TVZ_DKH) [41] implemented in the ORCA program. The third parameter of the factorial model was the Hamiltonian of the system; thus, the non-relativistic Hamiltonian was the first level, the ZORA Hamiltonian the second and the DKH Hamiltonian was established as the third level [47]. The Box–Behnken design [40, 48, 49] was developed as the twenty-seven (27) cases involving the three parameters and the three levels, which are reported in Table 2. In all the cases, the parameters were evaluated using the ANOVA analysis [50, 51] (Supplementary Material Table S2A), and the acceptance criteria was the Student’s t test [38–40].
The study of the ESIPT process was based on the absorption energy (E 1), the proton transfer energy (E 2), as well as the difference in the excited-state (S 1) energies between the keto and enol forms of the compounds. Finally, the fluorescence energy (F 2 or F 1), see Fig. 2, for the energy difference between the excited state (S 1) and ground state (S 0) was also evaluated. Those calculations were carried out at the DFT and TD-DFT levels for ground and excited states, respectively.

Energy diagram of the excited-state intramolecular proton transfer (ESIPT) for the four halogen derivative compounds for the 2-amino-1,4-naphthoquinone: a the diagram for the ANQ and AFNQ; b the diagram for the ACNQ, ABNQ and AINQ
3 Results and discussion
3.1 The Box–Behnken design
To our knowledge, there are only two compounds, 2-amino-3-chloro-1,4-naphthoquinone (ACNQ) [44], and 2-amino-3-Bromo-1,4-naphthoquinone (ABNQ) [43], for which experimental values of absorption have been reported, which can be used to develop our statistical model based on the Box–Behnken design [40]. Thus, the other two compounds used in this paper, 2-amino-3-fluor-1,4-naphthoquinone (AFNQ) and 2-amino-3-Iodo-1,4-naphthoquinone (AINQ), will have their energy barriers for the ESIPT process theoretically estimated. The Box–Behnken design for the four compounds, see Fig. 1, was evaluated according to the combination of the levels reported in Table 1, whose results are described in Table 2. Through the factorial model, three parameters have been investigated: (1) spin–orbit coupling (parameter A), (2) the basis set used (parameter B), and (3) the relativistic correction for the Hamiltonian of the system (parameter C). The lower level of each parameter was estimated without relativistic effects (no spin–orbit coupling, no relativistic basis set and no relativistic Hamiltonian), the medium level includes spin–orbit coupling SOC-ECN, ZORA basis set and ZORA Hamiltonian and at the high level spin–orbit coupling SOC-ENC-MFA, DKH basis set and DKH2 Hamiltonian. These levels are called levels −1, 0 and +1 on Tables 1 and 2, respectively. It is important to keep in mind that the factorial model 33 was developed with twenty-seven combinations from the three levels with three parameters and their interactions. The TD-DFT method and the COSMO model were employed to calculate the absorption and emission values for the four compounds as well as the values used for the statistical calculations. The selected model for the system was the quadratic model, and the significant effects selected were evaluated for statistical analyses, such as variance analysis (ANOVA), see Supplementary Material Table S2A. It is well known that with this model can get a better description of the interactions and the significance for the studied parameters [38].
3.2 The factorial model for the ACNQ and ABNQ
The experimental values of the absorption wavelength for ACNQ and ABQN are 476 nm [44] and 452 nm [43], respectively. The construction of the factorial design was performed using the error values from the difference between the experimental and theoretical values. According to ANOVA analysis [10, 52] reported in Supplementary Material (Table S2A), only the B and C parameters were significant for the factorial model for ACNQ, i.e., the basis set and Hamiltonian.
The variance analysis (ANOVA) is a statistical model used to employ the differences among group means and their associated methods [46, 49, 53]. In the ANOVA setting, the variance in a variable is divided into parts attributable to different fonts of variation. In other words, ANOVA can be useful for comparing (testing) three or more means (groups or variables) for statistical significance. In this context, Eqs. 1 and 2 represent statistical models to describe the dependence of a factor on the parameters A, B and C.
The value 9.35 is the average population of all factorial design responses in Eq. 1 and 5.20 is the value of the average population in Eq. 2. For the B parameter, when the nonrelativistic basis set (TVZ) is modified to the relativistic basis set (TVZ_DKH2), the error value decreases around 0.46 % according to Eq. 1. For the C parameter, the error value increases 0.10 % with the change of the Hamiltonian of the system ongoing from the nonrelativistic to the relativistic Hamiltonian (ZORA or DKH2). The factorial model equation exhibits significant value for the interaction between B and C parameters for ACNQ. An increase in the error value to 0.15 % when the lower level is modified to the high level was observed.
The factorial model for ABNQ showed that only the C parameter was significant. According to ANOVA analysis (see Supplementary Material (Table S2A)) when the level from lower to the higher was modified, the error value decreases 3.53 %, see Table 3. This feature showed that the relativistic Hamiltonian was essential for the absorption calculation for the bromide derivatives. Interestingly, the relativistic effects were more significant for the bromide derivatives than chloride derivatives. The B parameter was not relevant for the factorial model analysis for ABNQ compounds. However, it was important for the interaction between B and C parameters. Going on from the lowest level (−1) to the highest (+1), the error value decreases 2.85 % as reported in Table 3, indicating that the interaction was significant for the absorption calculation for ABNQ and ACNQ compounds. The equation generated for the ABNQ factorial model is described in Eq. 2. The Pareto graph shown in Supplementary Material Figure S1 exhibits the significant parameters according to Student’s t test. The analysis of those graphics indicates that in all cases the B and C parameters are always significant and for two compounds (ABNQ and AINQ), the interaction between parameter B and C was significant as well. The factorial model generated optimized values for the parameter B (TVZ_DKH) and C (DKH2 Hamiltonian). The parameters described will be used for the wavelength and the energy calculation of the ESIPT process in the third step.
3.3 The factorial model for AFNQ and AINQ
So far, experimental results for AFNQ and AINQ have still not been reported, for this reason, the factorial model used in these cases was developed in comparison with ACNQ and ABNQ. For AFNQ, the ANOVA analysis (in Supplementary Material Table S2A) showed that the B parameter is significant for the absorption calculation, but the C parameter is not. This feature indicated that the relativistic effects were not important for the fluoride derivatives. In fact, since fluoride is a small atom, the relativistic effects can be neglected for fluoride derivate compounds. Surprisingly, the C2 quadratic parameter is not neglected in this model. Comparing to the other two models, the relativistic contribution was important to the B parameter. In this case, ongoing from the lower level to the higher level, the wavelength value increases 5.02 %, (see Table 3). The B2 and C2 quadratic values increase the wavelength value of 4.83 and 2.90 %, respectively. Equation 3 shows the factorial model for AFNQ.
Just as the Eqs. 1 and 2, Eqs. 3 and 4 represent statistical models to describe the dependence of a factor on the parameters A, B and C.
In the factorial model described by Eq. 3, the interaction between B and C is not significant for the absorption calculation. This feature implies that only the isolated parameter was significant for the AFNQ compound. However, for other three models, the interaction between B and C parameters was significant.
For AINQ, for which experimental values are not available, the ANOVA analysis showed that the C parameter is significant according to Eq. 4. Ongoing from the lower level to the medium or higher level, the wavelength decreases 1.16 %. Comparatively, the ABNQ factorial model had a decrease in the wavelength value when going from the lower to the higher level, showing less error for ABNQ. In this way, the AINQ factorial model showed a lesser value for the wavelength and for comparison the lesser error when the DKH2 Hamiltonian is used. The bromide, chloride and iodine atoms have high influence on the relativistic Hamiltonian [35, 54]. The factorial model reinforced this finding. It is important note that the statistical models did not show that the basis set has a significant value for the three compounds; actually, only fluoride was significant. Therefore, the interaction between B and C was significant for ABNQ and AINQ. The interaction between B and C parameters decreased the wavelength values 2.85 and 2.35 %, respectively. ABNQ exhibits the lowest error values ongoing from the lower to the higher level. AINQ displays comparatively the same behavior. In this way, when the level goes from lower to the medium level, the AINQ factorial model decreases 2.35 %. In the other hand, going from the medium to the higher level, the value decreases another 2.35 %. All ANOVA analyses are shown in Supplementary Material Table S2A for the four compounds regarding the 33 factorial model.
3.4 Theoretical calculations for the ESIPT process
The factorial model confirmed that the ideal parameters for spectroscopic properties are the DKH2 Hamiltonian calculation and the relativistic basis set (TVZ_DKH). The spin–orbit coupling is not required. In this regard, we have applied this selected methodology and investigated the ESIPT process for all previous studied compounds and their halogen derivatives. The energy analysis considers three different energies: (1) the energy required for the keto form to reach the excited state, the absorption energy (E 1); (2) the energy needed for the conversion from the keto to enol form of the compounds in the excited state, the proton transfer energy (E 2); (3) the energy emitted when the enol form decays from the excited state to the ground state, emission (fluorescence) energy (F 2). The energy values are reported in Table 4. When the E 2 energy was positive, the enol form was more energetic than the keto form and the ESIPT process cannot occur. In this way, the compounds ACNQ, ABNQ and AINQ cannot exhibit the ESIPT process and the fluorescence occurs by the decaying of the keto form, emitting F1 energy. But, when the E 2 energy is negative or close to zero, the ESIPT process can occur emitting F 2 energy, the emission energy from the enol form is observed for ANQ and AFNQ. The two possibilities for the emission are showed in Fig. 2 where the first diagram shows the ESIPT process in which the keto form in the excited state is converted to an enol form before decaying to the ground state. In the second diagram, the conversion was not possible and the emission occurs in the keto form.
The absorption wavelength calculated showed lower error for ABNQ compared to ANQ, this feature indicated that the model is more accurate for the heavy atoms. The emission wavelength values (λ1) observed refers to emission energy (F1 or F 2, respectively) of each compound, and the value decreases with the increase in substituent mass, see Table 4. These results provide evidence that the increase in mass dispersed more energy into the environment promoting a bathochromic shift. The heavy atoms promoted better dispersion of the electronic density, and this decreases the possibility of the ESIPT process amino group less susceptible to transfer the proton. ANQ absorbed at 445 nm (the experimental value for ANQ is 471 nm) emitting a red color at 663.4 nm. AFNQ absorbed at 488 nm and ACNQ absorbed at 467 nm (the experimental value for ACNQ is 476 nm [44]). The blue emission was observed for ABNQ, which absorbed at 453 nm (the experimental value for ABNQ is 452 nm [43]) and AINQ absorbed at 460 nm.
4 Conclusion
The 33 factorial model used for the study of the influence of the relativistic effects on spectroscopic parameters showed adequate for the halogen derivatives of ANQ. The experimental design was adequate to evaluate the best theoretical strategy to be employed in the investigation of relativistic effects on spectroscopic properties. The model pointed out that DKH2, the relativistic basis set, as well as the interaction between them, are important parameters for the study. The study showed that the presence of the halogen atoms Cl, Br and I does not favor the ESIPT process. The two compounds exhibit the ESIPT process, ANQ and AFNQ, emitting the red and violet color, respectively. Other compounds do not exhibit ESIPT process, but emit violet (ACNQ) and blue color (ABNQ and AINQ) fluorescence for the keto decay. We believe that the current study can assist in understanding the ESIPT behavior of ANQ derivatives and why the relativistic effects affect the process, as well as what theoretical strategy for the theoretical calculations of this class of compounds is the most appropriate.
References
Zhao J, Ji S, Chen Y, et al (2012) Excited state intramolecular proton transfer (ESIPT): from principal photophysics to the development of new chromophores and applications in fluorescent molecular probes and luminescent materials. Phys Chem Chem Phys 14:8803–8817. doi:10.1039/c2cp23144a
Zhao G, Northrop BH, Han K, Stang PJ (2010) The effect of intermolecular hydrogen bonding on the fluorescence of a bimetallic platinum complex. J Phys Chem A 114:9007–9013. doi:10.1021/jp105009t
Levine BG, Martínez TJ (2007) Isomerization through conical intersections. Annu Rev Phys Chem 58:613–634. doi:10.1146/annurev.physchem.57.032905.104612
Park S, Kwon JE, Kim SH, et al (2009) A white-light-emitting molecule : frustrated energy transfer between constituent emitting centers. J Am Chem Soc 131:14043–14049. doi:10.1021/ja902533f
Dugave C, Demange L (2003) Cis− trans isomerization of organic molecules and biomolecules : implications and applications †. Chem Rev 103:2475–2532. doi:10.1021/cr0104375
Laurieri N, Egleton JE, Varney A et al (2013) A novel color change mechanism for breast cancer biomarker detection: naphthoquinones as specific ligands of human arylamine n-acetyltransferase 1. Plos One. doi:10.1371/journal.pone.0070600
Ferreira VF, Jorqueira A, Souza AMT et al (2006) Trypanocidal agents with low cytotoxicity to mammalian cell line: a comparison of the theoretical and biological features of lapachone derivatives. Bioorganic Med Chem 14:5459–5466. doi:10.1016/j.bmc.2006.04.046
Ramalho TC, Rocha EP (2016) Probing the ESIPT process in 2-amino-1,4-naphthoquinone: Thermodynamics properties, solvent effect and chemometric analysis. Theor Chem Acc 135:39. doi:10.1007/s00214-015-1786-4
Luo Y, Li Y, Qiu KM et al (2011) Metronidazole acid acyl sulfonamide: A novel class of anticancer agents and potential EGFR tyrosine kinase inhibitors. Bioorganic Med Chem 19:6069–6076. doi:10.1016/j.bmc.2011.08.038
Bermejo-Bescós P, Martín-Aragón S, Jiménez-Aliaga KL et al (2010) In vitro antiamyloidogenic properties of 1,4-naphthoquinones. Biochem Biophys Res Commun 400:169–174. doi:10.1016/j.bbrc.2010.08.038
Guzow K, Milewska M, Czaplewski C, Wiczk W (2010) A DFT/TD DFT study of the structure and spectroscopic properties of 5-methyl-2-(8-quinolinyl)benzoxazole and its complexes with Zn(II) ion. Spectrochim Acta - Part A Mol Biomol Spectrosc 75:773–781. doi:10.1016/j.saa.2009.11.053
Tucker SC, Honn KV (2013) Emerging targets in lipid-based therapy. Biochem Pharmacol 85:676–688. doi:10.1016/j.bcp.2012.11.028
Yang D, Zhao F, Zheng R et al (2015) A detailed theoretical investigation on the excited-state intramolecular proton-transfer mechanism of 3-BTHPB chemosensor. Theor Chem Acc 134:62. doi:10.1007/s00214-015-1664-0
López-de-Luzuriaga JM, Manso E, Monge M, Sampedro D (2015) Dual fluorescence of 4-(dimethylamino)-pyridine: a comparative linear response TDDFT versus state-specific CASSCF study including solvent with the PCM model. Theor Chem Acc 134:55. doi:10.1007/s00214-015-1659-x
Jana S, Dalapati S, Ghosh S, Guchhait N (2013) Excited state intramolecular charge transfer process in 5-(4-dimethylamino-phenyl)-penta-2,4-dienoic acid ethyl ester and effect of acceptor functional groups. J Photochem Photobiol A Chem 261:31–40. doi:10.1016/j.jphotochem.2013.04.005
Rocha MV, Carvalho HW, Lacerda LC et al (2014) Ionic desorption in PMMA-gamma-Fe2O3 hybrid materials induced by fast electrons: an experimental and theoretical investigation. Spectrochim Acta A Mol Biomol Spectrosc 117:276–283. doi:10.1016/j.saa.2013.08.029
Mancini DT, Sen K, Barbatti M et al (2015) Excited-state proton transfer can tune the color of protein fluorescent markers. ChemPhysChem 16:3444–3449. doi:10.1002/cphc.201500744
Brejc K, Sixma TK, Kitts PA et al (1997) Structural basis for dual excitation and photoisomerization of the Aequorea victoria green fluorescent protein. Proc Natl Acad Sci 94:2306–2311. doi:10.1073/pnas.94.6.2306
Gorin DJ, Toste FD (2007) Relativistic effects in homogeneous gold catalysis. Nature 446:395–403. doi:10.1038/nature05592
Philipsen P, van Lenthe E, Snijders J, Baerends E (1997) Relativistic calculations on the adsorption of CO on the (111) surfaces of Ni, Pd, and Pt within the zeroth-order regular approximation. Phys Rev B 56:13556–13562. doi:10.1103/PhysRevB.56.13556
Hemmilä I, Laitala V (2005) Progress in lanthanides as luminescent probes. J Fluoresc 15:529–542. doi:10.1007/s10895-005-2826-6
Dyall KG, van Lenthe E (1999) Relativistic regular approximations revisited: an infinite-order relativistic approximation. J Chem Phys 111:1366. doi:10.1063/1.479395
Kutzelnigg W (1997) Relativistic one-electron Hamiltonians “for electrons only” and the variational treatment of the Dirac equation. Chem Phys 225:203–222. doi:10.1016/S0301-0104(97)00240-1
Pyykkö P (2012) Relativistic effects in chemistry: more common than you thought. Annu Rev Phys Chem 63:45–64. doi:10.1146/annurev-physchem-032511-143755
Wolf A, Reiher M, Hess BA (2002) The generalized Douglas–Kroll transformation. J Chem Phys 117:9215. doi:10.1063/1.1515314
Cheng L, Stopkowicz S, Gauss J (2014) Analytic energy derivatives in relativistic quantum chemistry. Int J Quantum Chem 114:1108–1127. doi:10.1002/qua.24636
Wolff SK, Ziegler T, van Lenthe E, Baerends EJ (1999) Density functional calculations of nuclear magnetic shieldings using the zeroth-order regular approximation (ZORA) for relativistic effects: ZORA nuclear magnetic resonance. J Chem Phys 110:7689. doi:10.1063/1.478680
Green TFG, Yates JR (2014) Relativistic nuclear magnetic resonance J-coupling with ultrasoft pseudopotentials and the zeroth-order regular approximation. J Chem Phys 140:234106. doi:10.1063/1.4882678
Reiher M, Wolf A (2004) Exact decoupling of the Dirac Hamiltonian. II. The generalized Douglas–Kroll–Hess transformation up to arbitrary order. J Chem Phys 121:10945. doi:10.1063/1.1818681
Christiansen PA, Ermler WC, Pitzer KS (1985) Relativistic Effects in Chemical Systems. Annu Rev Phys Chem 36:407–432. doi:10.1146/annurev.pc.36.100185.002203
Bühl M, Reimann C, Pantazis DA et al (2008) Geometries of third-row transition-metal complexes from density-functional theory. J Chem Theory Comput 4:1449–1459. doi:10.1021/ct800172j
Pantazis DA, Chen X, Landis CR, Neese F (2008) All-electron scalar relativistic basis sets for third-row transition metal atoms. J Chem Theory Comput 4:908–919. doi:10.1021/ct800047t
Kubica A, Kowalewski J, Kruk D, Odelius M (2013) Zero-field splitting in nickel(II) complexes: a comparison of DFT and multi-configurational wavefunction calculations. J Chem Phys 138:064304. doi:10.1063/1.4790167
Arumugam K, Becker U (2014) Computational redox potential predictions: applications to inorganic and organic aqueous complexes, and complexes adsorbed to mineral surfaces. Minerals 4:345–387. doi:10.3390/min4020345
Elkechai A, Kias F, Talbi F, Boucekkine A (2014) Redox properties of biscyclopentadienyl uranium(V) imido-halide complexes: a relativistic DFT study. J Mol Model 20:2294. doi:10.1007/s00894-014-2294-5
Kühn M, Weigend F (2014) Phosphorescence lifetimes of organic light-emitting diodes from two-component time-dependent density functional theory. J Chem Phys 141:224302. doi:10.1063/1.4902013
Bonatsou S, Benítez A, Rodríguez-Gómez F et al (2015) Selection of yeasts with multifunctional features for application as starters in natural black table olive processing. Food Microbiol 46:66–73. doi:10.1016/j.fm.2014.07.011
de Azevedo ALMS, Neto BB, Scarminio IS et al (1996) A chemometric analysis of ab initio vibrational frequencies and infrared intensities of methyl fluoride. J Comput Chem 17:167–177. doi:10.1002/(SICI)1096-987X(19960130)17:2<167:AID-JCC4>3.0.CO;2-U
Ribeiro RLV, Grespan CB, Collins CH et al (1999) Optimization through Factorial Planning of the Use of Ethanol: Water as a Mobile Phase for Reversed Phase HPLC. J High Resolut Chromatogr 22:52–54. doi:10.1002/(SICI)1521-4168(19990101)22:1<52:AID-JHRC52>3.0.CO;2-T
Ferreira SLC, Bruns RE, Ferreira HS et al (2007) Box–Behnken design: An alternative for the optimization of analytical methods. Anal Chim Acta 597:179–186. doi:10.1016/j.aca.2007.07.011
Neese F (2012) The ORCA program system. Wiley Interdiscip Rev Comput Mol Sci 2:73–78. doi:10.1002/wcms.81
Pyykkö P (2004) Theoretical chemistry of gold. Angew Chem Int Ed Engl 43:4412–4456. doi:10.1002/anie.200300624
Pushpam S, Kottaisamy M, Ramakrishnan V (2013) Dynamic quenching study of 2-amino-3-bromo-1,4-naphthoquinone by titanium dioxide nano particles in solution (methanol). Spectrochim Acta - Part A Mol Biomol Spectrosc 114:272–276. doi:10.1016/j.saa.2013.05.038
Pal S, Jadhav M, Weyhermüller T et al (2013) Molecular structures and antiproliferative activity of side-chain saturated and homologated analogs of 2-chloro-3-(n-alkylamino)-1,4-napthoquinone. J Mol Struct 1049:355–361. doi:10.1016/j.molstruc.2013.06.062
Roemelt M, Beckwith MA, Duboc C et al (2012) Manganese K-edge X-ray absorption spectroscopy as a probe of the metal-ligand interactions in coordination compounds. Inorg Chem 51:680–687. doi:10.1021/ic202229b
Scherzer-Attali R, Farfara D, Cooper I et al (2012) Naphthoquinone-tryptophan reduces neurotoxic Aβ*56 levels and improves cognition in Alzheimer’s disease animal model. Neurobiol Dis 46:663–672. doi:10.1016/j.nbd.2012.03.005
Haiduke RLA, Comar M, da Silva ABF (2006) The employment of relativistic adapted Gaussian basis sets in Douglas–Kroll–Hess scalar calculations with diatomic molecules. Chem Phys 331:173–177. doi:10.1016/j.chemphys.2006.10.009
Aslan N, Cebeci Y (2007) Application of Box–Behnken design and response surface methodology for modeling of some Turkish coals. Fuel 86:90–97. doi:10.1016/j.fuel.2006.06.010
Ferreira SLC, Bruns RE, da Silva EGP et al (2007) Statistical designs and response surface techniques for the optimization of chromatographic systems. J Chromatogr A 1158:2–14. doi:10.1016/j.chroma.2007.03.051
Owens EA, Hyun H, Tawney JG, et al. (2015) Correlating Molecular Character of NIR Imaging Agents with Tissue-Specific Uptake. J Med Chem 58:4348–4356. doi:10.1021/acs.jmedchem.5b00475
Tiang JM, Butcher NJ, Minchin RF (2010) Small molecule inhibition of arylamine N-acetyltransferase Type I inhibits proliferation and invasiveness of MDA-MB-231 breast cancer cells. Biochem Biophys Res Commun 393:95–100. doi:10.1016/j.bbrc.2010.01.087
Di Rosso ME, Barreiro Arcos ML, Elingold I et al (2013) Novel o-naphthoquinones induce apoptosis of EL-4 T lymphoma cells through the increase of reactive oxygen species. Toxicol Vitr 27:2014–2094. doi:10.1016/j.tiv.2013.08.002
Bezerra MA, Santelli RE, Oliveira EP et al (2008) Response surface methodology (RSM) as a tool for optimization in analytical chemistry. Talanta 76:965–977. doi:10.1016/j.talanta.2008.05.019
Gourlaouen C, Eng J, Otsuka M et al (2015) Quantum chemical interpretation of ultrafast luminescence decay and intersystem crossings in rhenium(I) carbonyl bipyridine complexes. J Chem Theory Comput 11:99–110. doi:10.1021/ct500846n
Acknowledgments
The authors thank the Brazilian agencies FAPEMIG, CAPES, and CNPq for the financial support of this research and UFLA for infrastructure and encouragement in this work. T.C.R. thanks also the invited professor position at the Czech Republic Center for Basic and Applied research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published as part of the special collection of articles “CHITEL 2015 - Torino - Italy”.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
da Rocha, E.P., Castro, A.A., Ramalho, T.C. et al. Insights into the value of statistical models and relativistic effects for the investigation of halogenated derivatives of fluorescent probes. Theor Chem Acc 135, 135 (2016). https://doi.org/10.1007/s00214-016-1862-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-016-1862-4