
Abstract
Rationale
Combinations of mu and kappa-opioid receptor (KOR) agonists have been proposed as analgesic formulations with reduced abuse potential. The feasibility of this approach has been increased by the development of KOR agonists with biased signaling profiles that produce KOR-typical antinociception with fewer KOR-typical side effects.
Objective
The present study determined if the biased KOR agonists, nalfurafine and triazole 1.1, could reduce choice for oxycodone in rhesus monkeys as effectively as the typical KOR agonist, salvinorin A.
Methods
Adult male rhesus monkeys (N = 5) responded under a concurrent schedule of food delivery and intravenous cocaine injections (0.018 mg/kg/injection). Once trained, cocaine (0.018 mg/kg/injection) or oxycodone (0.0056 mg/kg/injection) was tested alone or in combination with contingent injections of salvinorin A (0.1–3.2 µg/kg/injection), nalfurafine (0.0032–0.1 µg/kg/injection), triazole 1.1 (3.2–100.0 µg/kg/injection), or vehicle. In each condition, the cocaine or oxycodone dose, as well as the food amount, was held constant across choice components, while the dose of the KOR agonist was increased across choice components.
Results
Cocaine and oxycodone were chosen over food on more than 80% of trials when administered alone or contingently with vehicle. When KOR agonists were administered contingently with either cocaine or oxycodone, drug choice decreased in a dose-dependent manner. Salvinorin A and triazole 1.1 decreased drug-reinforcer choice without altering total trials completed (i.e., choice allocation shifted to food), while nalfurafine dose dependently decreased total trials completed.
Conclusions
These results demonstrate that salvinorin A and triazole 1.1, but not nalfurafine, selectively reduce cocaine and oxycodone self-administration independent of nonspecific effects on behavior, suggesting that G-protein bias does not appear to be a moderating factor in this outcome. Triazole 1.1 represents an important prototypical compound for developing novel KOR agonists as deterrents for prescription opioid abuse.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Mu-opioid receptor (MOR) agonists (e.g., morphine, oxycodone) are highly effective treatments for moderate to severe pain but are limited by side effects (e.g., abuse liability; Chan et al. 2017). One strategy for reducing the abuse liability of prescription opioid medications is to co-formulate them with agents that deter misuse without affecting the therapeutic’s analgesic efficacy. Kappa-opioid receptor (KOR) agonists are reported to mitigate the abuse potential of MOR agonists (Bolanos et al. 1996; Freeman et al. 2014; Glick et al. 1995; Kaski et al. 2019; Negus et al. 2008; Townsend et al. 2017; Tsuji et al. 2001; Zamarripa et al. 2020a, b; Zamarripa et al. 2021) and have been proposed as candidate molecules to combine with MOR agonists to develop abuse-deterrent formulations (Townsend et al. 2017; Zamarripa et al. 2021). Furthermore, KOR agonists produce antinociceptive and antipruritic effects in rodents, nonhuman primates, and humans (Beck et al. 2019; Huskinson et al. 2020, 2022; Inan et al. 2019; Jones et al. 2016; Kamimura et al. 2017; Ko and Husbands 2009), which when combined with a MOR agonist, could reduce the amount of the MOR agonist required to treat pain and decrease itch produced by MOR agonist administration. However, the clinical feasibility of using KOR agonists for any indication has been limited due to untoward effects including dysphoria, psychotomimesis, and sedation (MacLean et al. 2013; Pfeiffer et al. 1986; Ranganathan et al. 2012).
In recent years, a novel series of KOR agonists have been discovered that are fully efficacious at the KOR but lack some or all of the adverse effects that are typical of this drug class such as psychotomimesis and dysphoria (for review, see Mores et al. 2019). One explanation for the atypical behavioral profiles of these drugs is “biased agonism,” typically defined for opioid agonists (at both the MOR and KOR) as a greater selectivity for activating the G-protein pathway relative to β-arrestin-2 recruitment (Mores et al. 2019; Rankovic et al. 2016; Urban et al. 2007). In the case of KOR agonists, G-protein signaling has been proposed as the principal mediator of antinociception, while KOR-mediated adverse effects such as dysphoria and psychotomimesis rely chiefly on the recruitment of β-arrestin-2 (Bedini et al. 2020; Brust et al. 2016). In support of this assertion, nalfurafine, which has been reported to be G-protein biased (Schattauer et al. 2017; Zhou et al. 2022), does not produce dysphoria or psychotomimesis in humans (Inui 2015) or place aversions in rodents within dose ranges that are behaviorally active in other measures (e.g., antinociception, anti-pruritus; Hasebe et al. 2004). Moreover, combining nalfurafine with oxycodone decreased self-administration of oxycodone in rats and monkeys (Townsend et al. 2017; Zamarripa et al. 2020a, 2021; Zhang and Kreek 2020) and blocked oxycodone-induced conditioned place preferences (CPP) in rats (Zamarripa et al. 2020b) and mice (Zhang and Kreek 2020) and morphine-induced CPP in mice (Kaski et al. 2019). However, other studies have reported that nalfurafine produces KOR-typical disruptions in motor coordination in mice and species-typical behavior in nonhuman primates (Dunn et al. 2019; Huskinson et al. 2020, 2022; Liu et al. 2019; Kaski et al. 2019). In addition, nalfurafine, when contingently administered with fentanyl in a food vs. drug choice study in rats, reduced fentanyl choice only at a dose that also reduced overall response rates (Townsend 2021). Taken together, these findings suggest that the biased KOR agonist, nalfurafine, while lacking some KOR-mediated side effects (e.g., dysphoria) relative to typical KOR agonists like salvinorin A or U50-488H, produces some KOR-typical adverse effects that disrupt general behavior.
Another biased KOR agonist, triazole 1.1, was reported to be highly G-protein biased and produce antinociceptive and antipruritic effects at doses that did not alter brain dopamine levels within the nucleus accumbens or locomotor behavior in mice (Brust et al. 2016). Additionally, triazole 1.1 enhanced oxycodone-mediated thermal antinociception in rats (Zamarripa et al. 2021) and blocked oxycodone-induced scratch in nonhuman primates at doses that did not suppress species-typical behavior, unlike nalfurafine and the typical KOR agonists, U50-488H and salvinorin A (Huskinson et al. 2020, 2022). Importantly, triazole 1.1 attenuated the reinforcing effects of oxycodone under a PR schedule of reinforcement (Zamarripa et al. 2021). Together, these findings suggest that the atypical KOR agonist, triazole 1.1, may produce an improved therapeutic profile relative to other KOR agonists when combined with a MOR agonist.
The majority of the self-administration work described above used single-operant (i.e., only one active lever) schedules of reinforcement to investigate the effects of contingent administration of the atypical KOR agonists, nalfurafine and triazole 1.1, on oxycodone self-administration. This observation has been interpreted as punishment, a term used in behavioral science to describe the observation of a consequence of behavior reducing the probability of its reoccurrence (Azrin and Holz 1966). However, as discussed above, KOR agonists can produce sedative effects, which could reduce self-administration through nonspecific effects on behavior rather than an associative learning process like punishment. Drug vs. food choice procedures are useful for making this distinction because the measure of reinforcing effect (i.e., behavioral allocation between response options resulting in food or drug reinforcers) is not dependent on response rate (Banks and Negus 2017) and punishment can be distinguished from nonspecific effects on behavior as a determinant of reductions in drug self-administration. Determining the dose–response relationships for drugs as punishers of MOR agonist self-administration is vital for evaluating their potential as deterrents for prescription opioid abuse because their utility in abuse-deterrent formulations will require that the dose be low enough to not discourage appropriate clinical use but high enough to produce a deterring effect if the medication is misused (e.g., taken at higher doses or with greater inter-dosing frequency than prescribed).
The goal of the current study was to investigate the biased KOR agonists, nalfurafine and triazole 1.1, as punishers of drug choice and to compare their effects to the typical KOR agonist, salvinorin A. We have previously reported that salvinorin A punishes cocaine and remifentanil (an ultra-short acting MOR agonist) choice in a drug vs. drug choice procedure in rhesus macaques (Freeman et al. 2014). However, salvinorin A and nalfurafine have yet to be tested as punishers in nonhuman primates in drug vs. non-drug choice, and there are no studies to our knowledge that have tested triazole 1.1 as a punisher in a choice procedure of any kind. In the first experiment of the current study, we tested the punishing effects of salvinorin A and nalfurafine on cocaine choice when food was the alternative reinforcer in male rhesus monkeys. In the second experiment, we expanded the series of KOR agonists by adding triazole 1.1 and compared the KOR agonists as punishers of oxycodone choice using the same food vs. drug choice procedure. All KOR agonists were tested at multiple doses and up to a dose that either (A) reduced drug choice to less than 20% of total choices without altering total trials completed or (B) reduced overall trials completed. These outcomes were used to distinguish between a selective punishing effect (outcome A) and nonspecific effects on behavior (outcome B).
Methods and materials
Subjects and apparatus
Five adult male rhesus monkeys (Macaca mulatta) weighing 10–15 kg served as subjects. Two subjects (0994 and 8752) were experimentally naïve at the start of the experiment, and three subjects (1356, 1010, and 9512) had prior exposure to cocaine, oxycodone, salvinorin A, and nalfurafine (Zamarripa et al. 2020a). Subjects were housed individually in stainless steel enrichment-style cages (each unit: 0.76 m × 0.76 m × 0.86 m; Carter2 Systems, Inc., Beaverton, OR) that allowed visual and olfactory access to other subjects. Subjects had unrestricted access to water, and food was mildly restricted in consultation with veterinary staff to maintain subjects at stable body weights by food provided during the session (1-g BioServ flavored pellets) as well as supplemental feeding (Teklad 25% monkey diet; Harlan/Teklad, Madison, WI). Supplemental feeding always occurred at least 30 min after the choice session ended. Fresh fruit or vegetables and forage material (e.g., dried fruit, vegetables, nuts) were provided daily, and a multivitamin was given three times/week. All subjects were part of an environmental enrichment program that included access to radio, TV, toys, and foraging devices. Lights were maintained on a 14/10 light/dark cycle, with lights on at 0600 h. Subjects were fit with a mesh jacket and tether, and the catheter attached to a double-lumen swivel (Lomir Biomedical, Malone, NY) that attached to a custom-designed operant panel (Carter2 Systems, Inc.) mounted on the home cage. Each panel contained two response levers with stimulus lights above each lever and a food receptacle between each lever (Med Associates, Inc.). A 15-rpm syringe pump (flow rate = 0.18 ml/s), feeder that delivered 1-g BioServ flavored pellets, and a Med Associates connection panel were housed on the outside of each panel. PC computers with Med Associates were used to control experimental events and record data. Animal-use procedures were approved by the University of Mississippi Medical Center’s Institutional Animal Care and Use Committee and were conducted in accordance with the National Research Council’s Guide for Care and Use of Laboratory Animals (8th edition, 2011).
Surgery
Each subject had a double-lumen catheter (Cole-Parmer Co., Chicago, IL) implanted using aseptic technique similar to that described previously (Freeman et al. 2014). Subjects were injected with atropine sulfate (0.04 mg/kg, i.m.) and ketamine hydrochloride (5–20 mg/kg, i.m.) followed by inhaled isoflurane, preoperative antibiotics (cefazolin, 20–25 mg/kg, i.m.), and analgesics (carprofen, 2–4 mg/kg, s.c. and/or buprenorphine SR, 0.05 mg/kg, s.c.). A double-lumen silicon or polyvinyl chloride catheter was implanted into a major vein (jugular or femoral) with the tip terminating near the right atrium. The distal end of the catheter was passed subcutaneously to the mid-scapular region, where it exited the subject’s back, threaded through the tether, and connected to the swivel. Sensorcaine (0.25%) was locally applied to incision sites prior to suturing. Postoperative antibiotics (usually Keflex, 22.2 mg/kg, p.o. twice daily or i.m. once daily) were given when recommended by veterinary staff. Postoperative analgesics (carprofen, 4 mg/kg, p.o. or s.c.) were given for 3 days following surgery. Additional analgesics were given when recommended by veterinary staff. If a catheter became nonfunctional, it was removed, and the subject was removed from the study for at least 1 week and until health was verified by veterinary staff, after which a new catheter was implanted. The catheter was filled with 40–100 U/ml heparinized saline between sessions to prevent clotting.
Baseline procedure
Sessions were conducted daily, beginning at approximately 0900 h. At least 30 min prior to each session, each subject’s catheter was filled with drug and/or vehicle solutions corresponding to the baseline or test condition in an amount sufficient to make the solution available for infusion after completion of the corresponding response requirement. Sessions consisted of six, 20-min components, and each component was separated by a 5-min inter-component interval (ICI). One lever was associated with food, and the other lever was associated with cocaine (0.018 mg/kg/injection). The food- and cocaine-associated levers remained constant within a condition but varied irregularly across conditions and subjects. For all sessions, the first component was a sample component that consisted of 10 sample trials with 5 samples/lever. During sample trials, one lever was active, signaled by illumination of the corresponding stimulus light. The stimulus light for food trials was always white, and for drug trials was red. Following 10 consecutive responses (FR 10) on the active lever, the associated stimulus light darkened, a 60-s timeout period was initiated, and either 1 food pellet or an injection of cocaine was delivered, depending on which lever was active. The cocaine dose was constant across components and conditions and was selected because it was the smallest dose that consistently maintained ≥ 90% cocaine choice across all components. The active lever was randomly determined at the start of the session, and all 5 samples had to be completed on the active lever prior to the start of the subsequent 5 samples on the opposite lever. Component 1 ended after 20 min elapsed. If all trials were completed before 20 min, stimulus lights remained darkened, and a timeout period ensued until the end of the component.
Components 2–6 were choice components and consisted of 10 choice trials. During choice trials, the food- (white stimulus light) and cocaine- (red stimulus light) associated lights were illuminated. Following 10 consecutive responses (FR 10) on one of two levers, both stimulus lights darkened, a 60-s timeout period was initiated, and either 1 food pellet or an injection of cocaine was delivered, depending on which lever was pressed. Responses on either lever reset the response requirement on the opposite lever, ensuring that responses that resulted in either reinforcer were consecutive. If all trials were completed before 20 min elapsed, the stimulus lights remained darkened, and a timeout period ensued until the end of the component. Responding was considered stable when choice of the cocaine-associated lever was ≥ 90% across all choice components, and all trials (sample and choice) were completed within a session, for 3 consecutive sessions.
Test procedure
Once baseline cocaine choice was stable, test conditions began. Food- and drug-associated levers were the same as in the immediately preceding baseline condition but were irregular across subjects and test conditions. Test sessions were similar to baseline sessions with some exceptions. First, in separate conditions, a fixed dose of cocaine (0.018 mg/kg/injection) or oxycodone (0.0056 mg/kg/injection) was available across all components, delivered through one lumen of the double-lumen catheter. The dose of oxycodone was selected based on pilot data and was the smallest dose that reliably produced ≥ 90% drug choice across all choice components. Second, a KOR agonist (salvinorin A, nalfurafine, of triazole 1.1) or vehicle (saline or 80% saline, 10% ethanol, and 10% tween 80; SET) was delivered in the second lumen of the catheter. In the sample component (component 1) and the first choice component (component 2), nothing was delivered through the second lumen (i.e., 0 ml of the KOR agonist or vehicle). In the final four choice components (components 3–6), the volume of KOR agonist or vehicle injection increased across components by adjusting the injection duration (i.e., 0.096, 0.3, 0.96, 3.0 s/injection), resulting in half-log increments in drug dose across the session. Delivery of the KOR agonist or vehicle was signaled by increasing the flash frequency of a green stimulus light located above the drug-associated lever that flashed on and off in 3-s cycles (i.e., components 1 and 2: no green stimulus light; component 3: 0.1-s on, 2.9-s off; component 4: 0.3-s on, 2.7-s off; component 5: 1.0-s on, 2.0-s off; component 6: solid green stimulus light). Injections in the second lumen began immediately after the cocaine or oxycodone injection was delivered in the first lumen. Figure 1 is a schematic showing the timeline of the within-session choice procedure as it occurred during test conditions with a KOR agonist.

A schematic of the within-session choice procedure. Each session consisted of six, 20-min components separated by 5-min inter-component intervals. Component 1 was a sample component, in which five consecutive sample trials were programmed on one, randomly determined lever (i.e., drug or food lever). The reinforcer associated with the active lever was available following 10 consecutive responses. After completion of the first five samples, five subsequent sample trials were programmed on the opposite lever. Components 2–6 consisted of 10 choice trials, in which food or drug were available following 10 consecutive responses on the food- or drug-associated lever, respectively
The KOR agonists tested were salvinorin A (0.1–3.2 µg/kg/injection), nalfurafine (0.0032–0.1 µg/kg/injection), and triazole 1.1 (3.2–100.0 µg/kg/injection). Dose ranges for each KOR agonist were selected based on pilot data to include at least one dose that did not affect drug choice in component 3 and at least one dose that reduced drug choice or disrupted total trials completed across components 4–6, whichever occurred first. Saline and SET served as vehicle conditions and were tested in combination with oxycodone. Salvinorin A and nalfurafine were tested in combination with both cocaine and oxycodone, and triazole 1.1 was tested in combination with oxycodone. KOR agonist and vehicle conditions were tested in an irregular order across subjects. All test conditions were conducted for a minimum of 14 sessions and until the following stability criteria were met across 3 consecutive sessions: (1) completion of all sample trials in component 1, (2) ≥ 80% drug choice in the first choice component (i.e., component 2, when the KOR agonist dose or vehicle volume was 0), (3) percent total drug choice across choice components (components 2–6) was within 20% of the 3-session mean, and (4) no upward or downward trends in total drug choice. If stability criteria were not met within 21 sessions, the test condition was ended, and the final 7 sessions were averaged and used in the analyses. The 21-day criterion was applied to the following conditions: oxycodone/vehicle, oxycodone/salvinorin A, oxycodone/nalfurafine, and cocaine/nalfurafine for subject 1356; oxycodone/nalfurafine and cocaine/nalfurafine for subject 1010; oxycodone/nalfurafine and cocaine/nalfurafine for subject 0994; oxycodone/triazole 1.1 for subject 8752; and cocaine/salvinorin A for subject 9512. All test conditions were followed by at least three consecutive sessions under the baseline procedure with cocaine (described above) before proceeding to subsequent test conditions. This was done to ensure drug choice with cocaine alone returned to ≥ 90% drug choice and separate test conditions followed the same baseline. Responding during the return to baseline conditions was considered stable when choice on the drug-associated lever was ≥ 90% across all choice components, and all trials (sample and choice) were completed within a session, for 3 consecutive sessions.
Data analysis
Data from baseline conditions with food vs. cocaine alone or from the sample component (component 1) are not shown as stability criteria required 100% completion of all trials and ≥ 90% choice of the drug-associated lever. There were seven test conditions in which food was available on one lever and either cocaine + salvinorin A, cocaine + nalfurafine, oxycodone + saline, oxycodone + SET vehicle, oxycodone + salvinorin A, oxycodone + nalfurafine, or oxycodone + triazole 1.1 were available on the opposite lever. For all test conditions, the primary dependent measures were (1) percent drug choice (cocaine or oxycodone) during each choice component (components 2–6), defined as the number of trials completed on the drug-associated lever, divided by the total number of trials completed on both levers, multiplied by 100 and (2) percent of trials completed during each choice component, defined as the number of trials completed divided by total number of trials available (i.e., 10 trials available per component), multiplied by 100. For each measure in each test condition, the mean was determined for each choice component (components 2–6) from the last three or seven sessions, depending on which stability criteria were met. Each dependent measure was analyzed using a separate one-way repeated-measures analysis of variance (ANOVA) for each test condition, with KOR dose as a within-subject variable. Dunnett’s multiple comparisons were used to compare data points from the last four choice components (components 3–6), when increasing doses of the KOR agonist or increasing volumes of the vehicles were delivered, to the first choice component (component 2), when no KOR agonist or vehicle was delivered.
Drugs
Oxycodone hydrochloride and cocaine hydrochloride were generously provided by the National Institute on Drug Abuse (NIDA) drug supply program. Nalfurafine hydrochloride and salvinorin A were synthesized by Dr. Thomas E. Prisinzano. Triazole 1.1 [2-(4-(furan-2-ylmethyl)-5-((4-methyl-3-(trifluoromethyl)benzyl)thio)-4H-1,2,4-triazol-3-yl)pyridine] was synthesized by Dr. Bruce E. Blough. Cocaine, oxycodone, and nalfurafine were prepared in 0.9% sterile saline. Salvinorin A and triazole 1.1 were dissolved in a vehicle of 1:1:8 ethanol:Tween 80:sterile saline. All solutions were passed through a 0.22-µm Millipore filter prior to administration to an animal. Injections were delivered at a rate of 0.18 ml/sec.
Results
Supplementary Fig. 1 illustrates percent drug choice (squares) or trials completed (triangles) for test conditions that involved choice between food (1 pellet/delivery) vs. oxycodone (0.0056 mg/kg/injection) + increasing volumes of saline (panel A) or SET (panel B) vehicles that correspond to components 2–6 of the choice procedure and the associated green-light flash frequencies. Increasing volumes of saline or SET paired with oxycodone delivery had no significant effects on oxycodone choice [F(4,16) = 1.4, p = 0.27; F(4,16) = 1.8, p = 0.19, respectively] or on trials completed [F(4,16) = 1.0, p = 0.44; F(4,16) = 1.0, p = 0.44, respectively]. These data indicate that neither vehicle affected drug choice or trial completion on its own.
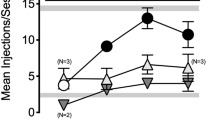
Figure 2 illustrates percent drug choice (circles) or trials completed (triangles) for test conditions that involved choice between food (1 pellet/delivery) vs. cocaine (0.018 mg/kg/injection) + increasing doses of a KOR agonist. Data were plotted as a function of salvinorin A (A) or nalfurafine (B) dose that correspond to components 2–6 of the choice procedure. Increasing doses of salvinorin A (A) paired with cocaine delivery significantly decreased cocaine choice [F(4,16) = 49.6, p < 0.05] without significant effects on percent of trials completed [F(4,16) = 2.6, p = 0.08], indicating that the decrease in drug choice also reflected an increase in choice of the food-associated lever. These effects of salvinorin A on cocaine choice were significantly different from cocaine alone during component 2 compared with components 5 and 6 when 1.0 and 3.2 µg/kg/injection of salvinorin A followed each cocaine injection (p’s < 0.05). Increasing doses of nalfurafine (B) paired with cocaine delivery also significantly decreased cocaine choice [F(4,16) = 7.1, p < 0.05]. Unlike salvinorin A, increasing doses of nalfurafine also resulted in significant reductions in percent of trials completed [F(4,16) = 3.7, p < 0.05], indicating an overall reduction in the number of trials completed rather than a reallocation of behavior from the drug-associated lever to the food-associated lever. These effects of nalfurafine on cocaine choice and trials completed were significantly different from cocaine alone (component 2) during component 6 when 0.1 µg/kg/injection of nalfurafine followed each cocaine injection (p’s < 0.05).

Food vs. cocaine + salvinorin A or nalfurafine choice. Mean percent drug choice (circles) and trials completed (upward triangle) under food vs. cocaine + a KOR agonist. Data are shown as a function of dose of salvinorin A (0–3.2 µg/kg/injection) (A) and nalfurafine (0–0.1 µg/kg/injection) (B). Error bars indicate ± SEM. Asterisks (*) indicate significant differences in cocaine choice during component 2, when choice was between food vs. cocaine alone, compared with cocaine choice during components 3–6, when increasing doses of the KOR agonist were combined with cocaine. Pound signs (#) indicate significant differences in trials completed during component 2 compared with trials completed during components 3–6
Figure 3 illustrates percent drug choice (squares) or trials completed (triangles) for test conditions that involved choice between food (1 pellet/delivery) vs. oxycodone (0.0056 mg/kg/injection) + increasing doses of a KOR agonist. Data paths are plotted as a function of salvinorin A (A), nalfurafine (B), or triazole 1.1 (C) dose that correspond to components 2–6 of the choice procedure as well as increasing delivery volumes and associated green-light flash frequencies. Similar to effects described above for cocaine, increasing doses of salvinorin A (A) paired with oxycodone significantly decreased oxycodone choice [F(4,16) = 36.7, p < 0.05] without significant effects on percent of trials completed [F(4,16) = 2.2, p = 0.11], indicating an increase in choice of the food-associated lever. Effects of salvinorin A on oxycodone choice were significantly different from oxycodone alone during component 2 compared with components 4–6 when 0.32–3.2 µg/kg/injection of salvinorin A followed each oxycodone injection (p’s < 0.05). Increasing doses of nalfurafine (B) paired with oxycodone delivery also significantly decreased oxycodone choice [F(4,16) = 7.7, p < 0.05]. As with cocaine, increasing doses of nalfurafine resulted in significant reductions in percent of trials completed [F(4,16) = 8.7, p < 0.05]. Effects of nalfurafine on oxycodone choice and trials completed were significantly different from oxycodone alone (component 2) during component 6 when 0.1 µg/kg/injection of nalfurafine followed each oxycodone injection. Finally, increasing doses of triazole 1.1 (C) paired with oxycodone significantly decreased oxycodone choice [F(4,16) = 30.0, p < 0.05] without significant effects on percent of trials completed [F(4,16) = 0.9, p = 0.48], similar to salvinorin A’s effects with cocaine and oxycodone. Effects of triazole 1.1 on oxycodone choice were significantly different from oxycodone alone during component 2 compared with components 4–6 when 10–100 µg/kg/injection of triazole 1.1 followed each oxycodone injection (p’s < 0.05).

Food vs. oxycodone + salvinorin A, nalfurafine, or triazole 1.1 choice. Mean percent drug choice (squares) and trials completed (upward triangle) under food vs. oxycodone + a KOR agonist. Data are shown as a function of dose of salvinorin A (0–3.2 µg/kg/injection) (A), nalfurafine (0–0.1 µg/kg/injection) (B), or triazole 1.1 (0–100 µg/kg/injection) (C). Error bars indicate ± SEM. Asterisks (*) indicate significant differences in oxycodone choice during component 2, when choice was between food vs. oxycodone alone, compared with oxycodone choice during components 3–6, when increasing doses of the KOR agonist were combined with oxycodone. Pound signs (#) indicate significant differences in trials completed during component 2 compared with trials completed during components 3–6
Discussion
The present study compared the effects of the atypical KOR agonists, nalfurafine and triazole 1.1, to the typical KOR agonist, salvinorin A, as punishers of drug choice in male rhesus monkeys. In the first study, both salvinorin A and nalfurafine reduced cocaine choice in a dose-dependent manner. However, only salvinorin A selectively reduced cocaine choice without altering the percent of trials completed. In the second study, salvinorin A and triazole 1.1 decreased oxycodone choice in a dose-dependent manner without altering the percent of trials completed. However, reductions in oxycodone choice produced by nalfurafine were accompanied by a reduction in percent of trials completed, particularly in the final component (i.e., at the highest nalfurafine dose).
Salvinorin A, nalfurafine, and triazole 1.1 have all been reported to reduce drug self-administration of oxycodone in either NHPs or rats under conditions of contingent administration using single-operant designs and PR schedules of reinforcement (Townsend et al. 2017; Zamarripa et al. 2020a, b; Zamarripa et al. 2021). The current results support the interpretation that the mitigating effects on oxycodone self-administration produced by salvinorin A and triazole 1.1 in prior studies were due to the KOR agonists functioning as punishers of oxycodone self-administration. However, the same conclusion cannot be made about nalfurafine. While it is possible that nalfurafine can function as a punisher, its effect in this regard may be accompanied by other nonspecific effects within dose ranges required to decrease drug self-administration, a supposition that is supported by recent work in rats (Townsend 2021).
Nalfurafine and triazole 1.1 have been reported to be G-protein–biased agonists, while salvinorin A produces a signaling profile more in line with prototypical KOR agonists such as U50-488H (Mores et al. 2019). It is notable that the two biased KOR agonists in the current study did not produce uniform effects. Rather, triazole 1.1 produced effects more similar to the unbiased KOR agonist, salvinorin A. One possible explanation is that nalfurafine’s selectivity for the KOR is not as high as salvinorin A and triazole 1.1. Salvinorin A and triazole 1.1 exhibit highly selective binding affinity and activity at KORs over MORs and delta opioid receptors (Cunningham et al. 2011; Zhou et al. 2013). However, nalfurafine exhibited MOR/KOR binding affinity ratios (Ki) ranging from 2.4 to 69 (i.e., moderate selectivity), and it functions as a partial agonist at the MOR (Zhou et al. 2022). Moreover, nalfurafine exhibited moderate affinity for the muscarinic M-1 receptor (Nakao and Mochizuki 2009), though its functional effects in relation to behavior at this site have not been reported to our knowledge. As such, nalfurafine’s departure from the behavioral profile exhibited by the other two KOR agonists in the current report may be due to actions at non-KOR sites. Probing different targets with selective antagonists will be a focus of future work.
Another potential reason for the observed differences for nalfurafine is its long duration of action. Salvinorin A and triazole 1.1 are relatively short-acting compounds with in vivo effects (antinociception, anti-pruritus) that are typically reported to last no longer than 30 min (Cunningham et al. 2011; Huskinson et al. 2022; Paton et al. 2017). However, nalfurafine’s in vivo effects, when administered intravenously as in the current study, have been reported to produce observable unconditioned effects for as long as 160 min in NHPs (Huskinson et al. 2020). Thus, it is possible that greater drug accumulation over repeated choice trials led to the observed nonspecific effects on trial completion for nalfurafine. Future work will investigate this issue by using protypical KOR agonists with longer durations of action (e.g., U50-488H) to determine if duration affects the selectivity of punishing effects in the current procedure.
A number of biased KOR agonists have been introduced in recent years (Mores et al. 2019), but the work conducted with triazole 1.1 has set it apart for a few reasons. First, triazole 1.1 produces antinociception at doses that do not reduce locomotor activity (Brust et al. 2016), a result that is in contrast with the observation in the same report that antinociceptive doses of U50-488H also reduce locomotor activity. Second, triazole 1.1 is the only KOR agonist that has been demonstrated to produce analgesic effects in an assay of pain-depressed behavior (Brust et al. 2016). Testing putative analgesics in models of pain-depressed behavior is important because these measures are not confounded by the potential false positives that sedative drugs can render in reflexive measures of pain such as tail-flick or hot plate (Negus et al. 2010). Also, unlike prototypical KOR agonists, triazole 1.1 does not reduce brain dopamine levels at a relatively high, behaviorally active dose (Brust et al. 2016). This latter finding raises the intriguing possibility that certain KOR agonists may be able to counteract the abuse-related effects of MOR agonists without producing counter reductions in brain dopamine that could cause negative affective outcomes and limit clinical feasibility. Future work focusing on the neurochemical changes associated with punished operant responding with triazole 1.1 and other KOR agonists will be needed to make firm conclusions on the relation between punishment by KOR agonists and corresponding dopamine activity.
A major goal of the current study was to determine full dose–response relations for the KOR agonists under study. Meeting this goal required that numerous conditions be run in all subjects, and the time required to obtain data for each of dose of each KOR agonist was 2–3 weeks with additional time between tests for returns to baseline. In order to focus on the dose–effect relations for the KOR agonists, the doses of the reinforcers (cocaine and oxycodone) were held constant. A limitation of this approach is the uncertainty of whether the observed effects with the KOR agonists will hold if they are combined with other doses of the reinforcers. While it is possible that raising the doses of the reinforcers would require higher doses of the KOR agonists to reduce drug choice, our prediction is that the relative potencies of the KOR agonists would not change. Work is planned to address this question in a future study.
In conclusion, a number of studies have reported that KOR agonists reduce the abuse-related effects of drug reinforcers (Freeman et al. 2014; Negus et al. 2008; Townsend et al. 2017; Zamarripa et al. 2020a, 2021), but interpretations of these effects in single-operant procedures have been somewhat obfuscated by the fact that KOR agonists can produce nonspecific effects on behavior that may result in reductions in operant responding. The current results demonstrate that at least two KOR agonists selectively reduce self-administration via reallocation of behavior toward the food alternative rather than by overall attenuation of behavior. Moreover, the ability of KOR agonists to punish drug taking is not limited to prototypical KOR agonists because triazole 1.1 was as effective as salvinorin A at punishing oxycodone choice. As such, triazole 1.1 represents an important prototypical compound for developing novel KOR agonists as deterrents for prescription opioid abuse.
References
Azrin NH, Holz WC (1966) Punishment. In: Honig WK (ed) Operant behavior: areas of research and application. Prentice-Hall, Englewood Cliffs, 380–447
Banks ML, Negus SS (2017) Insights from preclinical choice models on treating drug addiction. Trends Pharmacol Sci 38:181–194
Beck TC, Hapstack MA, Beck KR, Dix TA (2019) Therapeutic potential of kappa opioid agonist. Pharmaceuticals 12:95
Bedini A, Di Cesare Mannelli L, Micheli L, Baiula M, Vaca G, De Marco R, Gentilucci L, Ghelardini C, Spampinato S (2020) Functional selectivity and antinociceptive effects of a novel KOPr agonist. Front Pharmacol 11: 188 10.3389
Bolanos CA, Garmsen GM, Clair MA, McDougall SA (1996) Effects of the kappa-opioid receptor agonist U-50,488 on morphine-induced place preference conditioning in the developing rat. Eur J Pharmacol 317:1–8
Brust TF, Morgenweck J, Kim SA, Rose JH, Locke JL, Schmid CL, Zhou L, Stahl EL, Cameron MD, Scarry SM, Aube J, Jones SR, Martin TJ, Bohn LM (2016) Biased agonists of the kappa opioid receptor suppress pain and itch without causing sedation or dysphoria. Sci Signal 9: ra117
Chan HCS, McCarthy D, Li J, Palczewski K, Yuan S (2017) Designing safer analgesics via mu-opioid receptor pathways. Trends Pharmacol Sci 38:1016–1037
Cunningham CW, Rothman RB, Prisinzano TE (2011) Neuropharmacology of the naturally occurring kappa-opioid hallucinogen salvinorin A. Pharmacol Rev 63:316–347
Dunn AD, Reed B, Erazo J, Ben-Ezra A, Kreek MJ (2019) Signaling properties of structurally diverse kappa opioid receptor ligands: toward in vitro models of in vivo responses. ACS Chem Neurosci 10:3590–3600
Freeman KB, Naylor JE, Prisinzano TE, Woolverton WL (2014) Assessment of the kappa opioid agonist, salvinorin A, as a punisher of drug self-administration in monkeys. Psychopharmacology 231:2751–2758
Glick SD, Maisonneuve IM, Raucci J, Archer S (1995) Kappa opioid inhibition of morphine and cocaine self-administration in rats. Brain Res 681:147–152
Hasebe K, Kawai K, Suzuki T, Kawamura K, Tanaka T, Narita M et al (2004) Possible pharmacotherapy of the opioid kappa receptor agonist for drug dependence. Ann N Y Acad Sci 1025:404–413
Huskinson SL, Platt DM, Brasfield M, Follett ME, Prisinzano TE, Blough BE, Freeman KB (2020) Quantification of observable behaviors induced by typical and atypical kappa-opioid receptor agonists in male rhesus monkeys. Psychopharmacology 237:2075–2087
Huskinson SL, Platt DM, Zamarripa CA, Dunaway K, Brasfield M, Prisinzano TE, Blough BE, Freeman KB (2022) The G-protein biased kappa opioid agonists, triazole 1.1 and nalfurafine, produce non-uniform behavioral effects in male rhesus monkeys. Pharmacol Biochem Behav 217:173394
Inan S, Huerta AT, Jensen LE, Dun NJ, Cowan A (2019) Nalbuphine, a kappa opioid receptor agonist and mu opioid receptor antagonist attenuates pruritus, decreases IL-31, and increases IL-10 in mice with contact dermatitis. Eur J Pharmacol 864:172702
Inui S (2015) Nalfurafine hydrochloride to treat pruritus: a review. Clin Cosmet Investig Dermatol 8:249–255
Jones MR, Kaye AD, Kaye AJ, Urman RD (2016) The emerging therapeutic roles of kappa-opioid agonists. J Opioid Manag 12:101–107
Kaski SW, White AN, Gross JD, Trexler KR, Wix K, Harland AA et al (2019) Preclinical testing of nalfurafine as an opioid-sparing adjuvant that potentiates analgesia by the mu opioid receptor-targeting agonist morphine. J Pharmacol Exp Ther 371:487–499
Ko MC, Husbands SM (2009) Effects of atypical kappa-opioid receptor agonists on intrathecal morphine-induced itch and analgesia in primates. J Pharmacol Exp Ther 328:193–200
Kamimura K, Yokoo T, Kamimura H, Sakamaki A, Abe S, Tsuchiya A, Takamura M, Kawai H, Yamagiwa S, Terai S (2017) Long-term efficacy and safety of nalfurafine hydrochloride on pruritus in chronic liver disease patients: patient-reported outcome based analyses. PLoS ONE 12:e017899
Liu JJ, Chiu YT, DiMattio KM, Chen C, Huang P, Gentile TA, Muschamp JW, Cowan A, Mann M, Liu-Chen LY (2019) Phosphoproteomic approach for agonist-specific signaling in mouse brains: mTOR pathway is involved in κ opioid aversion. Neuropsychopharmacology 44:939–949
MacLean KA, Johnson MW, Reissig CJ, Prisinzano TE, Griffiths RR (2013) Dose-related effects of salvinorin A in humans: dissociative, hallucinogenic, and memory effects. Psychopharmacology 226:381–392
Mores KL, Cummins BR, Cassell RJ, van Rijn RM (2019) A review of the therapeutic potential of recently developed g protein-biased kappa agonists. Front Pharmacol 10:407
Nakao K, Mochizuki H (2009) Nalfurafine hydrochloride: a new drug for the treatment of uremic pruritus in hemodialysis patients. Drugs Today (barc) 45:323–329
Negus SS, Bilsky EJ, Do Carmo GP, Stevenson GW (2010) Rationale and methods for assessment of pain-depressed behavior in preclinical assays of pain and analgesia. Methods Mol Biol 617:79–91
Negus SS, Schrode K, Stevenson GW (2008) Micro/kappa opioid interactions in rhesus monkeys: implications for analgesia and abuse liability. Exp Clin Psychopharmacol 16:386–399
Paton KF, Kumar N, Crowley RS, Harper JL, Prisinzano TE, Kivell BM (2017) The analgesic and anti-inflammatory effects of Salvinorin A analogue β-tetrahydropyran salvinorin B in mice. Eur J Pain 21:1039–1050
Pfeiffer A, Brantl V, Herz A, Emrich HM (1986) Psychotomimesis mediated by kappa opiate receptors. Science 233:774–776
Ranganathan M, Schnakenberg A, Skosnik PD, Cohen BM, Pittman B, Sewell RA et al (2012) Dose-related behavioral, subjective, endocrine, and psychophysiological effects of the kappa opioid agonist salvinorin A in humans. Biol Psychiatry 72:871–879
Rankovic Z, Brust TF, Bohn LM (2016) Biased agonism: an emerging paradigm in GPCR drug discovery. Bioorg Med Chem Lett 26:241–250
Schattauer SS, Kuhar JR, Song A, Chavkin C (2017) Nalfurafine is a G-protein biased agonist having significantly greater bias at the human than rodent form of the kappa opioid receptor. Cell Signal 32:59–65
Townsend EA (2021) Effects of kappa opioid receptor agonists on fentanyl vs. food choice in male and female rats: contingent vs. non-contingent administration. Psychopharmacology 238:1017–1028
Townsend EA, Naylor JE, Negus SS, Edwards SR, Qureshi HN, McLendon HW, McCurdy CR, Kapanda CN, do Carmo JM, da Silva FS, Hall JE, Sufka KJ, Freeman KB (2017) Effects of nalfurafine on the reinforcing, thermal antinociceptive, and respiratory-depressant effects of oxycodone: modeling an abuse-deterrent opioid analgesic in rats. Psychopharmacology (Berl) 234: 2597-2605
Tsuji M, Takeda H, Matsumiya T, Nagase H, Narita M, Suzuki T (2001) The novel kappa-opioid receptor agonist TRK-820 suppresses the rewarding and locomotor-enhancing effects of morphine in mice. Life Sci 68:1717–1725
Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, Miller KJ, Spedding M, Mailman RB (2007) Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther 320(1–13):10
Zamarripa CA, Naylor JE, Huskinson SL, Townsend EA, Prisinzano TE, Freeman KB (2020a) Kappa opioid agonists reduce oxycodone self-administration in male rhesus monkeys. Psychopharmacology 237:1471–1480
Zamarripa CA, Pareek T, Schrock HM, Prisinzano TE, Blough BE, Sufka KJ, Freeman KB (2021) The kappa-opioid receptor agonist, triazole 1.1, reduces oxycodone self-administration and enhances oxycodone-induced thermal antinociception in male rats. Psychopharmacology 238:3463–3476
Zamarripa CA, Patel TR, Williams BC, Pareek T, Schrock HM, Prisinzano TE, Freeman KB (2020b) The kappa-opioid receptor agonist, nalfurafine, blocks acquisition of oxycodone self-administration and oxycodone’s conditioned rewarding effects in male rats. Behav Pharmacol 31:792–797
Zhang Y, Kreek MJ (2020) Nalfurafine modulates the reinforcing effects of oxycodone in male and female adolescent C57BL/6J mice. Neuropharmacology 176:108244
Zhou L, Lovell KM, Frankowski KJ, Slauson SR, Phillips AM, Streicher JM, Stahl E, Schmid CL, Hodder P, Madoux F, Cameron MD, Prisinzano TE, Aube J, Bohn LM (2013) Development of functionally selective, small molecule agonists at kappa opioid receptors. J Biol Chem 288:36703–36716
Zhou Y, Freeman K, Setola V, Cao D, Kaski S, Kreek MJ, Liu-Chen LY (2022) Preclinical studies on nalfurafine (TRK-820), a clinically used KOR agonist. Handb Exp Pharmacol 271:137–162
Acknowledgements
The authors would like to thank Josh Woods, Jessica Howard, Zachary Smith, Kandace Farmer, and Kristen Dunaway for their technical assistance. The authors would also like to thank Drs. Steve Negus and Matthew Banks for consultation on the programming code used for the choice procedure.
Funding
This research and manuscript preparation were supported by National Institute on Drug Abuse grants DA039167 to K.B.F., DA048586 to C.A.Z., DA018151 to T.E.P., DA045011 to S.L.H, and DA043204 to J.K.R.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zamarripa, C.A., Huskinson, S.L., Townsend, E.A. et al. Contingent administration of typical and biased kappa opioid agonists reduces cocaine and oxycodone choice in a drug vs. food choice procedure in male rhesus monkeys. Psychopharmacology 241, 305–314 (2024). https://doi.org/10.1007/s00213-023-06486-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-023-06486-5