
Abstract
Rationale
Benzodiazepines are known to evoke changes in cortical electrophysiological activity that can be correlated with action at distinct γ-aminobutyric acid type A (GABAA) receptor subtypes.
Objectives
We used electroencephalography (EEG) paired with electromyography (EMG) to evaluate the role of α1 subunit-containing GABAA receptors (α1GABAARs) in benzodiazepine-induced sedation and changes in EEG band frequencies during the active phase of the light/dark cycle.
Methods
Male Sprague–Dawley rats (N = 4/drug) were surgically instrumented with EEG/EMG electrodes. The rats were injected i.p. with zolpidem, an α1GABAAR-preferring compound, or L-838,417, which has selective efficacy for α2/3/5 subunit-containing GABAARs (i.e., α1GABAAR-sparing compound), in comparison with the non-selective benzodiazepine, triazolam.
Results
All ligands evaluated induced changes in sleep–wake states during the active phase consistent with an increase in slow-wave sleep (SWS). The degree of SWS increase appeared to be related to the magnitude of delta power band changes induced by the ligands, with the strongest effects engendered by the α1GABAAR-preferring drug zolpidem and the weakest effects by the α1GABAAR-sparing compound, L-838,417. Consistent with other research, a selective increase in beta band power was observed with L-838,417, which may be associated with α2GABAAR-mediated anxiolysis.
Conclusions
Overall, these findings support the establishment of pharmaco-EEG “signatures” for identifying subtype-selective GABAA modulators in vivo.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The behavioral effects of benzodiazepines are produced via their interactions with γ-aminobutyric acid (GABA) type A receptors (GABAAR). GABAARs are pentamers constituted from structurally distinct proteins, with each protein family comprised of different subunits (for reviews, see Ghit et al. (2021); Knoflach and Bertrand (2021)). The majority of GABAARs consist of two α, two β, and a single γ subunit, and GABAARs in the central nervous system can be identified based on different subtypes of the α subunits (α1– α6). Moreover, benzodiazepines are positive allosteric modulators (“modulators”) at GABAARs containing α1, α2, α3, and α5 subunits (α1GABAAR, α2GABAAR, α3GABAAR, and α5GABAAR, respectively) only. Accruing evidence over the past several decades suggests that different behavioral effects of benzodiazepines (e.g., anxiolysis and sedation) may be attributed to specific GABAAR subtypes (for review, see Engin et al. (2018)).
Benzodiazepines are known to evoke changes in cortical electrophysiological activity that can be correlated with behavioral phenomena (e.g., vigilance states, sedation, and sleep; Drinkenburg et al. 2015a, b). A useful in vivo approach to evaluating cortical electrophysiological activity is the use of electroencephalography (EEG) paired with electromyography (EMG). The use of EEG recordings in benzodiazepine research has the advantage of being highly translatable, with essentially similar benzodiazepine-induced EEG changes having been described in rodents (e.g., Leiser et al. 2011), non-human primates (Berro et al. 2021), and humans (e.g., Saletu et al. 2006; Gilles and Luthringer 2007). Specifically, low and non-sedating doses of benzodiazepines and other GABAAR modulators are known to increase power in higher frequency bands (i.e., beta/gamma), which has been proposed as a translatable biomarker for anxiolysis (Coenen and van Luijtelaar 1991; Jongsma et al. 2000; van Lier et al. 2004; Christian et al. 2015; Berro et al. 2021). Conversely, relatively high and sedating doses of benzodiazepines and other GABAAR modulators increase power in lower frequency bands (e.g. delta), and these increases have been correlated with non-EEG assessments of sedation (e.g., Liu et al. 1996; Berro et al. 2021). Importantly, benzodiazepine-induced changes in EEG band frequency seem to be state-dependent (wake vs sleep, or active vs. inactive phase). Specifically, benzodiazepines have been shown to increase delta band frequency when administered during the active phase, while inhibiting EEG delta power when administered during the inactive phase (Buchsbaum et al. 1985; Dijk et al. 1989; Davis et al. 2011).
Considerable evidence has accrued that implicates the α1GABAAR subtype as a key mediator of behaviorally measured sedation induced by benzodiazepines (Engin et al. 2018). This observation is based, in part, on studies with compounds lacking activity at the α1GABAAR (“α1GABAAR-sparing compounds”) which show a lack of sedative effects over a range of doses and procedures (e.g., McKernan et al. 2000; Duke et al. 2018). Because EEG-derived delta power increases are largely absent with α1GABAAR-sparing compounds, the active phase modulation of delta power has been proposed to be linked with behavioral measures of sedation and to be mediated by the α1GABAAR. However, studies using point mutations in mice in which the α1GABAAR has been rendered insensitive to benzodiazepines suggest that behavioral sedation and delta power changes may be dissociable (Tobler et al. 2001). In fact, delta power changes induced by a benzodiazepine during sleep (in the inactive phase) appear not to involve the α1GABAAR (Kopp et al. 2003, 2004). Therefore, the precise role of GABAAR receptors in mediating sleep vs. sedation, in relation to spectral power band changes, remains unclear.
In the present study, we used a pharmaco-EEG approach to evaluate the role of α1GABAAR subtypes in benzodiazepine-induced sedation and changes in EEG band frequencies in rats. Sedation in this study was assessed by standard analysis of sleep–wake states (scoring of wake, slow-wave sleep, and REM sleep) during the active phase after administration of benzodiazepine-type ligands. Note that we are assuming that there is a relationship between sedation assessed during the active phase and EEG/EMG-defined sleep, but we are not proposing that these two phenomena are the same process. Instead, we are using sleep-state analysis to quantify a state that may (or may not) overlap with sedation, yet involves increased slow-wave EEG activity. It is also important to note that this definition differs from the often-used definition of sedation based on decreases in locomotor motor activity in rodents. Using this approach, we assessed the effects of acute injections of the α1GABAAR-preferring compound zolpidem and the α1GABAAR-sparing compound L-838,417, which is an antagonist at α1 subunit-containing GABAA receptors but a partial modulator at other GABAAR subtypes (McKernan et al. 2000), in comparison with the non-selective classical benzodiazepine triazolam, on EEG spectral power and EEG-based sedation (i.e., analysis of sleep-wake states during the active phase).
Material and methods
Subjects
Subjects were 5 adult male Sprague–Dawley rats (Harlan, Indianapolis, IN) weighing 300–380 g at the beginning of the experiment. The rats were maintained on a 12-hour light/dark schedule with lights on at 6:00 AM. Rats initially were pair-housed in standard shoebox home cages until surgeries, and all rats had ad libitum access to Teklad Rodent Diet (Envigo, Indianapolis, IN) and water throughout the study. Following surgery, rats were housed individually to protect instrumentation. All experiments were conducted under a protocol approved by the University of Mississippi Medical Center’s Institutional Animal Care and Use Committee and were conducted in accordance with the National Research Council’s Guide for Care and Use of Laboratory Animals (8th edition, 2011).
Surgical procedures
The rats were implanted with sleep recording electrodes (Plastics One, Roanoke, VA) one week after arrival in the UMMC animal facilities. Surgeries were performed under aseptic conditions with isoflurane as an anesthetic (5% inhalation for 5 minutes to induce and 2.5% inhalation to maintain) during surgery. Body temperature was maintained at 37 °C with a homeothermic blanket. Subjects were surgically instrumented for EEG recording using standard stereotaxic techniques. Three stainless steel screws were placed in the skull to anchor the implant and served as cortical surface electrodes for EEG acquisition. Electrode coordinates were as follows: 1) centro-frontal screw: (Bregma): anterior–posterior (A-P): − 4.5 mm, L (left): 1.0 mm; 2) temporo-parietal screw: A-P: − 4.5 mm; L (left): 5.5 mm; and 3) occipital reference screw: A-P: − 10 mm, L: 0 mm. Two additional stainless steel electrodes were implanted into the trapezius neck muscle and served as intramuscular electrodes for EMG acquisition. All electrode screws were connected to a Teflon connector (Plastics One, Roanoke, VA) that was insulated and fixed to the skull with acrylic dental cement. Subjects were allowed two weeks of recovery in home cages before testing began.
In vivo electroencephalography and electromyography recording
After surgery, rats were handled for 1 week prior to the start of the experiment and were given saline i.p. injections for 1 week prior to the initiation of drug testing to assure that subjects were habituated to the conditions of the study. Four rats were used for the triazolam (vehicle, 0.1, 0.3, or 1.0 mg/kg) and zolpidem (vehicle, 1.0, 3.0, or 10.0 mg/kg) studies. At the end of these studies, one of these subjects lost its EEG/EMG connector. Therefore, three of these rats, as well as an additional new rat, were used for the L-838,417 (vehicle, 1.0, 3.0, or 10.0 mg/kg) studies. All EEG recordings were conducted in individual animal recording chambers (custom-designed: 1 × 1 × 3 m) that were light-controlled, air ventilated/sound-proofed, and video-monitored. Animals were attached by lightweight shielded cables to counterbalanced swing-arms fitted with 32-lead electrical commutators (Aireflite Electronics, Bayonne, NJ) that connected to recording equipment in an adjacent control room. All subjects were allowed three days to acclimate to the recording chambers before baseline recordings were taken. On drug testing days, EEG/EMG recording commenced 20 min before the onset of the dark phase and lasted 24 hours. Drugs were administered i.p. 10 min before the start of the dark phase. Doses of each drug and its vehicle were administered to subjects in a random order, and all doses of a given drug were studied before moving to the next drug.
Drug preparation
The base forms of L-838,417 (7-tert-butyl-3-(2,5-difluoro-phenyl)-6-(2-methyl-2H-[1,2,4]triazol-3-ylmethoxy)-[1,2,4]triazolo[4,3-b]pyridazine); Merck, Sharp, and Dohme Research Laboratories; Harlow Essex, UK) and zolpidem (Sigma; St. Louis, MO) were prepared in a vehicle of 50% propylene glycol and 50% sterile water (Fisher Scientific; Suwanee, GA). The base form of triazolam (Tocris Bioscience, Minneapolis, MN) was prepared in a vehicle of 20% propylene glycol and 80% sterile water. Injections were administered at a volume of 1.0 ml/kg (body weight).
Data analysis
In order to capture drug/compound effects when the rats were normally awake under vehicle conditions and the compounds were at maximum exposure levels, sleep–wake state analyses were performed on 30 min of EEG/EMG recordings starting 10 min after injections (i.e., at the start of the dark phase). This 30-min recording period also was used for EEG spectral power analyses, but divided into three 10-min recording periods to capture any time-dependent effects. Preliminary analysis of sleep–wake states and spectral power revealed no differences from vehicle for any ligand beyond the first 30 min of recording.
Sleep–wake state analysis
Each 30-min EEG/EMG recording was divided into 15-s epochs. The 15-s epochs were visually assessed for artifacts, and those with artifacts were omitted from further analysis. Then, epochs were scored as either wake, slow-wave sleep (SWS), or rapid-eye movement sleep (REM) using a combination of visual analysis and semi-automatic threshold scoring (Sirenia Sleep Pro, Pinnacle Technology, Inc. Lawrence KS). All epochs scored with semi-automatic cluster scoring were visually verified for accuracy. Epochs displaying mixed frequencies with small amplitudes in the EEG and high muscle tone in the EMG for more than 50% of the epoch were scored as “wake.” SWS was scored when an epoch displayed low-frequency and large amplitudes in the EEG (i.e., delta activity) in the presence of low muscle tone in the EMG for at least 50% of the epoch. Epochs predominantly showing mixed frequencies and low amplitude and the absence of muscle tone in EMG were scored as REM sleep.
The sleep–wake states of the 30-min recording following drug treatments was compared to the sleep–wake states of a 30-min recording following vehicle treatments. The total number of minutes spent in each sleep stage (wake, SWS, and REM sleep) was transformed into the percentage of the 30-min recording for drug and vehicle administration. For statistical analysis, the dependent measure was the average percent of total minutes spent in each sleep stage. The data were analyzed using separate one-way repeated measures ANOVA and pre-planned Dunnett’s tests comparing each dose with vehicle (i.e., separate analyses per sleep stage). Significance (alpha) was set at p ≤ 0.05. All statistical analyses were performed using GraphPad Prism (v 8.0.01) or SPSS (v 28).
EEG spectral power analysis
The 30-min drug treatment period was divided into three 10-min periods, which were each separated into 15-s epochs and transformed into a time series for Fast Fourier transformations. Spectral power bands were computed from a 1–50 Hz range with a resolution of 0.068 Hz for each transform. The EEG spectral power was partitioned into bands in accordance with the International Pharmacological EEG Group Guidelines (see Versavel et al. 1995) as follows: delta, 1–5.5 Hz; theta, 5.5–8.5 Hz; alpha, 8.5–12.5 Hz; beta, 12.5–30 Hz; and gamma, 30–50 Hz. Data were analyzed as relative power (raw EEG power, μV2/Hz, in each separate band as a percent of the absolute power summed over the five frequency bands for each 15-s epoch). Individual frequency bands were analyzed using separate one-way repeated measures ANOVAs and pre-planned Dunnett’s tests comparing each dose to its respective vehicle. Significance (alpha) was set at p ≤ 0.05. All statistical analyses were performed using GraphPad Prism (v 8.0.01).
Treatment effect size analysis
All experiments were conducted with n = 4 rats using a within-subjects design. In order to evaluate the relationship of effect size vs. p value (i.e., power), we computed partial eta squared values (η2p) which provides an estimate of effect based on treatment variance. In order to determine the effect size associated with alpha level for these experiments (p = 0.05), the ability of effect size to predict p value was evaluated by non-linear regression using a series of quadratic equations (1st, 2nd, and 3rd polynomial) and Akaike’s Information Criterion (AIC) to determine the best fit. In addition, we plotted the effect sizes for relative power for each power band and test compound across the three 10-min periods, since this measure provides spectral band analysis adjusted for changes across the entire power band spectrum.
Results
Sleep–wake states for triazolam
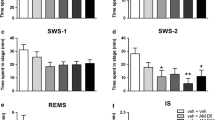
The 30-min analysis of the time spent in each sleep stage after triazolam administration is shown in Fig. 1, top panel. For triazolam, there was a significant effect of dose during the wake state (F(3,9) = 17.27, p = 0.011), with triazolam decreasing the percentage of time in the wake stage at all doses tested (p’s < 0.05, Dunnett’s test). This decrease was accompanied by a significant increase in time spent in SWS (F(3,9) = 18.91, p = 0.006), with all doses of triazolam significantly higher than vehicle (p’s < 0.05, Dunnett’s test). Administration of triazolam did not produce any significant changes in time spent in REM sleep (F(3,9) = 2.143, p = 0.239).

Sleep–wake state and EEG spectral power analyses for triazolam, a benzodiazepine non-selective for GABAA receptor subtypes. For sleep–wake state analysis, the average percent time spent in each sleep stage (wake, slow-wave sleep (SWS), or rapid eye movement sleep (REM)) is shown for the 30 min of recordings beginning at the start of the dark (active) phase. For the EEG spectral power analysis, data for the first, second, and third 10 min of recording after “lights off” were presented as relative power (percentage of total spectral power) for each frequency band. Data are shown as mean ± SEM (N = 4; *p ≤ 0.05 compared to vehicle, Dunnett’s tests)
Relative spectral power for triazolam
Results for all the raw spectral power (μV2) data are provided as Supplemental Materials (Tables S1–S3). Relative spectral power (% of raw power vs. power across all bands) for each frequency band is shown in Fig. 1. For triazolam during the first 10-min period (second panel from top), repeated measures ANOVA showed a significant effect for relative theta power (F (3,9) = 13.92, p = 0.001). Dunnett’s test showed that all three doses of triazolam significantly decreased relative theta power compared to vehicle administration (p’s < 0.05). For relative beta power, repeated measures ANOVA showed a significant treatment effect (F(3,9) = 8.702, p = 0.005). Administration of the two highest doses of triazolam (0.3 and 1.0 mg/kg) significantly increased relative beta power compared to vehicle conditions (p < 0.05, Dunnett’s test). No other power band effects were significant for the first 10-min period. For the second 10-min period (third panel from the top), only the relative theta power was significant (F(3,9) = 8.122, p = 0.006) with all three doses significantly decreasing this measure (p < 0.05, Dunnett’s test). For the third 10-min period (bottom panel), the theta band decreases were again significant (F(3,9) = 5.752, p = 0.0177) but with only the two highest doses significantly different from vehicle (p < 0.05, Dunnett’s test). Interestingly, the beta band increases returned to significance at the third 10-min period (F(3,9) = 10.27, p = 0.0029), with the 0.3 and 1.0 mg/kg doses higher than vehicle (p < 0.05, Dunnett’s test). No other effects of triazolam on power bands were significant at the third 10-min period.
Sleep–wake states for zolpidem
The sleep–wake states during the 30-min analysis period following vehicle and zolpidem administration are shown in Fig. 2, top panel. Although there appeared to be a trend for zolpidem to decrease time spent in wake, the ANOVA and Dunnett’s tests were not significant (F(3,9) = 1.933, p = 0.256). Time spent in SWS approached but did not achieve significance (F(3,9) = 5.889, p = 0.068), but pre-planned Dunnett’s tests showed a significant increase from vehicle at 10 mg/kg (p < 0.05). As with triazolam, the time spent in REM sleep did not significantly change with any of the doses of zolpidem tested (F(3,9) = 0.527, p = 0.553).

Sleep–wake state and EEG spectral power analyses for zolpidem, a ligand with selective affinity for α1 subunit-containing GABAA receptors (“α1GABAAR-preferring”). For sleep–wake state analysis, the average percent time spent in each sleep stage (wake, slow-wave sleep (SWS), or rapid eye movement sleep (REM)) is shown for the 30 min of recordings beginning at the start of the dark (active) phase. For the EEG spectral power analysis, data for the first, second, and third 10 min of recording after “lights off” were presented as relative power (percentage of total spectral power) for each frequency band. Data are shown as mean ± SEM (N = 4; *p ≤ 0.05 compared to vehicle, Dunnett’s tests)
Relative spectral power for zolpidem
Results for all the raw spectral power (μV2) data are provided as Supplemental Materials (Tables S1–S3). Relative spectral power (% of raw power vs. power across all bands) for each frequency band is shown in Fig. 2. The effects of zolpidem administration on EEG spectral power during the first 10 min of the 30 min recording session are shown in the second panel from the top of Fig. 2. For relative delta power, repeated measures ANOVA indicated a significant treatment effect (F(3,9) = 21.58, p < 0.001). Dunnett’s tests showed that all three zolpidem doses increased relative delta power significantly above vehicle (p’s < 0.05). Repeated measures ANOVA indicated significant treatment effects for relative theta power (F(3,9) = 28.98, p < 0.0001). All three doses of zolpidem significantly decreased relative theta power compared to vehicle (p’s < 0.05, Dunnett’s tests). No other significant treatment effects were observed at the first 10-min interval. A similar pattern of effects was observed for the second 10-min interval (Fig. 2, third panel from the top), with a significant ANOVA result for delta power (F(3,9) = 16.81, p = 0.0005) and theta power (F(3,9) = 12.52, p = 0.0015). Multiple comparison tests showed that all three doses of zolpidem induced increases in delta power and decreases in theta power relative to vehicle (p < 0.05, Dunnett’s tests). Evidence that the effects of zolpidem were decreasing with time was obtained in the third 10-min period (Fig. 2, bottom panel) for delta power. In this regard, the ANOVA approached, but did not achieve significance for this power band (F(3,9) = 3.723, p = 0.0545), although pre-planned Dunnett’s tests showed that the 1.0 mg/kg dose increased delta power above vehicle levels. In contrast, the effects on theta power were similar to the prior 10-min periods (F(3,9) = 19.59, p = 0.0003) with all three zolpidem doses showing lower theta power compared with vehicle (p < 0.05, Dunnett’s tests).
Sleep–wake states for L-838,417
The sleep–wake states during the 10-min analysis period following vehicle and L-838,417 administration are shown in Fig. 3, top panel. There was a significant effect of L-838,417 dose for percent of time in wake (F(3,9) = 6.064, p = 0.041), with the highest dose of L-838,417 (10.0 mg/kg) inducing a significant decrease compared with vehicle (p < 0.05, Dunnett’s test). Time in SWS also was significantly changed (F(3,9) = 8.343, p = 0.023), with the 10 mg/kg dose increasing the percentage of time spent in SWS compared with vehicle (p < 0.05, Dunnett’s test). As with the other drugs, the sleep-wake state analysis showed no changes in the percentage of time spent in REM sleep after L-838, 417 administration (F(3,9) = 0.738, p = 0.469).

Sleep–wake state and EEG spectral power analyses for L-838,417, a ligand that is an antagonist at α1 subunit-containing GABAA receptors and a partial modulator at other GABAAR subtypes (“α1GABAAR-sparing”). For sleep–wake state analysis, the average percent time spent in each sleep stage (wake, slow-wave sleep (SWS), or rapid eye movement sleep (REM)) is shown for the 30 min of recordings beginning at the start of the dark (active) phase. For the EEG spectral power analysis, data for the first, second, and third 10 min of recording after “lights off” were presented as relative power (percentage of total spectral power) for each frequency band. Data are shown as mean ± SEM (N = 4; *p ≤ 0.05 compared to vehicle, Dunnett’s tests)
Relative spectral power for L-838,417
Results for all the raw spectral power (μV2) data are provided as Supplemental Materials (Tables S1–S3). The EEG spectral power data for L-838,417 at the first 10-min time period are shown in Fig. 3, second panel from the top. In contrast to the other drugs, L-838,417 had no significant effects during this time period. However, at the second 10-min period (third panel from the top), a significant treatment effect was observed for relative beta power (F(3,9) = 4.447, p = 0.0354)). Dunnett’s tests showed that the 10.0 mg/kg dose of L-838,417 significantly increased relative beta power compared to vehicle (p < 0.05, Dunnett’s tests). No other power band effects were significant at this time period. A different pattern of results was observed during the third 10-min period (Fig. 3, bottom panel), with a significant ANOVA result for delta (F(3,9) = 8.720, p = 0.005). Uniquely, the 1.0 and 3.0 mg/kg doses, but not the highest dose of 10 mg/kg, were decreased significantly compared to vehicle. No other significant effects were observed during the third 10-min time period.
Effect size
The effect sizes (η2p) for all repeated measures ANOVAs performed on sleep–wake states are provided in supplemental Table S4. The effect sizes ranged from 0.144 to 0.862. For the larger spectral power dataset, we evaluated how closely effect size matched p values via non-linear regression analysis, based on quadratic equations ranging from 1st-order polynomial (straight line) to 3rd-order polynomial (cubic). Using AIC analysis, the strongest fit was the third-order polynomial (see supplemental Fig. S1). Five outliers were identified, however, even when included, the resulting goodness-of-fit value was 0.9991 with reasonably distributed data (QQ plot demonstrating a linear residuals relationship shown in Fig. S1). Based on the resulting equation, we calculated that for p = 0.05 level of effect (alpha), the predicted effect size was 0.559 for the entire data set.
Effect size values for each ligand and the corresponding power bands across the three time periods are shown in Fig. 4. For triazolam, the distribution of effect sizes was essentially in the middle frequency bands, with the delta and gamma band effect sizes overall having the lowest values. The distribution for zolpidem clearly favored the lower power bands, with delta and theta effect sizes the most robust of any condition. In contrast, the effect sizes for L-838,417 tended to be more variable across time, but in general favored the higher frequency bands, with beta power in most cases being the strongest effect. The clear exception was the third 10-min period, in which a relative robust effect size was observed for delta power bands, resulting in a bimodal distribution.

Treatment effect size (𝜂2p values) based on repeated measures ANOVAs computed for each power band for each ligand and based on relative spectral band power obtained for the first, second, and third 10-min period after initiation of the dark phase. Test ligands, indicated above each set of power bands, were triazolam (non-selective for GABAA receptor subtypes), zolpidem (selective affinity for α1 subunit-containing GABAA receptors, i.e., α1GABAAR-preferring), and L-838,417 (antagonist at α1 subunit-containing GABAA receptors, partial modulator at other GABAAR subtypes, i.e., α1GABAAR-sparing). Bars with cross-hatch patterns/lighter color indicate a decrease from vehicle, whereas solid bars represent increases from vehicle for the respective power band. Note that the horizontal dotted line indicates the lowest effect size for ANOVAs achieving the pre-determined significance level (alpha) of p = 0.05
Discussion
Use of benzodiazepines to treat anxiety and other disorders has been associated with unwanted side effects, such as significant sedation and ataxia. Intensive efforts in drug discovery and development have focused on leveraging GABAAR pharmacology to develop drug candidates lacking in these side effects (for review, see Cerne et al. 2022). Available evidence has implicated the α1GABAAR subtype in mediating the sedative–motor effects of benzodiazepines (Engin et al. 2018; Cerne et al. 2022), although sedative effects may involve other subtypes, depending on how sedation is measured (Behlke et al. 2016; Duke et al. 2018).
The present study used a pharmaco-EEG approach to evaluate the role of α1GABAAR subtypes in EEG-based sedation measures and EEG spectral power during the active phase of a rat’s light/dark cycle. The conventional benzodiazepine, triazolam, engendered an expected dose-related decrease in time spent in wake, with a concomitant increase in time spent in SWS. Similarly, the α1GABAAR subtype-preferring drug, zolpidem, demonstrated a trend for decreased time in the wake stage and showed a significant increase in SWS. Surprisingly, a similar pattern of effects was seen with the α1GABAAR-sparing ligand, L-838,417, which significantly reduced wake time and increased SWS at the highest dose tested. Of note, the magnitude of the effect of L-838,417 on SWS (mean = 34.4%) was smaller than observed with either triazolam (55.4%) or zolpidem (44.0%).
These results raise the possibility that an investigational α1GABAAR-sparing compound may have sedative effects at high enough doses, even in the absence of measurable in vitro activity at α1GABAAR subtypes. In fact, all such compounds tested to date in human subjects have shown some degree of sedation, albeit relatively mild in quality and/or magnitude. In this regard, compounds with similar receptor selectivity have resulted in reports of “somnolence” and “dizziness” in clinical trials, including TPA023B (Atack et al. 2011), AZD7325 (Chen et al. 2014), and darigabat (formerly PF-06372865; Nickolls et al. 2018). Moreover, L-838,417 and other α1GABAAR-sparing compounds have been shown to induce a behavioral measure referred to as “rest/sleep posture” in non-human primates, considered to be a mild form of sedative effects (Rowlett et al. 2005; Duke et al. 2018; Berro et al. 2019). Consistent with these pharmacological findings, Behlke et al. (2016) demonstrated that in transgenic mice with receptors other than α3GABAAR rendered insensitive to benzodiazepines, a behavioral measure of sedation (decrease in locomotor activity) was observed with diazepam administration, consistent with the idea that subtypes other than the α1GABAAR may play a role in sedative effects of these ligands.
Previous studies have distinguished α1GABAAR-sparing compounds from non-selective and α1GABAAR-preferring benzodiazepine-type drugs based on their profile of effects on EEG-measured spectral power (e.g., Christian et al. 2015). Consistent with this idea of a spectral power “signature,” we found that compared with the non-selective benzodiazepine triazolam, the α1GABAAR-preferring ligand zolpidem had relatively more robust effects on delta band power, whereas the α1GABAAR-sparing compound, L-838,417, only significantly increased beta band power. Triazolam and zolpidem, but not L-838,417, significantly decreased theta band power. These findings are notable in the context of sleep–wake state analyses, in which L-838,417 significantly increased SWS despite not significantly increasing delta power. Of note, the sedative effects of L-838,417 were less robust compared to those of triazolam and zolpidem, in terms of SWS magnitude of effect, and the treatment effect sizes for zolpidem for delta band increases were strikingly more robust than those of either triazolam or L-838,417. Regardless, these results demonstrate that sedation can occur in the context of sleep measures during the active phase of a light/dark cycle, even in the absence of statistically reliable increases in relative delta power bands.
Based on relative power, triazolam and L-838,417 significantly enhanced higher frequency bands, with the most reliable findings in the beta frequency range. These findings are consistent with previous pharmaco-EEG research across multiple species (e.g., Saletu et al. 2006; Christian et al. 2015; Berro et al. 2021). Moreover, this beta frequency increase has been proposed extensively as a quantitative biomarker of GABAA receptor modulation (Visser et al. 2003). Consistent with our findings with L-838,417, other α1GABAAR-sparing compounds also increase beta power with relatively few, if any, effects on lower frequency bands (Christian et al. 2015; Nickolls et al. 2018). Collectively, our findings and the previous research with α1GABAAR-sparing compounds support the idea that a selective increase in beta power may represent a “signature” for selectivity at α2GABAAR, α3GABAAR, and/or α5GABAAR subtypes. This signature was evident in the pattern of treatment effect sizes, with the non-selective triazolam showing strongest effects in the middle frequency bands (theta to beta), the α1GABAAR-preferring zolpidem showing strongest effects at the lower frequency power bands, and the α1GABAAR-sparing L-838,417 showing strongest effects at the higher frequencies (alpha to gamma). Evaluation of treatment effect sizes may provide a novel summary metric for identifying GABAAR signatures in vivo.
In general, the effects of all three ligands on relative power, as well as effect sizes per power band, were relatively consistent across the 30-min recording period, albeit with some evidence of the effects waning by the third 10-min session. Indeed, effects of the ligands were not statistically significant following the 30-min recording period, indicating that these ligands were relatively short-acting. The most striking effect that was time-dependent was a decrease in delta power band in the third 10-min period by L-838,417. This effect varied by dose, with only the 1.0 and 3.0 mg/kg doses demonstrating this decrease significantly. Characteristically, benzodiazepines tend to inhibit delta power when administered during the inactive phase (Buchsbaum et al. 1985; Dijk et al. 1989; Davis et al. 2011), suggesting that a time-dependent, active phase-based decrease in delta power may be another signature of α1-sparing compounds.
Strong support for the idea that selective enhancement of higher spectral band frequencies may be a signature for α2/3/5GABAAR selectivity is provided by Christian et al. (2015). In their paper, Christian et al. (2015) demonstrated a similar pattern of beta power increases with three α1GABAAR-sparing compounds: TPA023, AZD7325, and AZD6280. These compounds have lower α5GABAAR activity than L-838,417, raising the possibility that α5GABAARs are not involved in the GABAA modulator-induced increase in beta power. One difference between our results and those of Christian et al. (2015) is that these authors found significant increases in the higher frequency gamma bands, whereas we found no effects on these power bands with L-838,417. As mentioned, a major difference between L-838,417 and the ligands tested by Christian et al. (2015) is their lack of α5GABAAR efficacy; however, gamma band power was also increased by darigabat, a compound with α5GABAAR activity very similar to that of L-838,417 (Nickolls et al. 2018). Therefore, the differences in results likely do not represent pharmacological differences and may, instead, point to methodological differences. With Christian et al. (2015), the pharmaco-EEG recordings occurred while the rats were performing an operant responding-based task. However, Nickolls et al. (2018) also showed significant increases in gamma power with darigabat in subjects not performing a behavioral task. Another potentially relevant methodological detail of note is light/dark cycle: Nickolls et al. (2018) recorded spectral activity during the beginning of the inactive (light) phase, suggesting the possibility that gamma power is sensitive to α1GABAAR-sparing compounds during the beginning of the sleep cycle. While understanding these differences awaits further study, these findings nonetheless suggest that benzodiazepines and selective ligands can provide an EEG “signature” for receptor selectivity, although the environmental context (e.g., whether or not a behavioral task is performed, active vs. inactive phase of the sleep cycle) of the experiment must be considered carefully.
In addition to providing a pharmaco-EEG signature for benzodiazepine action, increases in beta power have been proposed over the years as having predictive validity for anxiolytic activity. A growing amount of evidence from both rodent and non-human primate studies suggests that α2GABAAR subtypes mediate benzodiazepine-induced anxiolysis (e.g., Engin et al. 2018; Meng et al. 2020). In parallel, studies using transgenic technology have shown that, in comparison with wild-type mice, mice engineered with benzodiazepine-insensitive α2GABAARs demonstrated no beta band increases when tested with diazepam (Kopp et al. 2004). Collectively, these findings provide support for the proposal that selective beta power increases may reflect selective activity at α2GABAARs, which, if borne out, provides a powerful in vivo approach for identifying anxiolysis associated with α2GABAARs.
In the present study, we also used the treatment effect size data to examine the robustness of our data set for statistical significance. Based on many criteria (e.g., Cohen’s rule of thumb), the level of effect needed to result in significance was relatively large at 0.559, resulting in an experimental approach that was somewhat conservative. However, as shown in the supplemental materials, the relationship of effect size and p value was strong, with relatively few outliers. We also used a single-subject design, relying on a priori comparisons to a vehicle control, which increases power (and reduces the number of animals needed). Most importantly, our data were highly consistent with existing literature, with the exception of the gamma power band results that may reflect methodological factors, providing a degree of external validity.
In summary, the present study found that all ligands evaluated induced changes in sleep–wake states during the active phase consistent with a decrease in wake and an increase in SWS, but no REM sleep changes, providing a measure of sedation based not on activity but brain electrophysiological changes. The degree of wake/SWS changes appeared to be related to the magnitude of delta power band increases induced by the ligands, with the strongest effect sizes engendered by the α1GABAAR-preferring drug zolpidem and the weakest effect sizes by the α1GABAAR-sparing compound, L-838,417. Consistent with other research, a selective increase in beta band power was obtained with L-838,417, which may be associated with α2GABAAR action as well as anxiolysis. Important for drug discovery and development efforts is the potential presence of putatively mild sedative effects with an α1GABAAR-sparing compound. However, as suggested by clinical trials with similar compounds, α1GABAAR-sparing compounds may have at least reduced sedation compared with available anxiolytic benzodiazepines, reflecting a clinically significant advantage for this approach to anxiolytic development.
References
Atack JR, Hallett DJ, Tye S, Wafford KA, Ryan C, Sanabria-Bohórquez SM, Eng WS, Gibson RE, Burns HD, Dawson GR, Carling RW, Street LJ, Pike A, De Lepeleire I, Van Laere K, Bormans G, de Hoon JN, Van Hecken A, McKernan RM et al (2011) Preclinical and clinical pharmacology of TPA023B, a GABAA receptor α2/α3 subtype-selective partial agonist. J Psychopharmacol 25:329–344
Behlke LM, Foster RA, Liu J, Benke D, Benham RS, Nathanson AJ, Yee BK, Zeilhofer HU, Engin E, Rudolph U (2016) A pharmacogenetic “restriction-of-function” approach reveals evidence for anxiolytic-like actions mediated by α5-containing GABAA receptors in mice. Neuropsychopharmacology 41:2492–2501
Berro LF, Overton JS, Reeves-Darby JA, Rowlett JK (2021) Alprazolam-induced EEG spectral power changes in rhesus monkeys: a translational model for the evaluation of the behavioral effects of benzodiazepines. Psychopharmacology (Berl) 238:1373–1386
Berro LF, Rüedi-Bettschen D, Cook JE, Golani LK, Li G, Jahan R, Rashid F, Cook JM, Rowlett JK, Platt DM (2019) GABAA receptor subtypes and the abuse-related effects of ethanol in rhesus monkeys: experiments with selective positive allosteric modulators. Alcohol Clin Exp Res 43:791–802
Buchsbaum MS, Hazlett E, Sicotte N, Stein M, Wu J, Zetin M (1985) Topographic EEG changes with benzodiazepine administration in generalized anxiety disorder. Biol Psychiatry 20:832–842
Cerne R, Lippa A, Poe MM, Smith JL, Jin X, Ping X, Golani LK, Cook JM, Witkin JM (2022) GABAkines—advances in the discovery, development, and commercialization of positive allosteric modulators of GABAA receptors. Pharmacol Ther 234:108035
Chen X, Jacobs G, de Kam M, Jaeger J, Lappalainen J, Maruff P, Smith MA, Cross AJ, Cohen A, van Gerven J (2014) The central nervous system effects of the partial GABA-Aα2,3 -selective receptor modulator AZD7325 in comparison with lorazepam in healthy males. Br J Clin Pharmacol 78:1298–1314
Christian EP, Snyder DH, Song W, Gurley DA, Smolka J, Maier DL, Ding M, Gharahdaghi F, Liu XF, Chopra M, Ribadeneira M, Chapdelaine MJ, Dudley A, Arriza JL, Maciag C, Quirk MC, Doherty JJ (2015) E EG-β/γ spectral power elevation in rat: a translatable biomarker elicited by GABAAα2/3-positive allosteric modulators at non-sedating anxiolytic doses. J Neurophysiol 113:116–131
Coenen AM, van Luijtelaar EL (1991) Pharmacological dissociation of EEG and behavior: a basic problem in sleep-wake classification. Sleep 14:464–465
Davis CJ, Clinton JM, Jewett KA, Zielinski MR, Krueger JM (2011) Delta wave power: an independent sleep phenotype or epiphenomenon? J Clin Sleep Med 15(5 Suppl):S16–S18
Dijk DJ, Beersma DG, Daan S, van den Hoofdakker RH (1989) Effects of seganserin, a 5-HT2 antagonist, and temazepam on human sleep stages and EEG power spectra. Eur J Pharmacol 171:207–218
Drinkenburg WH, Ahnaou A, Ruigt GS (2015a) Pharmaco-EEG studies in animals: a history-based introduction to contemporary translational applications. Neuropsychobiology 72:139–150
Drinkenburg WHIM, Ruigt GSF, Ahnaou A (2015b) Pharmaco-EEG studies in animals: an overview of contemporary translational applications. Neuropsychobiology 72:151–164
Duke AN, Meng Z, Platt DM, Atack JR, Dawson GR, Reynolds DS, Tiruveedhula VVNPB, Li G, Stephen MR, Sieghart W, Cook JM, Rowlett JK (2018) Evidence that sedative effects of benzodiazepines involve unexpected GABAA receptor subtypes: quantitative observation studies in rhesus monkeys. J Pharmacol Exp Ther 366:145–157
Engin E, Benham RS, Rudolph U (2018) An emerging circuit pharmacology of GABAA receptors. Trends Pharmacol Sci 39:710–732
Ghit A, Assal D, Al-Shami AS, Hussein DEE (2021) GABAA receptors: structure, function, pharmacology, and related disorders. J Gen Eng Biotech 19:1–15
Gilles C, Luthringer R (2007) Pharmacological models in healthy volunteers: their use in the clinical development of psychotropic drugs. J Psychopharmacol (Oxf) 21:272–282
Jongsma ML, van Rijn CM, van Egmond J, van Schaijk WJ, Sambeth A, Coenen AM (2000) The influence of diazepam on the electroencephalogram-evoked potential interrelation in rats. Neurosci Lett 293:83–86
Knoflach F, Bertrand D (2021) Pharmacological modulation of GABAA receptors. Curr Op Pharmacol 59:3–10
Kopp C, Rudolph U, Keist R, Tobler I (2003) Diazepam-induced changes on sleep and the EEG spectrum in mice: role of α3-GABAA receptor subtype. Eur J Neurosci 17:2226–2230
Kopp C, Rudolph U, Löwe K, Tobler I (2004) Modulation of rhythmic brain activity by diazepam: GABAA receptor subtype and state specificity. Proc Natl Acad Sci USA 101:3674–3679
Leiser SC, Dunlop J, Bowlby MR, Devilbliss DM (2011) Aligning strategies for using EEG as a surrogate biomarker: a review of preclinical and clinical research. Biochem Pharmacol 81:1408–1421
Liu J, Singh H, White PF (1996) Electroencephalogram bispectral analysis predicts the depth of midazolam-induced sedation. Anesthesiology 84:64–69
McKernan RM, Rosahl TW, Reynolds DS, Sur C, Wafford KA, Atack JR, Farrar S, Myers J, Cook G, Ferris P, Garrett L, Bristow L, Marshall G, Macaulay A, Brown N, Howell O, Moore KW, Carling RW, Street LJ et al (2000) Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABA(A) receptor alpha1 subtype. Nat Neurosci 3:587–592
Meng Z, Berro LF, Sawyer EK, Rüedi-Bettschen D, Cook JE, Li G, Platt DM, Cook JM, Rowlett JK (2020) Evaluation of the anti-conflict, reinforcing, and sedative effects of YT-III-31, a ligand functionally selective for α3 subunit-containing GABAA receptors. J Psychopharmacol 34:348–357
Nickolls SA, Gurrell R, van Amerongen G, Kammonen J, Cao L, Brown AR, Stead C, Mead A, Watson C, Hsu C, Owen RM, Pike A, Fish RL, Chen L, Qiu R, Morris ED, Feng G, Whitlock M, Gorman D et al (2018) Pharmacology in translation: the preclinical and early clinical profile of the novel α2/3 functionally selective GABAA receptor positive allosteric modulator PF-06372865. Br J Pharmacol 175:708–725
Rowlett JK, Platt DM, Lelas S, Atack JR, Dawson GR (2005) Different GABAA receptor subtypes mediate the anxiolytic, abuse-related, and motor effects of benzodiazepine-like drugs in primates. Proc Natl Acad Sci USA 102:915–920
Saletu B, Anderer P, Saletu-Zyhlarz GM (2006) EEG topography and tomography (LORETA) in the classification and evaluation of the pharmacodynamics of psychotropic drugs. Clin EEG Neurosci 37:66–80
Tobler I, Kopp C, Deboer T, Rudolph W (2001) Diazepam-induced changes in sleep: role of the α1GABAA receptor subtype. Proc Natl Acad Sci USA 98:6464–6469
van Lier H, Drinkenburg WH, van Eeten YJ, Coenen AM (2004) Effects of diazepam and zolpidem on EEG beta frequencies are behavior-specific in rats. Neuropharmacology 47:163–174
Versavel M, Leonard JP, Herrmann WM (1995) Standard operating procedure for the registration and computer-supported evaluation of pharmaco-EEG data. 'EEG in Phase I' of the Collegium Internationale Psychiatriae Scalarum (CIPS). Neuropsychobiology 32:166–170
Visser SAG, Wolters FLC, Gubbens-Stibbe JM, Tukker E, Van der Graaf PH, Peletier LA, Danhof M (2003) Mechanism-based PK/PD modelling of the EEG effects of GABAA receptor modulators: in vitro/in vivo correlations. J Pharmacol Exp Ther 304:88–101
Funding
This research was supported by USPHS grants DA011792, DA043204, DA049886, DA052801, AA029023, AA029306, and GM121334.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOCX 83 kb)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Reeves-Darby, J.A., Berro, L.F., Platt, D.M. et al. Pharmaco-EEG analysis of ligands varying in selectivity for α1 subunit-containing GABAA receptors during the active phase in rats. Psychopharmacology 240, 2561–2571 (2023). https://doi.org/10.1007/s00213-023-06450-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-023-06450-3