
Abstract
Social Signal Transduction Theory of Depression is a biologically plausible, multi-level theory that describes neural, physiologic, molecular, and genomic mechanisms that link experiences of social-environmental adversity with internal biological processes that drive depression pathogenesis, maintenance, and recurrence. Central to this theory is the hypothesis that interpersonal stressors involving social threat (e.g., social conflict, evaluation, rejection, isolation, and exclusion) upregulate inflammatory processes that can induce several depressive symptoms, including sad mood, anhedonia, fatigue, psychomotor retardation, and social-behavioral withdrawal. The original article describing this formulation (Psychol Bull 140:774–815, 2014) addressed critical questions involving depression onset and recurrence, as well as why depression is strongly predicted by early life stress and comorbid with anxiety disorders and certain physical disease conditions, such as asthma, rheumatoid arthritis, chronic pain, and cardiovascular disease. Here, we extend the theory to help explain sex differences in depression prevalence, which is a defining feature of this disorder. Central to this extension is research demonstrating that ovarian hormone fluctuations modulate women’s susceptibility to stress, brain structure and function, and inflammatory activity and reactivity. These effects are evident at multiple levels and are highly context-dependent, varying as a function of several factors including sex, age, reproductive state, endogenous versus exogenous hormones, and hormone administration mode and dose. Together, these effects help explain why women are at greater risk for developing inflammation-related depressed mood and other neuropsychiatric, neurodevelopmental, and neurodegenerative disorders during the reproductive years, especially for those already at heightened risk for depression or in the midst of a hormonal transition period.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Depression is a serious psychiatric condition that often emerges early in life and exerts a substantial social and economic impact on society that exceeds $200 billion per year in the USA alone (Greenberg et al. 2015). Nearly 17% of all men and 25% of all women experience a major depressive episode during their lifetime (Kessler et al. 2010), and although not all individuals develop multiple episodes, recurrence is common with up to 60% of people experiencing more than one lifetime depressive episode (Monroe et al. 2014b). This impact is further compounded by the fact that depression is associated with increased risk for several somatic health conditions that have an immunologic basis and can greatly affect wellbeing, including asthma, rheumatoid arthritis, chronic pain, cardiovascular disease, certain cancers, and neurodegenerative disorders (Slavich in press).
Given the serious implications of depression for overall health, one might expect that researchers have long possessed biologically plausible theories of major depressive disorder (MDD) that could be used to develop effective treatments. Progress on this front has been slow, though, due in part to the high cost historically associated with assessing neural, immunologic, genetic, and genomic processes underlying depression in humans. However, these methods have become considerably more affordable in recent years, leading to a proliferation of exciting new data revealing how social-environmental adversity influences biological processes that promote risk for depression and related diseases.
One of the most recent and important insights from this work has involved the realization that psychological stress can strongly upregulate components of the immune system involved in inflammation (Segerstrom and Miller 2004; Steptoe et al. 2007). Inflammation, in turn, is known to induce depression-like behavior in animals and several symptoms of depression in many (although not all) individuals (Dantzer et al. 2008; Miller et al. 2009). Inflammatory mediators, particularly pro-inflammatory cytokines, can also promote anxiety symptoms and increase risk for the types of physical health problems that frequently co-occur with MDD (O’Donovan et al. 2013), thus making a focus on inflammation critically important for developing a more complete understanding of the full range of clinical symptoms and somatic ailments that frequently accompany this disorder.
Although studies on stress and inflammation, and inflammation and depression, have largely represented distinct lines of enquiry, Slavich and Irwin (2014) recently integrated these literatures to describe how experiences of life stress influence neural, physiologic, molecular, and genomic processes that drive depression pathogenesis, maintenance, and recurrence. The resulting Social Signal Transduction Theory of Depression represents the first biologically plausible, multi-level theory of MDD. In the original article describing this formulation, the authors addressed several important questions in depression research, including how depression develops, why it frequently recurs, why it is strongly predicted by early life stress, and why it often co-occurs with symptoms of anxiety and with certain physical disease conditions. In the present paper, we extend this discussion to help explain one of the most striking features of depression—namely, the dramatic increase in risk for MDD in women relative to men following the pubertal transition in adolescence. To accomplish this goal, we first summarize Social Signal Transduction Theory of Depression and existing research supporting this theory. Second, we describe known sex differences in depression and inflammation, and how sex hormones influence immunologic, neuroinflammatory, and neuroendocrine processes that affect depression risk. Third, we discuss the relevance of these findings for updating Social Signal Transduction Theory of Depression. Finally, we suggest some possible avenues for future clarification and research.
Social Signal Transduction Theory of Depression
Several contemporary theories have attempted to explain how life stress increases risk for depression by affecting social-environmental (e.g., Brown and Harris 1978), psychodynamic (e.g., Blatt 2004), and cognitive (e.g., Beck 1967) processes. Although these formulations have served as invaluable blueprints for studying factors that contribute to and affect depression, their ability to provide a complete biopsychosocial picture of the disorder has been limited by three main issues. First, these theories have accounted for some symptoms and features of MDD—particularly those involving social, psychological, and cognitive processes—but they have not provided satisfactory explanations for somatic or neurovegetative aspects of the disorder. Second, they have not typically included biological data and, consequently, have not described mechanisms that are most proximally responsible for generating depressive symptoms, let alone the full clinical and biobehavioral phenotype. Finally, these theories have generally characterized depression at one major level of analysis (e.g., cognition) but have not drawn convincing links with other up- or down-stream processes that are implicated in this disorder.
Social Signal Transduction Theory of Depression addresses these issues by spanning several major levels of analysis to generate a biologically plausible explanation for both cognitive and somatic symptoms of MDD, as well as several other clinical phenomena that are frequently experienced by many depressed individuals (e.g., anxiety, chronic pain) but not presently regarded as symptoms of the disorder. The theory is grounded in the basic understanding that the primary purpose of the human brain and immune system is to keep the body safe from physical and biological threats that could cause illness or death if left unaddressed (Slavich in press; Slavich and Auerbach 2018). To accomplish this task, the brain continually surveys the social environment for threats that historically increased an individual’s risk of physical harm or wounding. These social-environmental stressors, which include things like social conflict, isolation, rejection, and exclusion, get represented in brain regions that play a role in processing experiences of social and physical threat, pain, and interoceptive cues. These brain regions in turn project to lower-level regions that upregulate systemic inflammatory activity via several non-mutually exclusive pathways, including the sympathetic nervous system (SNS), hypothalamic-pituitary-adrenal (HPA) axis, vagus nerve, and meningeal lymphatic vessels (Irwin and Cole 2011; Slavich and Cole 2013; Slavich and Irwin 2014).
This multi-level neurobiological response to social threat, which we have previously called the Conserved Transcriptional Response to Adversity (CTRA; Slavich and Cole 2013; Slavich and Irwin 2014), is characterized by a systematic shifting of immune system resources toward an increased pro-inflammatory and reduced anti-viral (i.e., type I interferon response) state. Small soluble proteins called cytokines play a central role in this response and are critically relevant for understanding the pathogenesis of depression and depression-related physical disease states, as they exert numerous physiologic, cognitive, and behavioral effects that can strongly affect disease risk (Hennessy et al. 2009; Medzhitov 2001; Medzhitov 2008). At specific sites of tissue damage or infection, for example, cytokines can promote redness, heat, swelling, and pain that help accelerate wound healing, limit the spread of infection, and heighten a host’s awareness of an injury (Murphy 2011). At a more systemic level, cytokines can induce the production of the acute-phase protein C-reactive protein (CRP), which acts together with different cytokines to increase body temperature, heart rate, and respiratory rate (Irwin and Slavich 2017).
These biobehavioral effects are critical for survival as they combine to kill pathogens, promote wound healing, and help the body conserve energy during times of injury and infection (Poon et al. 2013; Ricciotti and FitzGerald 2011). Cytokines can also communicate with the central nervous system to induce a constellation of behaviors called sickness behaviors, which closely resemble several “official” depressive symptoms, but also many other symptoms and somatic states that frequently accompany depression (Dantzer et al. 2008; Hart 1988). Included in this list of behaviors are anhedonia, fatigue, psychomotor retardation, and social-behavioral withdrawal, in addition to increased pain and threat sensitivity. Anhedonia and fatigue help individuals recuperate and recover from injury; psychomotor retardation and social-behavioral withdrawal reduce the likelihood that an infection will spread to nearby conspecifics; and pain and threat sensitivity draw an individual’s attention to bodily injury and potential threats in the environment. This multi-level biobehavioral response thus has notable benefits and costs. On the plus side, it dramatically increases a person’s likelihood of survival during times of actual physical injury or threat, which explains why it was highly conserved over the course of evolution. The downside, however, is that this response can also increase a person’s risk of developing MDD and other inflammation-related conditions, especially when it is prolonged or repeatedly engaged.
Summary of supporting evidence
Seven lines of research presently support Social Signal Transduction Theory of Depression. First, the specific types of life stressors that are most strongly associated with onset of MDD are those that involve social threat in general, and a combination of interpersonal loss and social rejection in particular (Kendler et al. 2003; Slavich et al. 2010a; Slavich et al. 2011). Previously, we have called these stressors targeted rejection life events (Slavich et al. 2009). Second, a large literature including both naturalistic and laboratory-based experimental studies has shown that these same social stressors are the strongest triggers of the CTRA (e.g., Murphy et al. 2013; Murphy et al. 2015; Takizawa et al. 2015; for reviews, see Kiecolt-Glaser et al. 2010; Marsland et al. 2017; Segerstrom and Miller 2004; Slavich and Irwin 2014). Third, depression is well-known to frequently co-occur with several somatic disorders and physical disease conditions that have an inflammatory basis, including rheumatoid arthritis, inflammatory bowel disease, metabolic syndrome, coronary heart disease, and chronic pain (Goodwin et al. 2004; Graff et al. 2009; Poole et al. 2009).
Fourth, basal levels of systemic inflammatory activity are elevated in a substantial proportion of individuals with depression, as indexed by CRP and the pro-inflammatory cytokines interleukin (IL)-1, IL-6, and tumor necrosis factor (TNF)-α (Howren et al. 2009). Moreover, there is evidence that elevations in these biomarkers precede the development of MDD (Gimeno et al. 2009) and that at least some symptoms of depression that are associated with inflammation can be alleviated with antidepressant medications (Hernández et al. 2008). Fifth, three different experimental inflammatory challenges—namely, interferon-α (IFN-α) administration, typhoid vaccination, and endotoxin administration—have been found to evoke depressive-like behaviors in animal models of depression and diagnosable forms of MDD in approximately 45–50% of individuals exposed to these challenges (Capuron and Miller 2004; Raison et al. 2006). In addition, depressive symptoms elicited by these challenges can be blocked with cytokine synthesis blockers or cytokine antagonists in animals and potentially alleviated with antidepressants in humans (Hannestad et al. 2011). Sixth, the three inflammatory challenges mentioned above have been found to alter neural or metabolic activity in the basal ganglia, cerebellum, anterior cingulate cortex, and ventral striatum, all of which have been implicated in depression (Capuron and Miller 2011; Zunszain et al. 2011). Moreover, several studies have suggested that neural activity in brain regions that have been implicated in MDD is associated with individuals’ inflammatory reactivity to social stress (Muscatell et al. 2015; Muscatell et al. 2016; Slavich et al. 2010b; see also Kumar et al. 2015). Finally, several double-blind, randomized, placebo-controlled trials have suggested that three anti-inflammatory agents—namely, the COX-2 inhibitor, celecoxib, and the TNF-α antagonists, etanercept and infliximab—can alleviate depressive symptoms in at least some individuals (Müller et al. 2006; Raison et al. 2013; Tyring et al. 2006).
Sex differences in risk for depression
Despite this large body of evidence supporting Social Signal Transduction Theory of Depression, one feature of MDD not addressed in the original theoretical formulation involves that of sex differences in risk for depression. This is notable given that sex-based differences in lifetime rates of MDD are a defining characteristic of the disorder (Thapar et al. 2012). Prior to the pubertal transition, for example, rates of depression are similar and relatively low, with approximately 3% of children meeting diagnostic criteria for MDD over the past year (Merikangas et al. 2010). Following puberty, though, rates of MDD increase nearly fivefold for both boys and girls—which itself is striking—but substantial sex differences also emerge such that adolescent girls become at least twice as likely as boys to develop depression (Avenevoli et al. 2015; Hankin and Abramson 1999). Several mechanisms have been proposed to explain this dramatic increase in risk for MDD for girls relative to boys, including stress generation, negative cognitive biases, and altered HPA axis reactivity to stress (e.g., Gold 2015; Hammen 2006; Hankin and Abramson 2001; Kuehner 2017). However, none of these mechanisms are directly capable of inducing depressive symptoms.
Highlighting possible interactions between sex hormones and Social Signal Transduction Theory of Depression may be useful in this regard, as it provides a framework for understanding how sex hormones can influence processes that are directly involved in depression pathogenesis, maintenance, and recurrence. Indeed, an interplay between sex hormones, psychosocial stress, and inflammation could be one pathway underpinning sex differences in MDD susceptibility. In the following sections, therefore, we first review data highlighting known sex differences in inflammation and the role that sex hormones play as mediators of the immune system. Second, we review data linking sex hormones with neuroinflammatory and neuroendocrine dynamics. Finally, we discuss how sex hormones can interact with psychosocial stress and neuroinflammatory processes to modulate mood and risk for depression.
Sex differences in inflammation
Sex differences in the human immune response and resistance to infection and inflammatory disease are well documented (Ferretti et al. 2018; Klein and Flanagan 2016; McClelland and Smith 2011; McCombe et al. 2009). Generally, females show greater susceptibility to autoimmune disease development and males show increased susceptibility to non-reproductive malignant cancers (Klein and Flanagan 2016). Sex differences have also been described for a variety of infectious diseases, but with a lesser magnitude (for a review, see Klein and Flanagan 2016).
Gender and sex both likely play a role in structuring these effects. For example, gender can influence a variety of risk factors for immune-related disorders, including exposure to infectious agents and environmental toxins, dietary habits, health behaviors, and healthcare-seeking behavior and access (Ngo et al. 2014). Our main interest here, though, are sex differences in inflammation-related health conditions that can result from environmental and preprogrammed hormonal or genetic cues, and from the complex interaction of environmental impacts mediated by epigenetic regulation driving gene expression for males and females (Deak et al. 2015; Hanamsagar and Bilbo 2016). Sex differences in immunologic activity can emerge early in life (e.g., during prenatal development) and lead to sex-based differences in disease risk during childhood and adulthood. Indeed, this literature is extensive and comprehensively reviewed elsewhere (see Goldstein et al. 2016; Klein and Flanagan 2016). Relevant research using animal model systems has also been summarized in detail elsewhere (e.g., see Deak et al. 2015). Given that age and reproductive status represent important determinants of sex-related differences in the immune response, below, we mainly examine sex differences in inflammation and inflammation-related health problems beginning in puberty, which is when sex differences in depression first emerge, and focus primarily on human studies, which are most directly relevant for understanding differences in risk for MDD in women versus men.
Sex hormones as hormonal mediators of the immune system
Estrogen
Estrone (E1), 17beta-estradiol (E2), estriol (E3), and estetrol (E4) are the four major types of endogenous estrogens (Thomas and Potter 2013). Estrogen production occurs primarily in the ovaries, and in the placenta during pregnancy, while small amounts are produced by other tissues such as the liver, pancreas, bone, adrenal glands, skin, brain, and adipose tissue (Hemsell et al. 1974). E2 is considered the most prevalent and potent form of circulating estrogen (Thomas and Potter 2013). E2 effects on the immune system are reflected not only by E2 concentration but also include density, distribution, and type of estrogen receptor (Thomas and Potter 2013). Two main subtypes have been described—namely, ERα and ERβ—and both exhibit different expression in various lymphoid tissue cells, such as lymphocytes, macrophages, and dendritic cells (Kovats 2015). For example, ERα is highly expressed in T cells and ERβ is highly expressed in B cells (Phiel et al. 2005). Furthermore, non-classical ER signaling, which allows for protein-protein interactions between ERs and ERE-independent transcription factors, also occurs in immune cells (Kovats 2015).
E2 affects both cell-mediated and humoral immune responses (Klein and Flanagan 2016). For example, whereas a low E2 state stimulates cell-mediated immunity and TH1-responses, which promote interferon gamma (IFN-γ)–driven pro-inflammatory activity that is responsible for killing intracellular parasites and perpetuating autoimmune responses, a high E2 state promotes humoral immune reactions and TH2-responses, which are driven by IL-4, IL-5, and IL-13 that promote the antibody immunoglobulin E (IgE) and eosinophilic responses in atopy, and by IL-10, which has anti-inflammatory effects (Berger 2000; Straub 2007). E2 can increase the neutrophilic cell count in both blood (Jilma et al. 1994) and the lungs (Robinson et al. 2014). Moreover, whereas in low concentrations E2 stimulates the production of pro-inflammatory cytokines, such as IL-1, IL-6, and TNF-α, in high concentrations, E2 impedes the production of these cytokines (Bouman et al. 2005). E2 can also initiate the differentiation from monocytes into inflammatory dendritic cells, which in turn leads to increased IFN-α production, stimulation of the TLR-signaling pathways, and activation of nuclear factor-κB (NF-κB), interferon regulatory factors (IRFs), or mitogen-activated protein (MAP) kinases (Kawasaki and Kawai 2014) to regulate the expression of cytokines, chemokines, and type I IFNs, as well as greater internalization and antigen presentation to naïve T cells (Seillet et al. 2012).
Progesterone
Progesterone (P4) is synthesized by the corpus luteum in the ovary during the second half of the menstrual cycle (Barbieri 2014) and by the placenta during pregnancy (Morel et al. 2016). P4 signaling is mainly mediated via progesterone receptors but can also occur via glucocorticoid and mineralocorticoid receptors. P4 also acts via S1-sigma receptors (Johannessen et al. 2011), as well as through its neurosteroid-active metabolites (e.g., 5α-dihydroprogesterone and allopregnanolone) as a modulator of the GABA-A receptor (Paul and Purdy 1992). P4 mostly promotes an anti-inflammatory state by decreasing IL-1ß and TNF production (Butts et al. 2007; Jones et al. 2010), inhibiting TLR and NF-κB-signaling (Hardy et al. 2006), and attenuating inducible nitric oxide synthase (iNOS) and nitric oxide (NO) production (Menzies et al. 2011).
Androgens
The most important androgens are testosterone (T); dihydrotestosterone (DHT), which is a metabolite of testosterone; and dehydroepiandrosterone (DHEA), which is the primary precursor of both testosterone and estrogens (Antoniou-Tsigkos et al. 2000). Testosterone can decrease NK cell activity; attenuate TNF, iNOS, and NO synthesis; and reduce the activity of NF-κB (Klein and Flanagan 2016). As a result, androgens are thought to mainly have immunosuppressive and anti-inflammatory effects (Klein and Flanagan 2016).
Summary
In summary, immune cells have sex hormone receptors and thus respond directly to changes in sex hormone levels (Buskiewicz et al. 2016). Sex hormones often do not act in isolation but rather in synergy; therefore, their effects on immune system activity are complex and not easy to characterize. In general, though, progesterone and testosterone have mostly been described as anti-inflammatory (Klein 2000). For estrogens, a U-curve pattern seems to best explain their effect on the immune system, wherein at low concentrations they are more often associated with pro-inflammatory function and at high concentrations they seem to promote an anti-inflammatory state (Klein 2000). These dynamics are directly related to risk for depression, as individual fluctuations across a woman’s own mean ovarian hormone levels greatly affect within-person vulnerability for MDD over time (Freeman et al. 2006; Frokjaer et al. 2015).
Sex hormones and regulation of neuroinflammation
Substantial support for a role for sex hormones in shaping inflammatory processes and risk for depression comes from research linking sex hormones with the regulation of neuroinflammatory activity, which has now been widely implicated in risk for MDD and several other neuropsychiatric disorders (Brites and Fernandes 2015; Kim et al. 2016). Research on this topic originates from the epidemiologic finding that women appear to be more vulnerable to developing neurodegenerative diseases across the lifespan than men, but especially during hormonal transition periods, as reviewed by Zsido et al. (2017). Over the past several decades, for example, it has become increasingly evident that sex hormones profoundly impact the occurrence and severity of neuropsychiatric and neurodegenerative disorders, often in conjunction with neuroinflammation (Villa et al. 2016). Indeed, sex hormones are now known to critically influence the activity of inflammatory mediators and immune dynamics within the central nervous system (CNS). These effects emerge early on in brain development and continue throughout adolescence (Juraska et al. 2013) and adulthood (Wise et al. 2008).
Mechanistically speaking, estrogen and progesterone both act via classical genomic receptors, as well as non-classical membrane-associated receptors. The estrogen receptors (ERα/β) (Gundlah et al. 2001; Mitra et al. 2003) and progesterone receptors (PRA/B) (Brinton et al. 2008) are highly expressed in brain areas involved in emotion and cognition, such as the amygdala and hippocampus (Barth et al. 2015). Moreover, ovarian hormones can act on multiple receptor types, such as voltage-gated ion channels including GABAA (Gulinello et al. 2001), NMDA (Foy et al. 1999), serotonin (Sumner and Fink 1998), and dopamine (Becker 1990) receptors.
Although these genomic actions of sex hormones require a comparably long time (e.g., ranging from minutes to hours) and are limited by the rate of protein biosynthesis, non-genomic modulation of membrane receptors is mostly faster and requires only milliseconds to seconds to exert an influence (Cornil et al. 2006; McEwen 1991). Ovarian hormones can also indirectly impact the CNS through the enhancement or mitigation of systemic processes that affect brain structure and function, such as inflammation. For example, both estrogen and progesterone exert acute effects on synaptic plasticity through the activation of multiple intracellular signaling pathways (Krebs et al. 2000; Minami et al. 1990; Singh 2001), including the mitogen-activated protein kinases/extracellular signal-regulated kinases (MAPK/ERK) pathway and the protein kinase B (Akt) pathway, both of which are involved in the promotion of cell survival (Singh 2001).
These dynamics have direct implications for neuropsychiatric disease risk, as synaptic plasticity and neuroinflammatory processes are tightly linked, and a dysregulation in their interplay has been proposed to underlie risk for depression as well as many neurodevelopmental and neurodegenerative disorders (Mottahedin et al. 2017). A key marker of neuroinflammation is microglial activation, for example, which is considered a hallmark of many CNS pathologies (Dheen et al. 2007). Additionally, most of these pathologies display a strong sexual dimorphism, including multiple sclerosis (Whitacre et al. 1999), anti-NMDA-receptor encephalitis (Dalmau et al. 2011), stroke (Bushnell 2008), and Alzheimer’s disease (Andersen et al. 1999). Another such disorder is autism, which affects boys nearly five times as often as girls (Werling and Geschwind 2013). Prenatal inflammation induced by maternal infection has been associated with increased risk for autism (Patterson 2011); abnormal levels of glial activation have been found in brains of autism patients postmortem (Vargas et al. 2005); and a candidate gene for autism has been identified that is responsive to sex hormones (Sarachana et al. 2011; see Baron-Cohen et al. 2005).
Importantly, a modulatory role for sex hormones has been described for all of the abovementioned pathologies. Estrogen receptors are present in microglial cells and estrogen clearly affects neuroinflammatory activity (Villa et al. 2016). Therefore, it should not be surprising that research involving cell lines, animal models, and humans all indicate a key role for sex hormones in regulating neuroinflammation. For example, in vitro experimental studies demonstrate that E2 can prevent morphological changes induced by lipopolysaccharide (LPS) and the subsequent synthesis of pro-inflammatory molecules (Vegeto et al. 2001). In addition, ovariectomized mice exhibit a greater inflammatory response to LPS stimulation than sham-operated mice (Bendusi et al., 2012). Moreover, tissue from the National Institute on Aging Alzheimer’s Disease brain banks has revealed upregulated macrophage-associated gene expression in brain samples from menopausal women as compared with those from younger, pre-menopausal women (Sarvari et al. 2012). Finally, estrogen appears to attenuate ischemic brain injury and inflammatory responding to LPS in females, especially when progesterone levels are high (Sunday et al. 2006). In sum, therefore, there is substantial evidence that sex hormones directly affect neuroinflammation, which in turn shapes lifespan risk for depression and other inflammation-related neuropsychiatric, neurodevelopmental, and neurodegenerative disorders.
Sex hormones, inflammation, and the hypothalamic-pituitary-adrenal axis
Additional support for the role of sex hormones in influencing inflammatory activity and risk for MDD comes from research demonstrating links between an imbalance of the HPA axis and pathologies related to inflammation in humans (Tsigos and Chrousos 2002; Yudkin et al. 2000). For example, the HPA axis is known to influence inflammatory responding and microglial activation across the lifespan (Sternberg 2006; Talbot et al. 2016), and as described above, neuroinflammatory processes play a central pathophysiologic role in depression, as well as in dysregulation of the autonomic nervous system and vascular, metabolic, and neurodegenerative pathologies (Slavich in press). Therefore, social-environmental stressors and exposure to other risk factors for dysregulated immune system activity occurring during critical periods of sexual differentiation of the human brain can strongly impact sexually dimorphic brain regions in sex-dependent ways (Goldstein et al. 2014). Consistent with this possibility, excesses in maternal glucocorticoids and abnormalities in immunologic activity have been found to have sex-dependent effects on fetal brain circuits that regulate mood, autonomic activity, blood pressure, and metabolism, resulting in recurrent MDD across the lifespan that is characterized by autonomic dysfunction, dysregulated immunologic stress reactivity, and cardiometabolic dysregulation later in life (Goldstein et al. 2018). Ultimately, an attenuated maternal HPA axis stress response may have important evolutionary value, as it can help protect the fetus from maternal stress exposure (Kajantie and Phillips 2006). At the same time, an estrogen-associated blunted HPA response has a distinct downside, as it confers increased risk for depression for the mother by lessening the natural anti-inflammatory effects of cortisol (Jarcho et al. 2013; Oldehinkel and Bouma 2011).
Interestingly, estrogen-related blunting of HPA axis activity appears to be relatively consistent across species. In rodents, for example, gonadectomy stimulates HPA axis response to endotoxin and IL-1 (Spinedi et al. 1992), and E2 administration has the opposite (i.e., attenuating) effect in both rats (Spinedi et al. 1992) and monkeys (Xia-Zhang et al. 1995). In humans, a similar pattern for a physiologic E2 dose appears in response to inflammatory challenge (Puder et al. 2001) and psychosocial stress (Komesaroff et al. 1999) in postmenopausal women. This effect may represent a healthy biological strategy for adapting to changing environmental circumstances during the reproductive years, as evidenced by menstrual cycle–associated modulation of neural activity in response to psychosocial stress (Goldstein et al. 2010) and E2 modulation of key stress-related circuitry in the brain (Jacobs et al. 2015). Given that women in remission from depression do not appear to show this hormonal capacity to regulate neural responsivity to stress (Jacobs et al. 2015), dysregulation of sex hormone–HPA axis dynamics may be a key trait of MDD.
Summary of evidence linking sex hormones with inflammatory and neuroendocrine processes
In summary, a wealth of research demonstrates that sex hormones regulate inflammatory activity in the CNS, where risk for depression is most directly constituted, and that they can affect the activity of the HPA axis, which has long been associated with risk for MDD. These effects are supported by findings highlighting sex-linked differences in inflammation and several inflammation-related disorders, by studies showing that sex hormones are associated with changes in inflammatory activity across the lifespan, and by mechanistic work demonstrating that sex hormones regulate inflammatory processes. These effects appear to be especially evident in females during different reproductive stages (Larson 2018), making hormonal transition periods a critical time for sex hormone-driven changes in risk for inflammation-related disorders such as depression (Eisenlohr-Moul et al. 2016; Owens and Eisenlohr-Moul 2018).
Stress, sex hormones, inflammation, and depression
Having presented evidence documenting a role for sex hormones in influencing inflammatory activity, we turn now to the relevance of these dynamics for depression. As alluded to earlier, life stress is strongly associated with risk for depression (Monroe et al. 2014a; Slavich 2016a; Slavich et al. 2014; Toussaint et al. 2016), and this effect is especially strong for women during most developmental periods but particularly earlier in life when first onsets of MDD typically occur (Harkness et al. 2010; see also Massing-Schaffer et al. 2019). Conversely, research has shown that greater social support during stress predicts less pronounced increases in depression for women but less so for men (Leifheit-Limson et al. 2010). Therefore, life stress exposure in general—and social stressors in particular—appear to be especially strongly related to MDD for women relative to men.
Such effects of life stress on mood have in turn been found to be modulated by fluctuations in ovarian hormones. For example, studies have shown that endogenous estradiol changes across the menstrual cycle alter mood and neural responses to psychosocial stress (Albert et al. 2015). In addition, perimenopausal variability in estradiol levels has been found to increase sensitivity to psychosocial stress, and the interplay of estradiol variability and psychosocial stress in turn predicts the subsequent development of depressed mood (Gordon et al. 2016). Finally, estradiol supplementation has been found to attenuate cortisol reactivity to acute social stress in perimenopausal women (Komesaroff et al. 1999), although the effects of estradiol appear to be highly context-dependent, as indicated by studies reporting enhanced negative mood to psychosocial stress after menopause (Newhouse et al. 2008).
Several theories of hormonal sensitivity to stress and depression have been proposed to elucidate mechanisms that may underlie these effects. The mechanisms identified have included estrogen-sensitive upregulation of monoaminergic metabolization (Rekkas et al. 2014; Sacher et al. 2015); differences at the cellular or molecular level in the actions of aromatase or in the ability of estradiol to regulate gene transcription (Mehta et al. 2014); the dynamic fluctuation in hippocampal estrogen receptor expression, which may be compromised by chronic stress (McEwen et al. 2012); linking estrogen-sensitive epigenetic mechanisms to serotonergic signaling changes (Mehta et al. in press); and the modulation of GABA-ergic transmission following changes in allopregnanolone (a progesterone-metabolite), induced by ovarian hormone fluctuations (Gordon et al. 2015). Moreover, emerging evidence suggests neurosteroid GABA-ergic inhibition of the release of pro-inflammatory mediators, such as TNF-α, via toll-like receptor 4 (TLR4) signal inhibition (Balan et al. 2019).
Support for the possibility that stress-related increases in inflammatory activity mediate sex-based differences in depression risk must, however, also be considered in light of studies showing male-specific associations between inflammatory markers (Ramsey et al., 2016)—in particular, CRP—and symptoms of anxiety and depression (Elovainio et al. 2009; Liukkonen et al. 2011; Vetter et al. 2013; Vogelzangs et al. 2012; see Raison et al. 2005). At the same time, exposure to social stress is associated with elevated basal levels of inflammatory activity (Shields and Slavich 2017; Slavich 2015; Slavich 2016b; Slavich in press) and with heightened inflammatory responses to social stress in women (Giletta et al. 2018). Second, increases in negative mood induced by inflammation have been well-documented in both healthy control participants (Strike et al. 2004; Wright et al. 2005) and in depressed patients (Niemegeers et al. 2016; Pace et al. 2006), and these effects are particularly strong for women (Irwin and Slavich 2017). Likewise, elevated basal levels of the pro-inflammatory cytokine IL-6 have been found to predict increases in depression among women at risk for MDD who have a history of childhood adversity (Miller and Cole 2012; see also Han et al. 2016). Third, forms of marital support that are known to mitigate the effects of stress on health are more strongly associated with reduced inflammatory activity for women than for men (Donoho et al. 2013). Finally, a recent longitudinal study that followed 2232 children from birth to 12 years old found that exposure to multiple forms of childhood adversity was associated with prospective increases in systemic inflammatory activity, but only for female youth (Baldwin et al. 2018).
Even stronger evidence linking sex-based differences in inflammatory processes with heightened depression risk in women comes from experimental studies examining these associations. First, some studies have shown that inflammatory challenges such as interferon-α administration lead to greater risk for MDD in women relative to men (Fontana et al. 2002; Gohier et al. 2003; Koskinas et al. 2002). Not all evidence consistently supports this finding and some research has found no sex differences in depressive symptoms following interferon-α treatment (Bonaccorso et al. 2002; Kraus et al. 2003; Miyaoka et al. 1999; see Raison et al. 2005). In contrast to these smaller individual studies, though, a recent meta-analysis of this literature that included 26 studies found that females are significantly more likely to develop MDD following interferon-α treatment (odds ratio = 1.40; 95% confidence interval = 1.02-1.91; Udina et al. 2012). This conclusion is consistent with prior consensus, which ranks depressed mood prior to treatment as the strongest predictor of developing depressive symptoms following interferon-α treatment, but views being female, a history of depression, and a substantial interferon-α dose or treatment duration as additional key risk factors.
Second, several experimental fMRI studies have shown that standardized inflammatory challenges alter associations between depression-relevant neural circuits, inflammation, and mood in a sex-dependent manner. In an early fMRI study on this topic, for example, LPS-induced increases in pro-inflammatory cytokine activity predicted changes in depressed mood for women but not for men; moreover, neural activity in brain areas associated with social pain (e.g., dorsal anterior cingulate cortex, anterior insula) mediated the relationship between changes in inflammatory activity and depressed mood but, again, only for women (Eisenberger et al. 2009). In a second study that used this same inflammatory challenge, increases in the pro-inflammatory cytokines TNF-α and IL-6 were strongly associated with feeling more socially disconnected for women but not for men (Moieni et al. 2015). In a third fMRI study that randomized male and female participants to receive either placebo or endotoxin, endotoxin (but not placebo) administration led to reduced activity in the ventral striatum, which mediates reward-related behavior, and this effect was found for females but not for males. Additionally, decreases in ventral striatum activity in the endotoxin condition were related to increases in inflammatory activity for females but not for males (Moieni et al. 2019). Finally, a fourth fMRI study that recruited a large proportion (70%) of depressed females found that higher CRP levels were associated with decreased functional connectivity within reward-related circuits including the ventral striatum and ventromedial prefrontal cortex, which was in turn related to greater anhedonia. Moreover, these effects mediated associations between patients’ CRP and anhedonia levels (Felger et al., 2016).
Inflammatory reactivity to endotoxin is not always related to changes in anxiety or depressed mood (Engler et al. 2016). In fact, some studies have found that inflammatory responses are more strongly associated with anxiety and depressive disorders in men than in women, particularly later in life (Vogelzangs et al. 2012) and that links between CRP, depression, and anxiety are more consistent in men (Elovainio et al. 2009; Liukkonen et al. 2011; Ramsey et al. 2016; Vetter et al., 2013) than in women (Ma et al. 2010). Therefore, although women may mount a greater inflammatory response to such challenges, other vulnerability factors may exist that render certain women more susceptible to experiencing depression during hormonal transition periods and immune system challenges. Because most fMRI studies in this area of research have been conducted in either all-male or all-female samples, there is a pressing need for additional research investigating sex-specific differences that includes both males and females.
Summary
In sum, these data argue for a context-dependent role of sex hormones in shaping depression susceptibility; highlight social stress as a potent risk factor for depression, particularly for women with heightened sensitivity to hormonal fluctuation; and provide evidence of sex-specific interconnections between life stress exposure, inflammation, and MDD. Overall, women appear to exhibit relatively greater risk for inflammation-related depressed mood than men, and this is especially true for those already at heightened risk for mood disorders or in the midst of a hormonal transition period. Male-specific associations between elevated inflammatory activity and depression have also been described. One possible conceptual consolidation of these seemingly inconsistent findings proposes that women may exhibit a stronger low-grade inflammatory response (Ahonen et al. 2012), whereas men may exhibit higher levels of more general inflammatory activation markers (e.g., CRP), specifically later in life (Vogelzangs et al. 2012). A recent review further argues that although women appear to be more vulnerable to the depressogenic effects of chronic stress and inflammation than men (Bekhbat et al. 2018), most research on this topic has been done in males. Given these lingering questions, additional longitudinal studies examining sex differences in stress-related disorders, neural and immune reactivity following stress exposure, and the negative effects that these processes have for mood and behavior are needed.
Social Signal Transduction Theory of Depression revisited
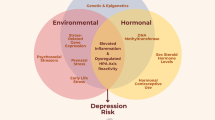
The original formulation of Social Signal Transduction Theory of Depression (Slavich and Irwin 2014) did not tackle the issue of sex differences in depression because, at that point in time, it was not sufficiently clear how sex hormones regulate the social signal transduction pathways described. The evidence presented here addresses this important issue and begins to explain how sex moderates activity at each of the major levels of analysis outlined in the theory. As reviewed above, for example, women have a greater likelihood of experiencing the types of major life stressors that most strongly trigger inflammatory activity; are more likely to exhibit sex-hormone linked elevations in neuroinflammation, especially during major hormonal transition periods; can experience estrogen-associated blunting of HPA axis reactivity, which lessens anti-inflammatory signaling; and may demonstrate enhanced stimulation of intracellular inflammatory signaling pathways by 17beta-estradiol (E2). Together, these and other effects may lead to greater basal levels of several pro-inflammatory cytokines, including IL-1, IL-6, TNF-α, and IFN-α, as well as to greater pro-inflammatory cytokine reactivity to social-environmental adversity and immunologic challenges that are known to heightened depression susceptibility. These responses possess critical adaptive value, as they help promote survival for women and their offspring by providing immunological protection during times of pathogenic threat. If repeatedly engaged or sustained, however, these biological dynamics can also increase women’s risk for depression, in addition to a wide range of other neuropsychiatric, neurodegenerative, metabolic, and somatic disorders that have an inflammatory component. These processes are depicted graphically in Fig. 1.

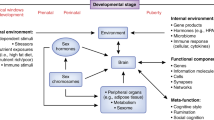
The role of sex hormones in Social Signal Transduction Theory of Depression. Social Signal Transduction Theory of Depression describes neural, physiologic, molecular, and genomic mechanisms that link experiences of social-environmental adversity with internal biological processes that drive depression pathogenesis, maintenance, and recurrence, as well as the co-occurrence of depression with other inflammation-related neuropsychiatric, neurodevelopmental, and neurodegenerative disorders. a Briefly, social-environmental circumstances that are neurocognitively appraised as threats are represented in the brain, which in turn influences the activity of the SNS, HPA axis, and vagus nerve. Neuroeffector molecules from these systems engage specific, highly conserved gene transcription programs in differing target cells. In leukocytes, for example, SNS and HPA signaling generally suppress innate antiviral genes (e.g., IFNA, IFNB), whereas SNS signaling activates—and HPA signaling generally inhibits—pro-inflammatory cytokine genes (e.g., IL1B, IL6, IL8, TNF). These changes represent a systematic shifting of immune system resources toward an increased pro-inflammatory and reduced anti-viral (i.e., type I interferon response) state, with corresponding effects for several immune-mediated health outcomes including depression. b These pathways can also be depicted conceptually, highlighting the fact that experiences of social-environmental adversity can become self-promoting via both external social recursion and internal physiologic recursion. Sex hormones can modulate these processes on several levels. For example, sex hormones can affect external social recursion by altering social behavior patterns including the tendency to socially isolate or respond defensively or aggressively to perceived threat. Such behaviors can in turn alter the actions of conspecifics in the surrounding environment and give rise to social stressors (e.g., social conflict, evaluation, rejection, isolation, exclusion) that further heighten the actual level of adversity in the environment and, therefore, an individual’s perception of such threat. Sex hormones can also affect internal physiologic recursion at several levels, including: (i) by influencing the perception, processing, and regulation of threat and fear via brain regions rich in neurosteroid receptors, such as the amygdala, prefrontal cortex, and hippocampus, and the direct and indirect interaction with key targets in monoaminergic systems (e.g., MAO-A, 5-HTT) and the interplay of sex hormone metabolites with inhibitory neurotransmission, such as ALLO-facilitated GABA-ergic inhibition of pro-inflammatory mediators (e.g., TNF-α via TLR4 signal inhibition); (ii) via modulation of SNS and HPA axis reactivity, as sex hormones can influence parasympathetic/sympathetic activity in addition to cortisol responses to psychosocial stress given that estrogen can enhance the activity of choline uptake and acetycholine synthesis, affect vascular endothelial properties, and may blunt cortisol responsivity, whereas progesterone may increase HPA axis reactivity to stress; and (iii) by influencing the activity of several cellular signal transduction processes that directly modulate innate and adaptive immune system dynamics, including (a) genomic signaling: for example, estrogen can bind directly to estrogen response elements in gene promoters or serve as cofactors with other transcription factors (i.e., NF-κB, AP-1), thus modulating IFN-γ expression; (b) non-genomic signaling: sex hormones can exert rapid effects via neurosteroid receptors on immune cells (e.g., by affecting dendritic and B cell development and function via membrane-bound non-genomic signaling mechanisms and membrane-associated specific kinase signaling pathways); (c) post-translational modification of neurosteroid receptors by acetylation, lipophilic moieteism, and ubiquitination; and (d) possible epigenetic mechanisms (e.g., microRNAs, histone modifications, and acetylation). Importantly, the effects of sex hormones on all of these levels are highly context-dependent. Therefore, factors like sex, age, reproductive state, endogenous versus exogenous hormones, and hormone administration mode and dose need to be carefully considered to fully understand the effects that sex hormones have on risk for depression and other inflammation-related neuropsychiatric, neurodevelopmental, and neurodegenerative diseases (e.g., asthma, rheumatoid arthritis, chronic pain, metabolic syndrome, cardiovascular disease, Alzheimer’s disease, etc.). ACTH, adrenocorticotropin hormone; ADRB2, β2-adrenergic receptor; CRH, corticotrophin releasing hormone; PRR, pattern recognition receptor; SNS, sympathetic nervous system; HPA axis, hypothalamic-pituitary-adrenal axis; MAO-A, monoamine oxidase-A; 5-HTT, serotonin transporter; ALLO, allopregnanolone; GABA, gamma-aminobutyric acid; TLR4, toll-like receptor 4; ER, estrogen receptor; NF-κB, nuclear factor-κB; AP-1, activator protein 1; IFN-γ, interferon-γ; MDD, major depressive disorder. Adapted from Slavich and Cole 2013 and Slavich and Irwin 2014
Turning windows of vulnerability into opportunities for discovery
Looking forward, much more research is needed to fully understand how sex moderates the psychological and biological processes described in Social Signal Transduction Theory of Depression. Recent studies have provided evidence that endogenous sex hormone fluctuations across the menstrual cycle are associated with changes in brain network connectivity (Andreano 2018; Arelin et al. 2015), that changes in gray matter occur during pregnancy and postpartum (Hoekzema et al. 2017), and that changes in neurochemistry occur during the immediate postpartum period (Sacher et al. 2010; Sacher et al. 2015) and perimenopause (Rekkas et al. 2014). At the same time, the effects that these neurobiological changes have on immune system activity and risk for mood disorders likely last long past the hormonal transition period itself, highlighting the importance of better understanding the longitudinal nature of this phenomenon.
Research on this topic should continue by further quantifying specific neurobiological mechanisms that have the ability to directly evoke depressive symptoms. Positron emission tomography (PET) is one tool that can be used for this purpose, as it enables researchers to go beyond studying non-specific relative changes in structural and functional connectivity in the brain in order to quantitatively image neuroreceptor-ligand binding associated with specific hormone-mediated neurochemical changes that promote depression. In fact, this approach has already been used in studies investigating postpartum depression, which have recently identified monoaminergic loss caused by estrogen depletion as a potential mechanism explaining postpartum depressed mood (Sacher et al. 2010; Sacher et al. 2015). This finding has already led to clinical trials of a monoamine oxidase A inhibitor for postpartum depression and to the development of selective dietary supplements for preventing this disorder (Dowlati et al. 2017).
In vivo neuroreceptor ligand imaging with PET can also be applied to measure specific markers of neuroinflammation using (for example) translocator protein (TSPO), which is an 18-kDa protein located on the outer mitochondrial membranes in microglia. Increases in TSPO expression occur when microglia are activated during inflammatory events in the brain (Rupprecht et al. 2010), and increased TSPO density has been shown in MDD patients in the midst of a depressive episode (Setiawan et al. 2015). In addition to leveraging these recent radiotracer developments for research on psychosocial stress, neuroinflammation, and depression, we believe the field requires more targets that are specifically dedicated to ovarian hormone receptors in the CNS. In this context, [18F]-FES is a PET tracer that is used in clinical practice to image the estrogen receptor (ER) in breast cancer (Lin et al. 2017; Venema et al. 2017). However, its potential suitability for imaging of ER in the human brain remains unclear. The ER consists of two isoforms—namely, ERα and ERβ—that have distinct biologic functions. Whereas activation of ERα stimulates cell proliferation and cell survival, ERβ promotes apoptosis. A potential ERβ-selective PET tracer—namely, 2-18F-fluoro-6-(6-hydroxynaphthalen-2-yl)pyridin-3-ol (18F-FHNP)—has been recently introduced (Antunes et al. 2017), but has not yet been tested for human neuroimaging.
Two newer approaches for aromatase imaging are 11C-cetrozole and [N-methyl-11C]-vorozole. Aromatase converts androgens to estrogens, thus enabling us to control local estrogen synthesis. Because it is challenging to measure local concentrations of estrogen in the brain, suitable tracers for this enzyme may provide critical insight into the dynamics of estrogen synthesis. A recent study found that ovarian uptake of 11C-vorozole varies across the menstrual cycle in pre-menopausal women; however, the tracer did not reveal sensitivity to such subtle endogenous ovarian hormone changes in the brain (Biegon et al. 2015). 11C-cetrozole shows a promising signal-to-noise ratio in the amygdala, hypothalamus, and nucleus accumbens in the brain of non-human primates (Takahashi et al. 2014), but remains to be tested in humans.
More broadly, Social Signal Transduction Theory of Depression views MDD as a biopsychosocial disease state characterized by disruptions in multiple systems; therefore, multimodal methods are warranted. Integration of PET with other techniques and research methods would vastly improve the identification of abnormalities in brain function that increase risk for MDD. The MR-PET Hybrid scanner is an exceptional tool for accomplishing this goal, as it enables researchers to examine neurochemical, structural, and functional brain mechanisms simultaneously. This is especially relevant for studying ovarian hormone changes that occur during the menstrual cycle, as ovulation lasts approximately 24 h, thus providing only a short and dynamic time window for obtaining multiple brain scans. Furthermore, such an approach provides an unprecedented opportunity to test participants’ functional brain activation, associated dopaminergic response, and downstream HPA axis and peripheral inflammatory activity during different types of laboratory-based stressors, which can be done all at the same time and across different hormonal states (e.g., ovulation vs. pre-menstrual state) to more fully characterize changes that occur in social signal transduction dynamics over time.
Finally, although assessing life stress exposure and menstrual cycle status can be highly informative for quantifying women’s risk of developing mood disorders that are mediated by ovarian hormones, a majority of studies still utilize self-report scales for evaluating stress exposure and cycle phase that are highly imprecise (Monroe and Slavich in press; Slavich 2019; Wegienka and Baird 2005). To address these issues, we suggest using instruments for measuring life stress exposure that assess stressors occurring in several domains across the entire lifespan (e.g., Stress and Adversity Inventory; Slavich and Shields 2018; Slavich et al. in press). With respect to the menstrual cycle, we recommend adopting comprehensive cycle monitoring that includes a combination of (a) recording body temperature daily to assess the reproductive phase, as marked by an increase in core body temperature up to 1 degree, with (b) assessing ovarian hormone serum blood levels at each time point to measure fluctuation dynamics and menstrual cycle phase, and (c) confirming ovulation by lutropin surge in urine or (d) employing vaginal ultrasounds to confirm and determine time of ovulation by measuring follicle size. Employing such methods will greatly enhance the quantification of life stress exposure and staging of the menstrual cycle phase of women (Barth et al. 2016; Epel et al. 2018), which could ultimately enable personalized predictions of a particular individual’s illness trajectory, treatment response, and/or likelihood of relapse (i.e., precision medicine/precision health; Schüssler-Fiorenza Rose et al. 2019).
Conclusion
In conclusion, Social Signal Transduction Theory of Depression provides the first biologically plausible, multi-level account of neural, physiologic, molecular, and genomic mechanisms linking experiences of social-environmental adversity with internal biological processes that drive depression pathogenesis, maintenance, and recurrence. Here, we extended this theory by highlighting the growing body of evidence showing that ovarian hormone fluctuations influence a woman’s susceptibility to stress, brain structure and function, and inflammatory activity and reactivity, all of which play important roles in structuring risk for depression in addition to several other somatic and physical disease states that have an inflammatory component. MDD is an extremely burdensome disorder, but this burden is not equally distributed between the sexes (Gotlib and Hammen 2014). By studying the roles that sex hormones play in Social Signal Transduction Theory of Depression, our hope is that we may move one step closer to understanding why women become twice as likely to experience depression than men following puberty to ultimately reduce their depression risk and help improve women’s health.
References
Albert K, Pruessner J, Newhouse P (2015) Estradiol levels modulate brain activity and negative responses to psychosocial stress across the menstrual cycle. Psychoneuroendocrinology 59:14–24
Ahonen T, Vanhala M, Kautiainen H, Kumpusalo E, Saltevo J (2012) Sex differences in the association of adiponectin and low-grade inflammation with changes in the body mass index from youth to middle age. Gend Med 9:1–8
Andersen K, Launer LJ, Dewey ME, Letenneur L, Ott A, Copeland JR, Dartigues JF, Kragh-Sorensen P, Baldereschi M, Brayne C, Lobo A, Martinez-Lage JM, Stijnen T, Hofman A (1999) Gender differences in the incidence of AD and vascular dementia: The EURODEM Studies. EURODEM Incidence Research Group. Neurology 53:1992–1997
Andreano JM, Touroutoglou A, Dickerson B, Barrett LF (2018) Hormonal cycles, brain network connectivity, and windows of vulnerability to affective disorder. Trends Neurosci 41:660–666
Antoniou-Tsigkos A, Zapanti E, Ghizzoni L, Mastorakos G (2000) Adrenal Androgens. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP (eds) Endotext. MDText.com, Inc., South Dartmouth, MA
Antunes IF, van Waarde A, Dierckx RA, de Vries EG, Hospers GA, de Vries EF (2017) Synthesis and evaluation of the estrogen receptor beta-selective radioligand 2-(18)F-Fluoro-6-(6-hydroxynaphthalen-2-yl)pyridin-3-ol: comparison with 16alpha-(18)F-fluoro-17beta-estradiol. J Nucl Med 58:554–559
Arelin K, Mueller K, Barth C, Rekkas PV, Kratzsch J, Burmann I, Villringer A, Sacher J (2015) Progesterone mediates brain functional connectivity changes during the menstrual cycle-a pilot resting state MRI study. Front Neurosci 9:44
Avenevoli S, Swendsen J, He JP, Burstein M, Merikangas KR (2015) Major depression in the National Comorbidity Survey-Adolescent Supplement: prevalence, correlates, and treatment. J Am Acad Child Adolesc Psychiatry 54:37–44
Balan I, Beattie MC, O’Buckley TK, Aurelian L, Morrow AL (2019) Endogenous neurosteroid (3alpha,5alpha)3-hydroxypregnan-20-one inhibits toll-like-4 receptor activation and proinflammatory signaling in macrophages and brain. Sci Rep 9:1220
Baldwin JR, Arseneault L, Caspi A, Fisher HL, Moffitt TE, Odgers CL, Pariante C, Ambler A, Dove R, Kepa A, Matthews T, Menard A, Sugden K, Williams B, Danese A (2018) Childhood victimization and inflammation in young adulthood: a genetically sensitive cohort study. Brain Behav Immun 67:211–217
Barbieri RL (2014) The endocrinology of the menstrual cycle. Methods Mol Biol 1154:145–169
Baron-Cohen S, Knickmeyer RC, Belmonte MK (2005) Sex differences in the brain: implications for explaining autism. Science 310:819–823
Barth C, Villringer A, Sacher J (2015) Sex hormones affect neurotransmitters and shape the adult female brain during hormonal transition periods. Front Neurosci 9:37
Barth C, Steele CJ, Mueller K, Rekkas VP, Arelin K, Pampel A, Burmann I, Kratzsch J, Villringer A, Sacher J (2016) In-vivo dynamics of the human hippocampus across the menstrual cycle. Sci Rep 6:32833
Beck AT (1967) Depression: clinical, experimental, and theoretical aspects. Harper and Row, New York
Becker JB (1990) Estrogen rapidly potentiates amphetamine-induced striatal dopamine release and rotational behavior during microdialysis. Neurosci Lett 118:169–171
Bekhbat M, Neigh GN (2018) Sex differences in the neuro-immune consequences of stress: focus on depression and anxiety. Brain Behav Immun 67:1–12
Benedusi V, Meda C, Della Torre S, Monteleone G, Vegeto E, Maggi A (2012) A lack of ovarian function increases neuroinflammation in aged mice. Endocrinology 153:2777–2788
Berger A (2000) Th1 and Th2 responses: what are they? BMJ 321:424
Biegon A, Alexoff DL, Kim SW, Logan J, Pareto D, Schlyer D, Wang GJ, Fowler JS (2015) Aromatase imaging with [N-methyl-11C]vorozole PET in healthy men and women. J Nucl Med 56:580–585
Blatt SJ (2004) Experiences of depression: theoretical, clinical, and research perspectives. American Psychological Association, Washington, DC
Bonaccorso S, Marino V, Biondi M, Grimaldi F, Ippoliti F, Maes M (2002) Depression induced by treatment with interferon-alpha in patients affected by hepatitis C virus. J Affect Disord 72:237–241
Bouman A, Heineman MJ, Faas MM (2005) Sex hormones and the immune response in humans. Hum Reprod Update 11:411–423
Brinton RD, Thompson RF, Foy MR, Baudry M, Wang J, Finch CE, Morgan TE, Pike CJ, Mack WJ, Stanczyk FZ, Nilsen J (2008) Progesterone receptors: form and function in brain. Front Neuroendocrinol 29:313–339
Brites D, Fernandes A (2015) Neuroinflammation and depression: microglia activation, extracellular microvesicles and microRNA dysregulation. Front Cell Neurosci 9:476
Brown GW, Harris TO (1978) Social origins of depression: a study of psychiatric disorder in women. Free Press, New York
Bushnell CD (2008) Stroke and the female brain. Nat Clin Pract Neurol 4:22–33
Buskiewicz IA, Huber SA, Fairweather D (2016) Chapter 4–sex hormone receptor expression in the immune system A2. In: Neigh GN, Mitzelfelt MM (eds) Sex differences in physiology. Academic Press, New York, pp 45–60
Butts CL, Shukair SA, Duncan KM, Bowers E, Horn C, Belyavskaya E, Tonelli L, Sternberg EM (2007) Progesterone inhibits mature rat dendritic cells in a receptor-mediated fashion. Int Immunol 19:287–296
Capuron L, Miller AH (2004) Cytokines and psychopathology: Lessons from interferon-alpha. Biol Psychiatry 56:819–824
Capuron L, Miller AH (2011) Immune system to brain signaling: Neuropsychopharmacological implications. Pharmacol Ther 130:226–238
Cornil CA, Ball GF, Balthazart J (2006) Functional significance of the rapid regulation of brain estrogen action: where do the estrogens come from? Brain Res 1126:2–26
Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R (2011) Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol 10:63–74
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW (2008) From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9:46–56
Deak T, Quinn M, Cidlowski JA, Victoria NC, Murphy AZ, Sheridan JF (2015) Neuroimmune mechanisms of stress: Sex differences, developmental plasticity, and implications for pharmacotherapy of stress-related disease. Stress 18:367–380
Dheen ST, Kaur C, Ling EA (2007) Microglial activation and its implications in the brain diseases. Curr Med Chem 14:1189–1197
Donoho CJ, Crimmins EM, Seeman TE (2013) Marital quality, gender, and markers of inflammation in the MIDUS cohort. J Marriage Fam 75:127–141
Dowlati Y, Ravindran AV, Segal ZV, Stewart DE, Steiner M, Meyer JH (2017) Selective dietary supplementation in early postpartum is associated with high resilience against depressed mood. Proc Natl Acad Sci U S A 114:3509–3514
Eisenberger NI, Inagaki TK, Rameson LT, Mashal NM, Irwin MR (2009) An fMRI study of cytokine-induced depressed mood and social pain: the role of sex differences. Neuroimage 47:881–890
Eisenlohr-Moul TA, Rubinow DR, Schiller CE, Johnson JL, Leserman J, Girdler SS (2016) Histories of abuse predict stronger within-person covariation of ovarian steroids and mood symptoms in women with menstrually related mood disorder. Psychoneuroendocrinology 67:142–152
Elovainio M, Aalto AM, Kivimaki M, Pirkola S, Sundvall J, Lonnqvist J, Reunanen A (2009) Depression and C-reactive protein: population-based health 2000 study. Psychosom Med 71:423–430
Engler H, Benson S, Wegner A, Spreitzer I, Schedlowski M, Elsenbruch S (2016) Men and women differ in inflammatory and neuroendocrine responses to endotoxin but not in the severity of sickness symptoms. Brain Behav Immun 52:18–26
Epel ES, Crosswell AD, Mayer SE, Prather AA, Slavich GM, Puterman E, Mendes WB (2018) More than a feeling: a unified view of stress measurement for population science. Front Neuroendocrinol 49:146–169
Felger JC, Li Z, Haroon E, Woolwine BJ, Jung MY, Hu X, Miller AH (2016) Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol Psychiatry 21:1358–1365
Ferretti MT, Iulita MF, Cavedo E, Chiesa PA, Schumacher Dimech A, Santuccione Chadha A, Baracchi F, Girouard H, Misoch S, Giacobini E, Depypere H, Hampel H, Women’s Brain Project and the Alzheimer Precision Medicine Initiative (2018) Sex differences in Alzheimer disease—the gateway to precision medicine. Nat Rev Neurol 14:457-469.
Fontana RJ, Schwartz SM, Gebremariam A, Lok AS, Moyer CA (2002) Emotional distress during interferon-alpha-2B and ribavirin treatment of chronic hepatitis C. Psychosomatics 43:378–385
Foy MR, Xu J, Xie X, Brinton RD, Thompson RF, Berger TW (1999) 17beta-estradiol enhances NMDA receptor-mediated EPSPs and long-term potentiation. J Neurophysiol 81:925–929
Freeman EW, Sammel MD, Lin H, Nelson DB (2006) Associations of hormones and menopausal status with depressed mood in women with no history of depression. Arch Gen Psychiatry 63:375–382
Frokjaer VG, Pinborg A, Holst KK, Overgaard A, Henningsson S, Heede M, Larsen EC, Jensen PS, Agn M, Nielsen AP, Stenbaek DS, da Cunha-Bang S, Lehel S, Siebner HR, Mikkelsen JD, Svarer C, Knudsen GM (2015) Role of serotonin transporter changes in depressive responses to sex-steroid hormone manipulation: a positron emission tomography study. Biol Psychiatry 78:534–543
Giletta M, Slavich GM, Rudolph KD, Hastings PD, Nock MK, Prinstein MJ (2018) Peer victimization predicts heightened inflammatory reactivity to social stress in cognitively vulnerable adolescents. J Child Psychol Psychiatry 59:129–139
Gimeno D, Kivimäki M, Brunner EJ, Elovainio M, De Vogli R, Steptoe A, Kumari M, Lowe GD, Rumley A, Marmot MG, Ferrie JE (2009) Associations of C-reactive protein and interleukin-6 with cognitive symptoms of depression: 12-year follow-up of the Whitehall II study. Psychol Med 39:413–423
Gohier B, Goeb JL, Rannou-Dubas K, Fouchard I, Cales P, Garre JB (2003) Hepatitis C, alpha interferon, anxiety and depression disorders: a prospective study of 71 patients. World J Biol Psychiatry 4:115–118
Gold PW (2015) The organization of the stress system and its dysregulation in depressive illness. Mol Psychiatry 20:32–47
Goldstein JM, Jerram M, Abbs B, Whitfield-Gabrieli S, Makris N (2010) Sex differences in stress response circuitry activation dependent on female hormonal cycle. J Neurosci 30:431–438
Goldstein JM, Holsen L, Handa R, Tobet S (2014) Fetal hormonal programming of sex differences in depression: Linking women’s mental health with sex differences in the brain across the lifespan. Front Neurosci 8:247
Goldstein JM, Holsen L, Huang G, Hammond BD, James-Todd T, Cherkerzian S, Hale TM, Handa RJ (2016) Prenatal stress-immune programming of sex differences in comorbidity of depression and obesity/metabolic syndrome. Dialogues Clin Neurosci 18:425–436
Goldstein JM, Hale T, Foster SL, Tobet SA, Handa RJ (2018) Sex differences in major depression and comorbidity of cardiometabolic disorders: impact of prenatal stress and immune exposures. Neuropsychopharmacology 44:59–70
Goodwin RD, Fergusson DM, Horwood LJ (2004) Asthma and depressive and anxiety disorders among young persons in the community. Psychol Med 34:1465–1474
Gordon JL, Girdler SS, Meltzer-Brody SE, Stika CS, Thurston RC, Clark CT, Prairie BA, Moses-Kolko E, Joffe H, Wisner KL (2015) Ovarian hormone fluctuation, neurosteroids, and HPA axis dysregulation in perimenopausal depression: A novel heuristic model. Am J Psychiatry 172:227–236
Gordon JL, Rubinow DR, Eisenlohr-Moul TA, Leserman J, Girdler SS (2016) Estradiol variability, stressful life events, and the emergence of depressive symptomatology during the menopausal transition. Menopause 23:257–266
Gotlib IH, Hammen CL (eds) (2014) Handbook of depression, third edn. The Guilford Press, New York
Graff LA, Walker JR, Bernstein CN (2009) Depression and anxiety in inflammatory bowel disease: a review of comorbidity and management. Inflamm Bowel Dis 15:1105–1118
Greenberg PE, Fournier AA, Sisitsky T, Pike CT, Kessler RC (2015) The economic burden of adults with major depressive disorder in the United States (2005 and 2010). J Clin Psychiatry 76:155–162
Gulinello M, Gong QH, Li X, Smith SS (2001) Short-term exposure to a neuroactive steroid increases alpha4 GABA(A) receptor subunit levels in association with increased anxiety in the female rat. Brain Res 910:55–66
Gundlah C, Lu NZ, Mirkes SJ, Bethea CL (2001) Estrogen receptor beta (ERbeta) mRNA and protein in serotonin neurons of macaques. Brain Res Mol Brain Res 91:14–22
Hammen C (2006) Stress generation in depression: reflections on origins, research, and future directions. J Clin Psychol 62:1065–1082
Han TJ, Felger JC, Lee A, Mister D, Miller AH, Torres MA (2016) Association of childhood trauma with fatigue, depression, stress, and inflammation in breast cancer patients undergoing radiotherapy. Psychooncology 25:187–193
Hanamsagar R, Bilbo SD (2016) Sex differences in neurodevelopmental and neurodegenerative disorders: focus on microglial function and neuroinflammation during development. J Steroid Biochem Mol Biol 160:127–133
Hankin BL, Abramson LY (1999) Development of gender differences in depression: description and possible explanations. Ann Med 31:372–379
Hankin BL, Abramson LY (2001) Development of gender differences in depression: an elaborated cognitive vulnerability–transactional stress theory. Psychol Bull 127:773–796
Hannestad J, DellaGioia N, Ortiz N, Pittman B, Bhagwagar Z (2011) Citalopram reduces endotoxin-induced fatigue. Brain Behav Immun 25:256–259
Hardy DB, Janowski BA, Corey DR, Mendelson CR (2006) Progesterone receptor plays a major antiinflammatory role in human myometrial cells by antagonism of nuclear factor-kappaB activation of cyclooxygenase 2 expression. Mol Endocrinol 20:2724–2733
Harkness KL, Alavi N, Monroe SM, Slavich GM, Gotlib IH, Bagby RM (2010) Gender differences in life events prior to onset of major depressive disorder: the moderating effect of age. J Abnorm Psychol 119:791–803
Hart BL (1988) Biological basis of the behavior of sick animals. Neurosci Biobehav Rev 12:123–137
Hemsell DL, Grodin JM, Brenner PF, Siiteri PK, MacDonald PC (1974) Plasma precursors of estrogen. II. Correlation of the extent of conversion of plasma androstenedione to estrone with age. J Clin Endocrinol Metab 38:476–479
Hennessy MB, Schiml-Webb PA, Deak T (2009) Separation, sickness, and depression: a new perspective on an old animal model. Curr Dir Psychol Sci 18:227–231
Hernández ME, Mendieta D, Martínez-Fong D, Loría F, Moreno J, Estrada I, Bojalil R, Pavón L (2008) Variations in circulating cytokine levels during 52 week course of treatment with SSRI for major depressive disorder. Eur Neuropsychopharmacol 18:917–924
Hoekzema E, Barba-Muller E, Pozzobon C, Picado M, Lucco F, Garcia-Garcia D, Soliva JC, Tobena A, Desco M, Crone EA, Ballesteros A, Carmona S, Vilarroya O (2017) Pregnancy leads to long-lasting changes in human brain structure. Nat Neurosci 20:287–296
Howren MB, Lamkin DM, Suls J (2009) Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosom Med 71:171–186
Irwin MR, Cole SW (2011) Reciprocal regulation of the neural and innate immune systems. Nat Rev Immunol 11:625–632
Irwin MR, Slavich GM (2017) Psychoneuroimmunology. In: Cacioppo JT, Tassinary LG, Berntson GG (eds) Handbook of psychophysiology, fourth edn. Cambridge University Press, New York, pp 377–398
Jacobs EG, Holsen LM, Lancaster K, Makris N, Whitfield-Gabrieli S, Remington A, Weiss B, Buka S, Klibanski A, Goldstein JM (2015) 17beta-estradiol differentially regulates stress circuitry activity in healthy and depressed women. Neuropsychopharmacology 40:566–576
Jarcho MR, Slavich GM, Tylova-Stein H, Wolkowitz OM, Burke HM (2013) Dysregulated diurnal cortisol pattern is associated with glucocorticoid resistance in women with major depressive disorder. Biol Psychol 93:150–158
Jilma B, Eichler HG, Breiteneder H, Wolzt M, Aringer M, Graninger W, Rohrer C, Veitl M, Wagner OF (1994) Effects of 17 beta-estradiol on circulating adhesion molecules. J Clin Endocrinol Metab 79:1619–1624
Johannessen M, Fontanilla D, Mavlyutov T, Ruoho AE, Jackson MB (2011) Antagonist action of progesterone at sigma-receptors in the modulation of voltage-gated sodium channels. Am J Phys Cell Phys 300:328–337
Jones SC, Brahmakshatriya V, Huston G, Dibble J, Swain SL (2010) TLR-activated dendritic cells enhance the response of aged naive CD4 T cells via an IL-6-dependent mechanism. J Immunol 185:6783–6794
Juraska JM, Sisk CL, DonCarlos LL (2013) Sexual differentiation of the adolescent rodent brain: hormonal influences and developmental mechanisms. Horm Behav 64:203–210
Kajantie E, Phillips DI (2006) The effects of sex and hormonal status on the physiological response to acute psychosocial stress. Psychoneuroendocrinology 31:151–178
Kawasaki T, Kawai T (2014) Toll-like receptor signaling pathways. Front Immunol 5:461
Kendler KS, Hettema JM, Butera F, Gardner CO, Prescott CA (2003) Life event dimensions of loss, humiliation, entrapment, and danger in the prediction of onsets of major depression and generalized anxiety. Arch Gen Psychiatry 60:789–796
Kessler RC, Birnbaum H, Bromet E, Hwang I, Sampson N, Shahly V (2010) Age differences in major depression: results from the National Comorbidity Survey Replication (NCS-R). Psychol Med 40:225–237
Kiecolt-Glaser JK, Gouin JP, Hantsoo L (2010) Close relationships, inflammation, and health. Neurosci Biobehav Rev 35:33–38
Kim YK, Na KS, Myint AM, Leonard BE (2016) The role of pro-inflammatory cytokines in neuroinflammation, neurogenesis and the neuroendocrine system in major depression. Prog Neuro-Psychopharmacol Biol Psychiatry 64:277–284
Klein SL (2000) The effects of hormones on sex differences in infection: from genes to behavior. Neurosci Biobehav Rev 24:627–638
Klein SL, Flanagan KL (2016) Sex differences in immune responses. Nat Rev Immunol 16:626–638
Komesaroff PA, Esler MD, Sudhir K (1999) Estrogen supplementation attenuates glucocorticoid and catecholamine responses to mental stress in perimenopausal women. J Clin Endocrinol Metab 84:606–610
Koskinas J, Merkouraki P, Manesis E, Hadziyannis S (2002) Assessment of depression in patients with chronic hepatitis: effect of interferon treatment. Dig Dis 20:284–288
Kovats S (2015) Estrogen receptors regulate innate immune cells and signaling pathways. Cell Immunol 294:63–69
Kraus MR, Schafer A, Faller H, Csef H, Scheurlen M (2003) Psychiatric symptoms in patients with chronic hepatitis C receiving interferon alfa-2b therapy. J Clin Psychiatry 64:708–714
Krebs CJ, Jarvis ED, Chan J, Lydon JP, Ogawa S, Pfaff DW (2000) A membrane-associated progesterone-binding protein, 25-Dx, is regulated by progesterone in brain regions involved in female reproductive behaviors. Proc Natl Acad Sci U S A 97:12816–12821
Kuehner C (2017) Why is depression more common among women than among men? Lancet Psychiatry 4:146–158
Kumar P, Slavich GM, Berghorst LH, Treadway MT, Brooks NH, Dutra SJ, Greve DN, O’Donovan A, Bleil ME, Maninger N, Pizzagalli DA (2015) Perceived life stress exposure modulates reward-related medial prefrontal cortex responses to acute stress in depression. J Affect Disord 180:104–111
Larson TA (2018) Sex steroids, adult neurogenesis, and inflammation in CNS homeostasis, degeneration, and repair. Front Endocrinol 9:205
Leifheit-Limson EC, Reid KJ, Kasl SV, Lin H, Jones PG, Buchanan DM, Parashar S, Peterson PN, Spertus JA, Lichtman JH (2010) The role of social support in health status and depressive symptoms after acute myocardial infarction: evidence for a stronger relationship among women. Circ Cardiovasc Qual Outcomes 3:143–150
Lin FI, Gonzalez EM, Kummar S, Do K, Shih J, Adler S, Kurdziel KA, Ton A, Turkbey B, Jacobs PM, Bhattacharyya S, Chen AP, Collins JM, Doroshow JH, Choyke PL, Lindenberg ML (2017) Utility of (18)F-fluoroestradiol ((18)F-FES) PET/CT imaging as a pharmacodynamic marker in patients with refractory estrogen receptor-positive solid tumors receiving Z-endoxifen therapy. Eur J Nucl Med Mol Imaging 44:500–508
Liukkonen T, Rasanen P, Jokelainen J, Leinonen M, Jarvelin MR, Meyer-Rochow VB, Timonen M (2011) The association between anxiety and C-reactive protein (CRP) levels: Results from the Northern Finland 1966 birth cohort study. Eur Psychiatry 26:363–369
Ma Y, Chiriboga DE, Pagoto SL, Rosal MC, Li W, Merriam PA, Hebert JR, Whited MC, Ockene IS (2010) Association between depression and C-reactive protein. Cardiol Res Pract 2011:286509
Marsland AL, Walsh C, Lockwood K, John-Henderson NA (2017) The effects of acute psychological stress on circulating and stimulated inflammatory markers: a systematic review and meta-analysis. Brain Behav Immun 64:208–219
Massing-Schaffer M, Helms SW, Rudolph KD, Slavich GM, Hastings PD, Giletta M, Nock MK, Prinstein MJ (2019) Preliminary associations among relational victimization, targeted rejection, and suicidality in adolescents: a prospective study. J Clin Child Adolesc Psychol 48:288–295
McClelland EE, Smith JM (2011) Gender specific differences in the immune response to infection. Arch Immunol Ther Exp 59:203–213
McCombe PA, Greer JM, Mackay IR (2009) Sexual dimorphism in autoimmune disease. Curr Mol Med 9:1058–1079
McEwen BS (1991) Non-genomic and genomic effects of steroids on neural activity. Trends Pharmacol Sci 12:141–147
McEwen BS, Akama KT, Spencer-Segal JL, Milner TA, Waters EM (2012) Estrogen effects on the brain: actions beyond the hypothalamus via novel mechanisms. Behav Neurosci 126:4–16
Medzhitov R (2001) Toll-like receptors and innate immunity. Nat Rev Immunol 1:135–145
Medzhitov R (2008) Origin and physiological roles of inflammation. Nature 454:428–435
Mehta D, Newport DJ, Frishman G, Kraus L, Rex-Haffner M, Ritchie JC, Lori A, Knight BT, Stagnaro E, Ruepp A, Stowe ZN, Binder EB (2014) Early predictive biomarkers for postpartum depression point to a role for estrogen receptor signaling. Psychol Med 44:2309–2322
Mehta D, Rex-Haffner M, Sondergaard HB, Pinborg A, Binder EB, Frokjaer VG (in press) Evidence for oestrogen sensitivity in perinatal depression: pharmacological sex hormone manipulation study. Br J Psychiatry
Menzies FM, Henriquez FL, Alexander J, Roberts CW (2011) Selective inhibition and augmentation of alternative macrophage activation by progesterone. Immunology 134:281–291
Merikangas KR, He JP, Brody D, Fisher PW, Bourdon K, Koretz DS (2010) Prevalence and treatment of mental disorders among US children in the 2001-2004 NHANES. Pediatrics 125:75–81
Miller GE, Cole SW (2012) Clustering of depression and inflammation in adolescents previously exposed to childhood adversity. Biol Psychiatry 72:34–40
Miller AH, Maletic V, Raison CL (2009) Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry 65:732–741
Minami T, Oomura Y, Nabekura J, Fukuda A (1990) 17 beta-estradiol depolarization of hypothalamic neurons is mediated by cyclic AMP. Brain Res 519:301–307
Mitra SW, Hoskin E, Yudkovitz J, Pear L, Wilkinson HA, Hayashi S, Pfaff DW, Ogawa S, Rohrer SP, Schaeffer JM, McEwen BS, Alves SE (2003) Immunolocalization of estrogen receptor beta in the mouse brain: comparison with estrogen receptor alpha. Endocrinology 144:2055–2067
Miyaoka H, Otsubo T, Kamijima K, Ishii M, Onuki M, Mitamura K (1999) Depression from interferon therapy in patients with hepatitis C. Am J Psychiatry 156:1120
Moieni M, Irwin MR, Jevtic I, Olmstead R, Breen EC, Eisenberger NI (2015) Sex differences in depressive and socioemotional responses to an inflammatory challenge: implications for sex differences in depression. Neuropsychopharmacology 40:1709–1716
Moieni M, Tan KM, Inagaki TK, Muscatell KA, Dutcher JM, Jevtic I, Breen EC, Irwin MR, Eisenberger NI (2019) Sex differences in the relationship between inflammation and reward sensitivity: a randomized controlled trial of endotoxin. Biol Psychiatry Cogn Neurosci Neuroimaging 4:619–626
Monroe SM, Slavich GM (in press) Major life events: a review of conceptual, definitional, measurement issues, and practices. In: Harkness K, Hayden E (eds) The Oxford handbook of stress and mental health. Oxford University Press, New York
Monroe SM, Slavich GM, Georgiades K (2014a) The social environment and depression: the roles of life stress. In: Gotlib IH, Hammen CL (eds) Handbook of depression, third edn. The Guilford Press, New York, pp 296–314
Monroe SM, Slavich GM, Gotlib IH (2014b) Life stress and family history for depression: the moderating role of past depressive episodes. J Psychiatr Res 49:90–95
Morel Y, Roucher F, Plotton I, Goursaud C, Tardy V, Mallet D (2016) Evolution of steroids during pregnancy: maternal, placental and fetal synthesis. Ann Endocrinol 77:82–89
Mottahedin A, Ardalan M, Chumak T, Riebe I, Ek J, Mallard C (2017) Effect of neuroinflammation on synaptic organization and function in the developing brain: Implications for neurodevelopmental and neurodegenerative disorders. Front Cell Neurosci 11:190
Müller N, Schwarz MJ, Dehning S, Douhe A, Cerovecki A, Goldstein-Müller B, Spellmann I, Hetzel G, Maino K, Kleindienst N, Möller HJ, Arolt V, Riedel M (2006) The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry 11:680–684
Murphy K (2011) Janeway’s Immunobiology. Garland Science, New York
Murphy MLM, Slavich GM, Rohleder N, Miller GE (2013) Targeted rejection triggers differential pro- and anti-inflammatory gene expression in adolescents as a function of social status. Clin Psychol Sci 1:30–40
Murphy MLM, Slavich GM, Chen E, Miller GE (2015) Targeted rejection predicts decreased anti-inflammatory gene expression and increased symptom severity in youth with asthma. Psychol Sci 26:111–121
Muscatell KA, Dedovic K, Slavich GM, Jarcho MR, Breen EC, Bower JE, Irwin MR, Eisenberger NI (2015) Greater amygdala activity and dorsomedial prefrontal-amygdala coupling are associated with enhanced inflammatory responses to stress. Brain Behav Immun 43:46–53
Muscatell KA, Dedovic K, Slavich GM, Jarcho MR, Breen EC, Bower JE, Irwin MR, Eisenberger NI (2016) Neural mechanisms linking social status and inflammatory responses to social stress. Soc Cogn Affect Neurosci 11:915–922
Newhouse PA, Dumas J, Hancur-Bucci C, Naylor M, Sites CK, Benkelfat C, Young SN (2008) Estrogen administration negatively alters mood following monoaminergic depletion and psychosocial stress in postmenopausal women. Neuropsychopharmacology 33:1514–1527
Ngo ST, Steyn FJ, McCombe PA (2014) Gender differences in autoimmune disease. Front Neuroendocrinol 35:347–369
Niemegeers P, De Boer P, Dumont GJH, Van Den Eede F, Fransen E, Claes SJ, Morrens M, Sabbe BGC (2016) Differential effects of inflammatory and psychosocial stress on mood, hypothalamic-pituitary-adrenal axis, and inflammation in remitted depression. Neuropsychobiology 74:150–158
O’Donovan A, Slavich GM, Epel ES, Neylan TC (2013) Exaggerated neurobiological sensitivity to threat as a mechanism linking anxiety with increased risk for diseases of aging. Neurosci Biobehav Rev 37:96–108
Oldehinkel AJ, Bouma EM (2011) Sensitivity to the depressogenic effect of stress and HPA-axis reactivity in adolescence: a review of gender differences. Neurosci Biobehav Rev 35:1757–1770
Owens SA, Eisenlohr-Moul T (2018) Suicide risk and the menstrual cycle: a review of candidate RDoC mechanisms. Curr Psychiatry Rep 20:106
Pace TW, Mletzko TC, Alagbe O, Musselman DL, Nemeroff CB, Miller AH, Heim CM (2006) Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am J Psychiatry 163:1630–1633
Patterson PH (2011) Maternal infection and immune involvement in autism. Trends Mol Med 17:389–394
Paul SM, Purdy RH (1992) Neuroactive steroids. FASEB J 6:2311–2322
Phiel KL, Henderson RA, Adelman SJ, Elloso MM (2005) Differential estrogen receptor gene expression in human peripheral blood mononuclear cell populations. Immunol Lett 97:107–113
Poole H, White S, Blake C, Murphy P, Bramwell R (2009) Depression in chronic pain patients: prevalence and measurement. Pain Pract 9:173–180
Poon DC, Ho YS, Chiu K, Chang RC (2013) Cytokines: how important are they in mediating sickness? Neurosci Biobehav Rev 37:1–10
Puder JJ, Freda PU, Goland RS, Wardlaw SL (2001) Estrogen modulates the hypothalamic-pituitary-adrenal and inflammatory cytokine responses to endotoxin in women. J Clin Endocrinol Metab 86:2403–2408
Raison CL, Demetrashvili M, Capuron L, Miller AH (2005) Neuropsychiatric adverse effects of interferon-alpha: recognition and management. CNS Drugs 19:105–123
Raison CL, Capuron L, Miller AH (2006) Cytokines sing the blues: Inflammation and the pathogenesis of depression. Trends Immunol 27:24–31
Raison CL, Rutherford RE, Woolwine BJ, Shuo C, Schettler P, Drake DF, Haroon E, Miller AH (2013) A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry 70:31–41
Ramsey JM, Cooper JD, Bot M, Guest PC, Lamers F, Weickert CS, Penninx BW, Bahn S (2016) Sex differences in serum markers of major depressive disorder in the Netherlands Study of Depression and Anxiety (NESDA). PLoS One 11:e0156624
Rekkas PV, Wilson AA, Lee VW, Yogalingam P, Sacher J, Rusjan P, Houle S, Stewart DE, Kolla NJ, Kish S, Chiuccariello L, Meyer JH (2014) Greater monoamine oxidase a binding in perimenopausal age as measured with carbon 11-labeled harmine positron emission tomography. JAMA Psychiatry 71:873–879
Ricciotti E, FitzGerald GA (2011) Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol 31:986–1000
Robinson DP, Hall OJ, Nilles TL, Bream JH, Klein SL (2014) 17beta-estradiol protects females against influenza by recruiting neutrophils and increasing virus-specific CD8 T cell responses in the lungs. J Virol 88:4711–4720
Rupprecht R, Papadopoulos V, Rammes G, Baghai TC, Fan J, Akula N, Groyer G, Adams D, Schumacher M (2010) Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov 9:971–988
Sacher J, Wilson AA, Houle S, Rusjan P, Hassan S, Bloomfield PM, Stewart DE, Meyer JH (2010) Elevated brain monoamine oxidase A binding in the early postpartum period. Arch Gen Psychiatry 67:468–474
Sacher J, Rekkas PV, Wilson AA, Houle S, Romano L, Hamidi J, Rusjan P, Fan I, Stewart DE, Meyer JH (2015) Relationship of monoamine oxidase-A distribution volume to postpartum depression and postpartum crying. Neuropsychopharmacology 40:429–435
Sarachana T, Xu M, Wu RC, Hu VW (2011) Sex hormones in autism: androgens and estrogens differentially and reciprocally regulate RORA, a novel candidate gene for autism. PLoS One 6:e17116
Sarvari M, Hrabovszky E, Kallo I, Solymosi N, Liko I, Berchtold N, Cotman C, Liposits Z (2012) Menopause leads to elevated expression of macrophage-associated genes in the aging frontal cortex: rat and human studies identify strikingly similar changes. J Neuroinflammation 9:264
Schüssler-Fiorenza Rose SM, Contrepois K, Moneghetti KJ, Zhou W, Mishra T, Mataraso S, Dagan-Rosenfeld O, Ganz AB, Dunn J, Hornburg D, Rego S, Perelman D, Ahadi S, Sailani MR, Zhou Y, Leopold SR, Chen J, Ashland M, Christle JW, Avina M, Limcaoco P, Ruiz C, Tan M, Butte AJ, Weinstock GM, Slavich GM, Sodergren E, McLaughlin TL, Haddad F, Snyder MP (2019) A longitudinal big data approach for precision health. Nat Med 25:792–804
Segerstrom SC, Miller GE (2004) Psychological stress and the human immune system: a meta-analytic study of 30 years of inquiry. Psychol Bull 130:601–630
Seillet C, Laffont S, Tremollieres F, Rouquie N, Ribot C, Arnal JF, Douin-Echinard V, Gourdy P, Guery JC (2012) The TLR-mediated response of plasmacytoid dendritic cells is positively regulated by estradiol in vivo through cell-intrinsic estrogen receptor alpha signaling. Blood 119:454–464
Setiawan E, Wilson AA, Mizrahi R, Rusjan PM, Miler L, Rajkowska G, Suridjan I, Kennedy JL, Rekkas PV, Houle S, Meyer JH (2015) Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry 72:268–275
Shields GS, Slavich GM (2017) Lifetime stress exposure and health: a review of contemporary assessment methods and biological mechanisms. Soc Personal Psychol Compass 11(8):e12335
Singh M (2001) Ovarian hormones elicit phosphorylation of Akt and extracellular-signal regulated kinase in explants of the cerebral cortex. Endocrine 14:407–415
Slavich GM (2015) Understanding inflammation, its regulation, and relevance for health: A top scientific and public priority. Brain Behav Immun 45:13–14
Slavich GM (2016a) Life stress and health: a review of conceptual issues and recent findings. Teach Psychol 43:346–355
Slavich GM (2016b) Psychopathology and stress. In: Miller HL (ed) The SAGE encyclopedia of theory in psychology, first edn. SAGE Publications, Thousand Oaks, CA, pp 762–764
Slavich GM (2019) Stressnology: The primitive (and problematic) study of life stress exposure and pressing need for better measurement. Brain Behav Immun 75:3–5
Slavich GM (in press) Psychoneuroimmunology of stress and mental health. In: Harkness K, Hayden E (eds) The Oxford handbook of stress and mental health. Oxford University Press, New York
Slavich GM, Auerbach RP (2018) Stress and its sequelae: depression, suicide, inflammation, and physical illness. In: Butcher JN, Hooley JM (eds) APA handbook of psychopathology, Psychopathology: understanding, assessing, and treating adult mental disorders, vol 1. American Psychological Association, Washington, DC, pp 375–402
Slavich GM, Cole SW (2013) The emerging field of human social genomics. Clin Psychol Sci 1:331–348
Slavich GM, Irwin MR (2014) From stress to inflammation and major depressive disorder: a social signal transduction theory of depression. Psychol Bull 140:774–815
Slavich GM, Shields GS (2018) Assessing lifetime stress exposure using the Stress and Adversity Inventory for Adults (Adult STRAIN): an overview and initial validation. Psychosom Med 80:17–27
Slavich GM, Thornton T, Torres LD, Monroe SM, Gotlib IH (2009) Targeted rejection predicts hastened onset of major depression. J Soc Clin Psychol 28:223–243
Slavich GM, O’Donovan A, Epel ES, Kemeny ME (2010a) Black sheep get the blues: a psychobiological model of social rejection and depression. Neurosci Biobehav Rev 35:39–45
Slavich GM, Way BM, Eisenberger NI, Taylor SE (2010b) Neural sensitivity to social rejection is associated with inflammatory responses to social stress. Proc Natl Acad Sci U S A 107:14817–14822
Slavich GM, Monroe SM, Gotlib IH (2011) Early parental loss and depression history: associations with recent life stress in major depressive disorder. J Psychiatr Res 45:1146–1152
Slavich GM, Tartter MA, Brennan PA, Hammen C (2014) Endogenous opioid system influences depressive reactions to socially painful targeted rejection life events. Psychoneuroendocrinology 49:141–149
Slavich GM, Stewart JG, Esposito EC, Shields GS, Auerbach RP (in press) The Stress and Adversity Inventory for Adolescents (Adolescent STRAIN): associations with mental and physical health, risky behaviors, and psychiatric diagnoses in youth seeking treatment. J Child Psychol Psychiatry
Spinedi E, Suescun MO, Hadid R, Daneva T, Gaillard RC (1992) Effects of gonadectomy and sex hormone therapy on the endotoxin-stimulated hypothalamo-pituitary-adrenal axis: evidence for a neuroendocrine-immunological sexual dimorphism. Endocrinology 131:2430–2436
Steptoe A, Hamer M, Chida Y (2007) The effects of acute psychological stress on circulating inflammatory factors in humans: a review and meta-analysis. Brain Behav Immun 21:901–912
Sternberg EM (2006) Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol 6:318–328
Straub RH (2007) The complex role of estrogens in inflammation. Endocr Rev 28:521–574
Strike PC, Wardle J, Steptoe A (2004) Mild acute inflammatory stimulation induces transient negative mood. J Psychosom Res 57:189–194
Sumner BE, Fink G (1998) Testosterone as well as estrogen increases serotonin2A receptor mRNA and binding site densities in the male rat brain. Brain Res Mol Brain Res 59:205–214
Sunday L, Tran MM, Krause DN, Duckles SP (2006) Estrogen and progestagens differentially modulate vascular proinflammatory factors. Am J Physiol Endocrinol Metab 291:261–267
Takahashi K, Hosoya T, Onoe K, Doi H, Nagata H, Hiramatsu T, Li XL, Watanabe Y, Wada Y, Takashima T, Suzuki M, Onoe H, Watanabe Y (2014) 11C-cetrozole: an improved C-11C-methylated PET probe for aromatase imaging in the brain. J Nucl Med 55:852–857
Takizawa R, Danese A, Maughan B, Arseneault L (2015) Bullying victimization in childhood predicts inflammation and obesity at mid-life: a five-decade birth cohort study. Psychol Med 45:2705–2715
Talbot S, Foster SL, Woolf CJ (2016) Neuroimmunity: physiology and pathology. Annu Rev Immunol 34:421–447
Thapar A, Collishaw S, Pine DS, Thapar AK (2012) Depression in adolescence. Lancet 379:1056–1067
Thomas MP, Potter BV (2013) The structural biology of oestrogen metabolism. J Steroid Biochem Mol Biol 137:27–49
Toussaint L, Shields GS, Dorn G, Slavich GM (2016) Effects of lifetime stress exposure on mental and physical health in young adulthood: how stress degrades and forgiveness protects health. J Health Psychol 21:1004–1014
Tsigos C, Chrousos GP (2002) Hypothalamic-pituitary-adrenal axis, neuroendocrine factors and stress. J Psychosom Res 53:865–871
Tyring S, Gottlieb A, Papp K, Gordon K, Leonardi C, Wang A, Lalla D, Woolley M, Jahreis A, Zitnik R, Cella D, Krishnan R (2006) Etanercept and clinical outcomes, fatigue, and depression in psoriasis: Double-blind placebo-controlled randomised phase III trial. Lancet 367:29–35
Udina M, Castellvi P, Moreno-Espana J, Navines R, Valdes M, Forns X, Langohr K, Sola R, Vieta E, Martin-Santos R (2012) Interferon-induced depression in chronic hepatitis C: a systematic review and meta-analysis. J Clin Psychiatry 73:1128–1138
Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA (2005) Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol 57:67–81
Vegeto E, Bonincontro C, Pollio G, Sala A, Viappiani S, Nardi F, Brusadelli A, Viviani B, Ciana P, Maggi A (2001) Estrogen prevents the lipopolysaccharide-induced inflammatory response in microglia. J Neurosci 21:1809–1818
Venema CM, Mammatas LH, Schroder CP, van Kruchten M, Apollonio G, Glaudemans A, Bongaerts AHH, Hoekstra OS, Verheul HMW, Boven E, van der Vegt B, de Vries EFJ, de Vries EGE, Boellaard R, Menke van der Houven van Oordt CW, Hospers GAP (2017) Androgen and estrogen receptor imaging in metastatic breast cancer patients as a surrogate for tissue biopsies. J Nucl Med 58:1906–1912
Vetter ML, Wadden TA, Vinnard C, Moore RH, Khan Z, Volger S, Sarwer DB, Faulconbridge LF, Group P-UR (2013) Gender differences in the relationship between symptoms of depression and high-sensitivity CRP. Int J Obes 37(Suppl 1):S38–S43
Villa A, Vegeto E, Poletti A, Maggi A (2016) Estrogens, neuroinflammation, and neurodegeneration. Endocr Rev 37:372–402
Vogelzangs N, Duivis HE, Beekman AT, Kluft C, Neuteboom J, Hoogendijk W, Smit JH, de Jonge P, Penninx BW (2012) Association of depressive disorders, depression characteristics and antidepressant medication with inflammation. Transl Psychiatry 2:e79
Wegienka G, Baird DD (2005) A comparison of recalled date of last menstrual period with prospectively recorded dates. J Women's Health 14:248–252
Werling DM, Geschwind DH (2013) Sex differences in autism spectrum disorders. Curr Opin Neurol 26:146–153
Whitacre CC, Reingold SC, O’Looney PA (1999) A gender gap in autoimmunity. Science 283:1277–1278
Wise DD, Felker A, Stahl SM (2008) Tailoring treatment of depression for women across the reproductive lifecycle: The importance of pregnancy, vasomotor symptoms, and other estrogen-related events in psychopharmacology. CNS Spectr 13:647–662
Wright CE, Strike PC, Brydon L, Steptoe A (2005) Acute inflammation and negative mood: mediation by cytokine activation. Brain Behav Immun 19:345–350
Xia-Zhang L, Xiao E, Ferin M (1995) A 5-day estradiol therapy, in amounts reproducing concentrations of the early-mid follicular phase, prevents the activation of the hypothalamo-pituitary-adrenal axis by interleukin-1 alpha in the ovariectomized rhesus monkey. J Neuroendocrinol 7:387–392
Yudkin JS, Kumari M, Humphries SE, Mohamed-Ali V (2000) Inflammation, obesity, stress and coronary heart disease: is interleukin-6 the link? Atherosclerosis 148:209–214
Zsido RG, Villringer A, Sacher J (2017) Using positron emission tomography to investigate hormone-mediated neurochemical changes across the female lifespan: implications for depression. Int Rev Psychiatry 29:580–596
Zunszain PA, Anacker C, Cattaneo A, Carvalho LA, Pariante CM (2011) Glucocorticoids, cytokines and brain abnormalities in depression. Prog Neuropsychopharmacol Biol Psychiatry 35:722–729
Funding
Preparation of this article was supported by a Society in Science—Branco Weiss Fellowship, NARSAD Young Investigator Grant #23958 from the Brain & Behavior Research Foundation, and National Institutes of Health grant K08 MH103443 to George M. Slavich, and by a Society in Science—Branco Weiss Fellowship, NARSAD Young Investigator Grant #25032 from the Brain & Behavior Research Foundation, and a Minerva Research Group grant from the Max Planck Society to Julia Sacher.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article belongs to a Special Issue on Neuroimmune Signaling in Psychiatric Disease.
Rights and permissions
About this article

Cite this article
Slavich, G.M., Sacher, J. Stress, sex hormones, inflammation, and major depressive disorder: Extending Social Signal Transduction Theory of Depression to account for sex differences in mood disorders. Psychopharmacology 236, 3063–3079 (2019). https://doi.org/10.1007/s00213-019-05326-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-019-05326-9