
Abstract
Preclinical and clinical research supports a role for neuroactive steroids in the pathophysiology of posttraumatic stress disorder (PTSD). We investigated ganaxolone (a synthetic 3β-methylated derivative of allopregnanolone, a GABAergic neuroactive steroid) for treatment of PTSD in a proof-of-concept, multisite, double-blind, placebo-controlled trial. Veteran and non-veteran participants (n = 112) were randomized to ganaxolone or placebo at biweekly escalating doses of 200, 400, and 600 mg twice daily for 6 weeks. During an open-label 6-week extension phase, the initial ganaxolone group continued ganaxolone, while the placebo group crossed over to ganaxolone. Eighty-six and 59 participants, respectively, completed the placebo-controlled and open-label phases. A modified intent-to-treat mixed model repeated measures analysis revealed no significant differences between the effects of ganaxolone and placebo on Clinician Administered PTSD Symptom (CAPS) scores, global well-being, negative mood, or sleep. Dropout rates did not differ between groups, and ganaxolone was generally well tolerated. Trough blood levels of ganaxolone at the end of the double-blind phase were, however, lower than the anticipated therapeutic level of ganaxolone in >35% of participants on active drug. Pharmacokinetic profiling of the ganaxolone dose regimen used in the trial and adverse event sensitivity analyses suggest that under-dosing may have contributed to the failure of ganaxolone to out-perform placebo. Future investigations of ganaxolone may benefit from higher dosing, rigorous monitoring of dosing adherence, a longer length of placebo-controlled testing, and targeting of treatment to PTSD subpopulations with demonstrably dysregulated pre-treatment neuroactive steroid levels.
Clinicaltrials.gov identifier: NCT01339689.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Over the past 15 years, strides have been made in defining the neurobiological characteristics of PTSD (Pitman et al. 2012). Nevertheless, there remain limited efficacious pharmacotherapies for PTSD, either as stand-alone treatment or for augmentation of trauma-focused psychotherapies such as Prolonged Exposure or Cognitive Processing Therapy (Resick et al. 2014). Medications tested in PTSD have typically shown large effect sizes in small preliminary studies, but smaller effects when tested against placebo in large multisite trials. The serotonin selective reuptake inhibitors (SSRIs) sertraline and paroxetine, the only two FDA-approved medications for the treatment of PTSD, showed only moderate effect sizes in FDA registration trials (Brady et al. 2000; Davidson et al. 2001; Marshall et al. 2001; Tucker et al. 2001). Combat veterans, in particular, appeared to be resistant to SSRIs (e.g., Friedman et al. 2007), although variability in outcomes across veteran studies now suggests that ethnicity, age, or other subpopulation differences may play a role (Bernardy and Friedman 2015). The growing evidence for biological heterogeneity underlying the PTSD phenotype has therefore provoked a call for medications that address individually variable PTSD-specific pathophysiological processes (Rasmusson and Abdallah 2015; Friedman and Bernardy 2016).
As discussed below, preclinical and clinical evidence supports a role for neuroactive steroids with effects at gamma-amino-butyric acid (GABA)A receptors in the pathophysiology and treatment of PTSD in at least some subpopulations. Levels of allopregnanolone, a progesterone metabolite that positively and potently modulates effects of GABA at GABAA receptors, increase following administration of SSRIs, and appear to contribute to the therapeutic effects of SSRIs (Uzunova et al. 1998; Romeo et al. 1998). In healthy humans, increases in serum allopregnanolone levels during a study of the effects of pregnenolone (a precursor for allopregnanolone) on emotion regulation correlated negatively with amygdala activity; further, a higher ratio of allopregnanolone to pregnenolone after pregnenolone infusion was positively correlated with dorsomedial prefrontal cortex-amygdala connectivity, which in turn correlated negatively with anxiety (Sripada et al. 2013). In contrast, a reduction in the corticolimbic expression of allopregnanolone in male rodent models (Pibiri et al. 2008; Zhang et al. 2014) is associated with anxiety-like behaviors, increased aggression, enhanced contextual fear conditioning, and extinction deficits. In turn, administration of allopregnanolone, drugs that stimulate allopregnanolone synthesis, or an SSRI at doses well below that which block serotonin reuptake but that rectify allopregnanolone levels reverse these aberrant behaviors (Pinna et al. 2006; Pibiri et al. 2008; Zhang et al. 2014).
In women with PTSD, cerebrospinal fluid (CSF) levels of allopregnanolone and pregnanolone (the GABAergic 5β stereoisomer of allopregnanolone) were together decreased to 39% of levels seen in healthy women, correlated inversely with PTSD re-experiencing symptoms and negative mood, and were lowest in patients with comorbid major depressive disorder (MDD) (Rasmusson et al. 2006). The ratio of these GABAergic neuroactive steroids to the immediate allopregnanolone precursor, 5α-dihydroprogesterone, was also decreased, consistent with a block in their synthesis. Recent studies demonstrate a similar role for these neuroactive steroids in the pathophysiology of PTSD in men (Marx 2014; Rasmusson et al. 2016).
Allopregnanolone acts at benzodiazepine-sensitive synaptic GABAA receptors and more potently at benzodiazepine-insensitive, extrasynaptic GABA A receptors (Semyanov et al. 2004). Of note, Chhatwal et al. (2005) demonstrated downregulation of synaptic GABAA receptors in the amygdala after fear conditioning, thus providing a rationale for the poor efficacy in PTSD of benzodiazepines. In contrast, gabapentin and topiramate (which target extrasynaptic GABAA receptors) have shown efficacy in several PTSD trials (Bernardy and Friedman 2015). Thus, the potential utility of allopregnanolone in treating PTSD appears plausible.
Although allopregnanolone has been intravenously administered to healthy humans (Timby et al. 2006; van Broekhoven et al. 2007), and to patients with traumatic brain injury (ClinicalTrials.gov: NCT01673828), Alzheimer’s disease (ClinicalTrials.gov: NCT02221622), and postpartum depression (ClinicalTrials.gov Identifier: NCT02942004), an oral formulation of allopregnanolone is not yet available. Ganaxolone, a synthetic 3β-methylated analog of allopregnanolone, can be administered orally, however. Like allopregnanolone, ganaxolone is a positive allosteric modulator of GABAA receptors and demonstrates anxiolytic and anticonvulsant actions in rodent models (Kaminski et al. 2004; Reddy et al. 2004). The tolerability and safety of ganaxolone also has been demonstrated in phase 1 and phase 2 clinical trials in patients with refractory epilepsy (Laxer et al. 2000; Nohria and Giller 2007; Bialer et al. 2010). We therefore decided to investigate ganaxolone for the treatment of PTSD in a phase 2, proof-of-concept clinical trial. We hypothesized that ganaxolone would show an advantage over placebo in reducing PTSD symptoms and improving well-being. We also expected ganaxolone to be well tolerated and that adverse events would not differ between the ganaxolone and placebo treatments.
Methods
Study design
The trial was conducted between April 19, 2011 and January 9, 2014, at eight Department of Veterans Affairs Medical Centers. The trial consisted of a 6-week, randomized, double-blind, placebo-controlled phase, followed by a 6-week, open-label extension phase in which subjects initially randomized to ganaxolone continued ganaxolone (GNX-GNX), while placebo-treated subjects crossed over to ganaxolone (PLC-GNX). A 1-week taper of ganaxolone followed with safety assessments extending to week 15 (Fig. 1). Eligible participants were randomized in a 1:1 ratio based on a randomly permuted block design stratified by site and sex. The GNX-GNX group received biweekly escalating ganaxolone doses of 200, 400, and 600 mg twice a day; ganaxolone was continued at 600 mg twice a day during the 6-week extension phase. The PLC-GNX group received placebo and then ganaxolone according to the same titration schedule for the blinded and open-label phases, respectively. Subjects unable to tolerate the 400 or 600 mg twice a day doses were dropped back to the previous dose and remained at that dose for the remainder of the study.

Trial design. GNX-GNX group: randomized to ganaxolone for the first 6 weeks of the blinded phase of the study, with continuation of open-label active drug during the second 6 weeks; PLC-GNX group: randomized to placebo for the first 6 weeks and crossed over to open-label ganaxolone during the second 6 weeks
Study drug
Each gelatin capsule of ganaxolone contained 200 mg ganaxolone (3α-hydroxy-3β-methyl-5α-pregnan-20-one). The placebo formulation was comprised of sucrose spheres of comparable size to the ganaxolone spray-layered spheres encapsulated in a gelatin capsule of identical (00) size, weight, and appearance. Randomization and study drug dispensing were managed through an Interactive Web Response System (IWRS). The contents of each drug bottle were blinded; labels contained a unique bottle number corresponding to either ganaxolone or placebo. Upon completion of each participant’s baseline evaluation, the investigator or appropriate designee logged onto the IWRS to randomize the subject to treatment type and receive corresponding bottle numbers. Designated personnel at the clinical site matched the assigned bottle numbers with the correct bottles of study drug and distributed the bottles to the investigator or designee. Only the investigational drug supplier and an unblinded inventory manager had access to the bottle numbers and treatment assignment codes.
Dosing adherence
Adherence to the study drug-dosing regimen was assessed at all visits by counting the returned supplies. Of note, each bottle contained an unspecified greater number of capsules than needed for the expected duration of time between visits. The amount of drug dispensed/returned was recorded in the Drug Accountability Log. Subjects that fell below 80% compliance at two consecutive visits were withdrawn from the study. Sites were provided a “Compliance Table” to facilitate assessment of drug adherence.
Patient sample
Potential subjects participated in informed consent procedures approved by Institutional Review Boards for each performance site, the INTRuST Consortium at University of California, San Diego, and the Department of Defense Human Research Protection Office (HRPO).
To be included in the trial, veteran or civilian outpatients, aged 18–65 years,Footnote 1 had to be exposed to DSM-IV PTSD A1 and A2 criteria trauma at least 6 months prior to evaluation, and meet DSM-IV criteria for current PTSD (APA 1994). Exposure to potentially traumatic events was assessed using the Childhood Trauma Questionnaire (CTQ: Bernstein et al. 1994), the Life Events Checklist (LEC: Kubany et al. 2000; Gray et al. 2004) modified to ascertain whether endorsed items met the DSM-IV A2 criterion, and the Deployment Risk and Resilience Inventory (Combat Experiences Scale, Exposure to the Aftermath of Battle Scale, Perceived Threat Scale, and Sexual Harassment Scale) (DRRI: King et al. 2006). Participants also had to score at least a “1” for frequency and “2” for intensity on at least one Criterion B re-experiencing symptom, three Criterion C avoidance symptoms, and two Criterion D hyperarousal symptoms over the past month. A 1-month CAPS total score ≥50 and a past-week CAPS score ≥50 were required at the screening and baseline visits, respectively. Substance abuse and psychiatric diagnoses other than PTSD were assessed using the Mini-International Neuropsychiatric Interview (M.I.N.I.: Sheehan et al. 1998). Participants had to be otherwise in general good health as confirmed by medical history, physical examination, electrocardiogram, and screening laboratory tests, have a negative urine drug screen for benzodiazepines, opiates, barbiturates, phencyclidine, cocaine, amphetamines, and tetrahydrocannabinol [THC], and test negative for pregnancy and refrain from breastfeeding, if female. Participants with childbearing potential had to agree to use effective contraception.
Participants with clinically unstable medical conditions, a history of seizures (except childhood febrile seizures), and moderate to severe traumatic brain injury (TBI) (American Congress of Rehabilitation Medicine, Kay et al. 1993) were excluded. Also exclusionary were DSM-IV (APA 1994) diagnoses of current or past schizophrenia or other psychotic disorders (except psychosis NOS due to the presence of sensory hallucinations clearly related to trauma), bipolar type I disorder, dementia, substance abuse or dependence on drugs or alcohol within 6 months of study entry, unwillingness to abstain from alcohol during the study, suicidal or homicidal ideation necessitating clinical intervention, history of suicide attempt in the past 10 years, alanine or aspartate transferase levels greater than two times the upper limits of normal, and unwillingness to abstain from grapefruit products that inhibit the enzyme CYP3A4, which metabolizes ganaxolone. Participants taking psychotropic medications other than approved insomnia medications (zolpidem, zaleplon, eszopiclone, or trazodone up to 150 mg at a frequency of four times a week or less) were excluded, as were subjects in evidence-based trauma-focused treatment for PTSD within 6 weeks of the trial. Participation in other psychotherapeutic modalities maintained for 3 months before and during the trial was permitted.
Efficacy and safety assessments
The primary PTSD outcome measure was the CAPS (1-week version; Blake et al. 1995), which provides a continuous measure of PTSD symptom frequency and intensity. It was administered at baseline and biweekly through week 12. The secondary outcome measure was the clinician-assessed Clinical Global Impression rating of Improvement (CGI-I), which uses a seven-point Likert-like scale to measure overall health and function. It was administered biweekly beginning at week 2.
Ancillary symptom measures included the following: PTSD Check-List (PCL), a 17-item self-report of DSM-IV PTSD symptom severity (Conybeare et al. 2012); patient-rated CGI-I; Profile of Mood States (POMS: McNair et al. 1992), a 65-item self-report with 5-point measures of five factor analytically derived dimensions of mood (vigor/activity, anger/irritability, anxiety/tension, depression/dejection, confusion/bewilderment, and fatigue/inertia); Patient Health Questionnaire-9 (PHQ-9: Kroenke et al. 2001), a 9-item depression subscale of the Patient Health Questionnaire based on the nine DSM-IV diagnostic criteria for major depressive disorder; Insomnia Severity Index (ISI: Bastien et al. 2001), a 5-item 2-week assessment of self-perceived difficulty with sleep onset, middle of the night awakening, early morning awakening, and impairment in daily function attributed to insomnia; and Connor-Davidson Resilience Scale (CD-RISC: Connor and Davidson 2003), a 25-item scale that typically correlates with treatment response. The PCL was administered biweekly. The other ancillary ratings were administered at baseline, week 6, and week 12.
Safety assessments conducted at each planned visit (and at additional visits if clinically indicated) included the Columbia–Suicide Severity Rating Scale (CSSRS: Posner et al. 2011), a mental status examination by the study psychiatrist, vital signs including orthostatic blood pressure changes, weight, clinical blood tests, urinalysis, a urine toxicology screen, measurement of urine cotinine (a long-acting metabolite of nicotine), a pregnancy test, and an electrocardiogram. Physical and neurological examinations were conducted at screening, week 6, week 12, and additional visits if clinically indicated.
Statistical methods
Sample size was based on a two-sided, two-sample t test for change. Cohen’s d effects sizes for published, large, multicenter PTSD trials of venlafaxine, sertraline, and paroxetine range between 0.19 and 0.44, and are considered inadequate in relation to current satisfaction with these drugs. Hence, the study was powered to detect a somewhat larger effect size of 0.65.
Assuming 33% attrition, 76 completers (or approximately 112 subjects overall) were required to detect a 65% difference in the standardized change in treatment arms with 80% power.
An interim and final analyses were planned using the method of DeMets and Lan (1994) and the O’Brien-Fleming-type spending function; futility rules were based on conditional power approaches (Pepe and Anderson 1992). Results of the interim analysis were reviewed by an independent Data Monitoring Committee that recommended continuation of the trial. Statistical analyses were performed in R version 3.1.0. No adjustments were made for multiple comparisons; a p < 0.047 and p < 0.05 were considered to be statistically significant for the primary and secondary analyses, respectively.
The primary outcome (CAPS total score) was analyzed using a mixed model repeated measures (MMRM) approach in a modified intent-to-treat (mITT) population. The mITT population consisted of randomized subjects dispensed study medication and having at least one post-baseline data point during the 6-week, placebo-controlled phase of the study. The model included change from baseline in CAPS total score at each post-baseline visit during the blinded phase as the dependent variable. Independent variables included treatment type (placebo vs. active ganaxolone), study visit, the treatment-by-visit interaction, and CAPS total score at baseline. Visits (weeks 2, 4, and 6) were treated as a categorical variable. An unstructured variance-covariance structure was used. Pre-specified variables, including baseline age, sex, smoking status, comorbid depression, and history of mild TBI, were assessed as potential model covariates if the variable was unbalanced between the treatment groups and associated with outcome in the univariate analysis.
Secondary outcome CGI-I scores were collapsed into two categories (“Responders” if scored as 1 or 2, or “Non-responders” if scored between 3 and 7) and analyzed using a generalized estimating equation (GEE) model for binary data. The model included the binary responder status at weeks 2, 4, and 6 as the dependent variable; treatment, study visit, and the treatment-by-visit interaction were treated as independent variables. Ancillary efficacy outcomes were analyzed in a manner similar to the primary analysis if measured biweekly. An analysis of covariance (ANCOVA) model was used for outcomes measured only at baseline and week 6 during the blinded phase of the study. Descriptive analyses were performed to compare baseline data between treatment groups. Categorical variables were evaluated using Fisher’s exact test. Continuous variables were analyzed with Wilcoxon’s rank sum test. Safety data were summarized overall and by treatment group for the blinded and extended phases of the study. Fisher’s exact test was used for comparisons between groups.
Results
Demographic and clinical characteristics of sample
A total of 344 individuals consented to participate in the study; 230 screen-failed (see the CONSORT diagram, Fig. 2). One hundred twelve (112) participants with PTSD were randomized into the study (GNX-GNX group: n = 59; PLC-GNX group: n = 53); 86 completed the first 6-week, blinded, placebo-controlled phase of the trial (GNX: n = 42; PLC: n = 44); 53 participants remained in the study through the subsequent 6-week open-label phase, the 1-week taper, and the 2-week, post-taper follow-up period (GNX-GNX: n = 27; PLC-GNX: n = 26). There were no significant differences between participants randomized to the GNX-GNX and PLC-GNX groups with regard to demographic or baseline clinical characteristics, including age, sex, ethnicity, combat veteran status, smoking status, comorbid major depression, or presence of mild TBI (Table 1).

CONSORT flow diagram. Single asterisk means 114 subjects were randomized, but two randomizations violated the inclusion criteria and were removed from the randomized population; double asterisks mean other categorical reasons for screen failure included the following: study termination (3); unwillingness to follow birth control requirement (2); unreliable reporter (1); pending lawsuit (1); age (1). Triple asterisks mean the per-protocol analysis sample was distinguished, a priori, from the intent-to-treat sample by the following criteria: (a) available for the 6-week CAPS assessment, (b) at least 80% compliant with study medication during the blinded phase of the study, (c) received the correct intervention, (d) did not initiate prohibited medications during the blinded phase of the trial
Treatment efficacy
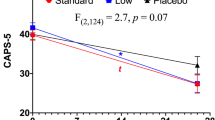
A total of 99 patients completed one post-randomization visit and were included in the primary mITT efficacy analysis (GNX: n = 49; PLC: n = 50). Over the 6-week, placebo-controlled phase, MMRM analysis revealed that the GNX group experienced a mean reduction of 17.6 points (95% CI −23.6, −11.6) in total CAPS score, while the PLC group experienced a mean reduction of 15.1 points (95% CI −21, −9.2). There was no significant difference between the two groups (p = 0.55, Fig. 3 and Table 2). There were also no significant differences between treatment groups for changes in CAPS Cluster B re-experiencing symptoms, Cluster C avoidance symptoms, or Cluster D hyperarousal symptoms (Table 2). In addition, there was not a group difference in clinician-rated CGI-I responder rates: odds ratio at week 6 comparing ganaxolone vs. placebo = 1.38 (95% CI 0.55, 3.49), p = 0.49.

Estimated change from baseline in Clinician Administered PTSD Scale (CAPS) total score during the first 6-week blinded phase of the trial, based on a modified intent-to-treat (mITT) mixed model repeated measure (MMRM) analysis
Reductions in PCL scores and other ancillary measures also showed no significant differences between the ganaxolone and placebo-treated groups (Table 3). Per-protocol analyses, which included only participants who adhered strictly to study procedures and completed the three biweekly visits following randomization, also revealed no significant group differences.
Over the full 12 weeks of the trial, the GNX-GNX group, which was continuously treated with ganaxolone, had a mean change in the CAPS total score of −28.6 points at week 12 (95% CI −34.7, −22.4) compared to −26.8 (95% CI −32.8, −20.9) for the PLC-GNX group, (p = 0.69). The changes in PCL scores at week 12 were −15.5 (95% CI −20.1, −10.9) and −13.6 (95% CI −18.0, −9.1), respectively, for the GNX-GNX vs. PLC-GNX groups (p = 0.55). There also were no significant differences between these two groups for the other measures collected in the study (data not shown).
Treatment tolerability and safety
There were no significant differences between groups in rates of early discontinuation during the blinded phase (GNX, 28.8% vs. PLC, 17.0%; Fisher’s exact test p = 0.18) or whole study (GNX-GNX, 54.2% vs PLC-GNX, 50.9%; Fisher’s exact test p = 0.85).
At the end of the placebo-controlled phase, all participants on active ganaxolone had been dispensed 1200 mg/day of study drug, except for one participant receiving 400 mg/day. At the end of the open-label extension phase, all participants had been dispensed ganaxolone at 1200 mg/day except for seven receiving 800 mg/day. The proportion of patients experiencing at least one adverse event after randomization did not differ by treatment group during either the blinded (GNX, 66.1% vs. PLC, 60.4%, p = 0.56) or extension (GNX-GNX, 42.9% vs. PLC-GNX, 45.4%, p = 0.83) phases. Adverse events reported by at least 10% of patients who completed the first 6 weeks of the trial were headache (13.6% (GNX) vs. 7.5% (PLC), p = 0.37) and somnolence (18.6% (GNX) vs. 13.2% (PLC), p = 0.45). Adverse events reported by at least 10% of patients who completed the open-label phase were headache (19% (GNX-GNX) vs. 9.1% (PLC-GNX), p = 0.22) and somnolence (7.1% (GNX-GNX) vs. 13.6% (PLC-GNX), p = 0.48). No patients discontinued treatment due to the emergence of laboratory abnormalities, and there were no treatment group differences in laboratory or EKG abnormalities or clinically significant changes. There were significantly greater reductions in sitting and standing pulse by week 6 in the GNX compared to PLC group (sitting: −2.9 ± 11.0 bpm vs. +1.1 ± 10.0 bpm, p = 0.035; standing: −4.3 ± 9.3 bpm vs. 0.6 ± 11.3 bpm, p = 0.011). The mean change in weight over the double-blind phase did not differ between groups: (GNX, 0.81 ± 6.6 lbs.; PLC, −0.17 ± 7.37 lbs., p = 0.56). There were three serious adverse events in patients taking ganaxolone during the first 6 weeks of the trial. One was unrelated to drug; the second involved an overnight hospital stay for dizziness, confusion, and balance problems that resolved with ganaxolone discontinuation. The third was an episode of suicidal ideation that resolved without change in study drug or other clinical intervention; the participant completed the study without further incident.
Sensitivity analyses based on therapeutic trough ganaxolone levels
As this was the first study to administer ganaxolone to PTSD patients, the target therapeutic trough level of ganaxolone was inferred from previous phase 2 clinical trials in patients with refractory partial complex seizures (Laxer et al. 2000; Bialer et al. 2010) and phase 1 inpatient pharmacokinetic (PK) studies in healthy volunteers taking single or multiple daily doses of an earlier ganaxolone formulation with a high fat meal under supervision (Monaghan et al. 1997; Marinus Pharmaceuticals, Inc. data on file). The PK study in healthy volunteers predicted trough values of ∼30 ng/ml or higher with adherent dosing of ganaxolone at 1200 mg/day.
While the current study was being conducted, PK data for the ganaxolone formulation and dosing regimen used in the study was collected in healthy male inpatient volunteers taking ganaxolone with a standard meal or snack at 200, 400, and 600 mg bid. Ganaxolone was rapidly absorbed with a T max of ∼2 h. The half-life (T 1/2) for ganaxolone was 7–10 h, with elimination occurring primarily in the feces. The mean ganaxolone trough level in 22 of the healthy volunteers taking the 600 mg bid dose after 3 days was 38.9 ± 16.9 ng/ml; this was about 25% lower than the trough level obtained when ganaxolone was taken with high fat meals. Based on this data, and assuming that participants took study medication with meals as instructed, we estimate that about 16% of study participants taking ganaxolone at 600 mg bid between weeks 4 and 6 of the study would have had trough levels of ≤22 ng/ml [i.e., mean trough level (38.9 ng/ml) minus 1 SD of the mean trough level (16.9 ng/ml)].
Plasma trough levels of ganaxolone were measured in the current study at week 6 in 72 of 86 participants who completed the placebo-controlled phase of the trial. Among the 34 patients on active ganaxolone, 41.2% (n = 14) had ganaxolone levels below 30 ng/ml; 35.3% (n = 12) had ganaxolone levels below the predicted therapeutic trough level of 20 ng/ml, and 23.5% (n = 8) had undetectable levels. Drug levels were also measured at week 12 in 46 of the 59 participants on active ganaxolone. Among these participants, 32.6% (n = 15) had ganaxolone trough levels below 30 ng/ml level; 26.1% (n = 12) had trough levels below 20 ng/ml, and 10.9% (n = 5) had undetectable levels. The extent of ganaxolone under-dosing (i.e., trough level < 20 ng/ml) may be underestimated, however, as 7 participants took their last ganaxolone dose in the morning within 5 h of blood sampling, thus confounding accurate measurement of trough drug levels in these cases.
We therefore conducted a sensitivity analysis comparing the 22 participants on active ganaxolone with trough drug levels ≥20 ng/ml to the controls. The MMRM efficacy analysis revealed no significant treatment group differences in the change in CAPS scores from baseline to week 6 (GNX: −16.7 (95% CI −25.6, −7.9); PLC: −15.0 (95% CI −21.1, −8.9); p = 0.75). We also re-assessed adverse event rates. When only those participants with ganaxolone levels ≥20 ng/ml were included in the comparison, a significantly larger proportion of the GNX group (91.0%) than the PLC group (60.4%) reported at least one adverse event (p = 0.01). The proportion of headaches was 23% (5/22) for patients in the GNX group compared to 7.5% (4/53) for the PLC group, p = 0.11. The proportion of somnolence was 27% (6/22) in the GNX group compared to 13.2% (7/53) in the PLC group, p = 0.18.
Comment
This multicenter proof-of-concept, placebo-controlled study represents the first attempt to evaluate ganaxolone, a synthetic derivative of allopregnanolone, for the treatment of PTSD in a population of male and female, veteran and civilian patients. The study found no significant differences in the impact of placebo and ganaxolone on total CAPS score over 6 weeks (the primary endpoint). Ganaxolone and placebo also had similar effects on general well-being, self-rated PTSD symptoms, negative mood, sleep, and resilience. Ganaxolone was generally well tolerated at the doses administered; the most frequent side effects were headache and mild somnolence that resolved with time or a reduction in the ganaxolone dose.
This study thus suggests that ganaxolone may not be effective in the treatment of PTSD, despite preclinical and clinical studies suggesting a possible role for deficits in allopregnanolone in the pathophysiology of PTSD. Although ganaxolone has GABAA receptor modulating properties and has shown efficacy in epilepsy, small variations in the structure of neurosteroids can profoundly alter their functional impact (Schüle et al. 2014). For example, the two 3β stereoisomers of allopregnanolone do not exhibit GABAA receptor activity, although their molecular weights are identical to those of allopregnanolone. It is also possible that administration of GABAergic neuroactive steroids is not helpful in PTSD.
Problems with ganaxolone dosing, however, may have contributed to the lack of difference in outcomes between the ganaxolone and placebo groups. Rates of adverse events were not different between participants randomized to active drug versus placebo during the blinded phase of the study. However, adverse events were higher among participants on ganaxolone with trough levels above the expected therapeutic trough level of 20 ng/ml compared to participants on placebo, suggesting that participants on ganaxolone with trough levels below 20 ng/ml were under-dosed. Nevertheless, sensitivity analyses excluding participants with trough levels below 20 ng/ml also did not suggest ganaxolone efficacy. These analyses were likely underpowered, however, given the omission of over one third of the participants randomized to ganaxolone. It is also possible that therapeutic dosing of ganaxolone in PTSD requires dosages that yield plasma levels higher than 22 ng/ml.
When selecting doses of ganaxolone for this proof-of-concept trial, the investigators hypothesized that effective doses in PTSD might be at the lower end of the dose range studied in epilepsy (1200–1800 mg/day), assuming that some ganaxolone effects would be mediated via high affinity, benzodiazepine-resistant extrasynaptic GABAA receptors (Lambert et al. 2003; Mody and Pearce 2004) and because treatment-resistant epilepsy patients generally have high drug tolerance. Therefore, although ganaxolone was successfully titrated to doses above 1200 mg/day in 2 weeks or less in epilepsy studies, the extended titration of ganaxolone in the current study to just 1200 mg/day was designed to maximize tolerability, minimize drop out, and collect evidence about possible effects of lower as well as higher doses of ganaxolone. As PK testing of the specific ganaxolone formulation and bid dosing regimen used in the study suggests that about 16% of individuals with low trough levels in the current study would be expectable, other factors likely contributed to under-dosing, including the following: non-adherence to the dosing regimen, including failure to take the medication with a meal (absorption of ganaxolone is decreased by about 50–66% in the fasting state), or increased ganaxolone metabolism in patients with PTSD. Stress steroids and other ligands, such as nicotine, activate the nuclear pregnane X receptor (PXR) (Lamba et al. 2004); PXR, in turn, increases expression of CYP3A4 (Sinz et al. 2008), which metabolizes ganaxolone.
Potential experimental design issues also should be considered in evaluating the outcome of the current study. The short duration of the controlled phase of the study and the short time at maximum drug dose (2 weeks) likely contributed to a failure to discern a potential ganaxolone advantage. Perusal of the mean PTSD symptom response curves between weeks 4 and 6 of the study suggests that the downward slope in the placebo group was flattening (with a CAPS score reduction of 2.3 points), while it was steepening in the ganaxolone group (CAPS scores reduction of 6.7 points) (Table 2). In addition, as can be seen in Fig. 3, CAPS scores in both treatment groups decreased in parallel over the first 4 weeks of the placebo-controlled phase. Given that ganaxolone levels in many patients may have been subtherapeutic at ganaxolone doses of 200 mg bid and 400 mg bid, these reductions in symptoms may largely represent placebo effects in both groups. Future studies thus might consider trial designs that help reduce placebo effects (Chen et al. 2011; Heger 2013).
It is also now clear that multiple, individually variable neurobiological factors contribute to the pathophysiology of PTSD (Rasmusson and Shalev 2014), suggesting a need for precision medicine approaches to PTSD therapeutics (Rasmusson and Abdallah 2015). For example, clinical research suggests that lower levels of ALLO in CSF are associated with more severe PTSD and negative mood or depressive symptoms (Uzunova et al. 1998; Rasmusson et al. 2006). Studies also have shown that individuals with PTSD/MDD generally have more severe PTSD (Breslau et al. 2000), and may recover less quickly or completely in response to exposure-based cognitive therapy for PTSD (Nishith et al. 2005). Moreover, extinction and extinction retention deficits in allopregnanolone deficient rodents were normalized after a single dose of ganaxolone administered at the end of the first extinction training session; extinction and extinction retention was not influenced by ganaxolone in rodents with normal allopregnanolone levels (Pinna and Rasmusson 2014). Together, these observations suggest that ganaxolone may specifically benefit individuals with demonstrable allopregnanolone deficits, which appear to be greater or most common among individuals with comorbid PTSD/MDD. The rigorous exclusion criteria for this outpatient study thus may have inadvertently excluded individuals more likely to be responsive to ganaxolone (Table 1). Finally, to the extent that allopregnanolone-like treatments may facilitate PTSD recovery by facilitating extinction learning or retention, it may be necessary to pair such treatments with structured extinction learning opportunities (such as those afforded by trauma-focused PTSD psychotherapies) in order to achieve clinical benefit. Alternatively, longer periods of drug administration and/or higher dosing may allow exposure-avoidant patients with chronic PTSD to encounter a sufficient number of natural re-exposure opportunities to enable effective extinction and recovery over time.
Consideration of other drug administration paradigms also may be indicated. The release of stress hormones such as allopregnanolone from peripheral and brain sources may have a diurnal rhythm, as do other stress hormones, as well as ultra-radian rhythms contingent on exposure to natural stressors. Steady state dosing of ganaxolone could potentially suppress critically timed neurobiological responses to stress and interfere with extinction learning or other processes inherent to PTSD recovery. In the rodent study highlighted above, a single dose of ganaxolone administered immediately after one session of extinction training markedly enhanced and stabilized extinction and extinction retention, suggesting that ganaxolone may block reconsolidation of conditioned fear or facilitate consolidation of new learning. Further experimental work to translate these preclinical observations into more effective uses of ganaxolone is warranted.
The enrollment period for this study, which achieved its pre-determined enrollment benchmarks, was comparable or better than many previously reported pharmacological intervention studies in PTSD. Nevertheless, there were several challenges to efficient recruitment. Although critical to prevent confounding of the trial outcome, the rigorous inclusion criteria contributed to recruitment challenges. Conduct of PTSD pharmacological trials in settings where substance abuse or use of other medications is less common (e.g., active duty settings) or where patients with more severe illness can be kept safe (e.g., inpatient units or residential programs) may address some of these challenges. Current research-related administrative procedures also contributed to trial length: 17 institutional review boards (IRBs) were involved in approving the study, 1 for the Department of Defense (DOD), and 2 for each site (one for the Veterans Administration and one for each site’s academic affiliate); non-lead sites had to wait for IRB approvals at the lead site and DOD before submitting study materials to their IRBs and the DOD. Use of a central IRB could reduce IRB-related delays in study implementation. There were complicated contracting procedures, which delayed hiring of study staff at trial outset. Mid-trial changes in recruitment strategies (requiring IRB approval) to address unanticipated lags in recruitment prolonged recruitment lags at several study sites; implementation of multiple recruitment strategies at trial outset could prevent such delays. Finally, a mid-trial halt in recruitment occurred across all sites while funding was sought for manufacture of an additional supply of study drug—reflecting the fiscal challenges of small pharmaceutical companies even when partnering with public sources of support. Addressing the ubiquitous inefficient practices embedded in the current drug development process is essential if the rational iterative research process required to develop and effectively target new PTSD therapeutics is to be successful.
In summary, this first phase 2 clinical trial of ganaxolone in PTSD showed that ganaxolone, as dosed, was safe but no more efficacious than placebo in reducing PTSD symptoms. Several possible reasons for this failure will need to be evaluated in future trials. Pharmacokinetic studies to evaluate metabolism of ganaxolone in PTSD patients may be helpful. Rigorous monitoring of adherence to study medication, such as use of electronically monitored bottles or video apps to confirm that capsules were taken, as well as measurement of ganaxolone levels, will be necessary to confirm compliance and adequacy of dosing. Lengthening the placebo-controlled phase of testing is indicated. In addition, future studies might consider use of novel precision medicine approaches, such as targeting of ganaxolone to patients with demonstrated deficits in GABAergic neuroactive steroids, or precise targeting of drug to specific neurocognitive processes critical to PTSD recovery. Future studies should also capture other study population characteristics that might influence therapeutic response in PTSD clinical trials, such as type and timing of trauma exposure, sex, and patterns of previous response to PTSD treatments.
Notes
Based on the observed general safety of ganaxolone, the upper end of the age range for study eligibility was increased from 55 to 65 years on February 22, 2013, after which an additional 18 participants included in the modified intent-to-treat sample were recruited.
References
American Psychiatric Association (1994) Diagnostic and Statistical Manual of Mental Disorders, 4th edn. Washington, DC, American Psychiatric Association
Bastien CH, Vallières A, Morin CM (2001) Validation of the Insomnia Severity Index as an outcome measure for insomnia research. Sleep Med 2:297–307. doi:10.1016/S1389-9457(00)00065-4
Bernardy NC, Friedman MJ (2015) Psychopharmacological strategies in the management of posttraumatic stress disorder (PTSD): what have we learned? Curr Psychiatry Rep 17:564. doi:10.1007/s11920-015-0564-2
Bernstein D, Fink L, Handelsman L, Foote J, Lovejoy M, Wenzel K et al (1994) Initial reliability and validity of a new retrospective measure of child abuse and neglect. Am J Psychiatry 151:1132–1136. doi:10.1176/ajp.151.8.1132
Bialer M, Johannessen SI, Levy RH, Perucca E, Tomson T, White HS (2010) Progress report on new antiepileptic drugs: a summary of the Tenth Eilat Conference (EILAT X). Epilepsy Res 92:89–124. doi:10.1016/j.eplepsyres.2010.09.001
Blake DD, Weathers FW, Nagy LM, Kaloupek DG, Gusman FD, Charney DS et al (1995) The development of a clinician-administered PTSD scale. J Traum Stress 8:75–90. doi:10.1002/jts.2490080106
Brady K, Pearlstein T, Asnis GM, Baker D, Rothbaum B, Sikes C et al (2000) Efficacy and safety of sertraline treatment of posttraumatic stress disorder. JAMA 283:1837–1844. doi:10.1001/jama.283.14.1837
Breslau N, Davis GC, Peterson EL, Schultz LR (2000) A second look at comorbidity in victims of trauma: the posttraumatic stress disorder-major depression connection. Biol Psychiatry 48:902–909
Chen YF, Yang Y, Hung HJ, Wang SJ (2011) Evaluation of performance of some enrichment designs dealing with high placebo response in psychiatric clinical trials. Contemp Clin Trials 32:592–604
Chhatwal JP, Myers KM, Ressler KJ, Davis M (2005) Regulation of gephyrin and GABAA receptor binding within the amygdala after fear acquisition and extinction. J Neurosci 25:502–506. doi:10.1523/JNEUROSCI.3301-04.2005
Connor KM, Davidson JR (2003) Development of a new resilience scale: the Connor-Davidson Resilience Scale (CD-RISC). Dep Anx 18:76–82. doi:10.1002/da.10113
Conybeare D, Behar E, Solomon A, Newman MG, Borkovec TD (2012) The PTSD Checklist-Civilian Version: reliability, validity, and factor structure in a nonclinical sample. J Clin Psychol 68:699–713. doi:10.1002/jclp.21845
Davidson JRT, Rothbaum BO, van der Kolk BA, Sikes CR, Farfel GM (2001) Multicenter, double-blind comparison of sertraline and placebo in the treatment of posttraumatic stress disorder. Arch Gen Psychiatry 58:485–492. doi:10.1001/archpsyc.58.5.485
DeMets DL, Lan KK (1994) Interim analysis: the alpha spending function approach. Stats Med 13:1341–1352. doi:10.1002/sim.4780131308
Friedman MJ, Bernardy NC (2016) Considering future pharmacotherapy for PTSD. J Neurosci Lett. doi:10.1016/j.neulet.2016.11.048
Friedman MJ, Davidson JRT (2014) Pharmacotherapy for PTSD. In: Friedman M, Keane T, Resick P (eds) Handbook of PTSD, 2nd edn. Guilford Publications, Inc., New York, pp 482–501
Friedman MJ, Marmar CR, Baker DG, Sikes CR, Farfel GM (2007) Randomized, double-blind comparison of sertraline and placebo for posttraumatic stress disorder in a Department of Veterans Affairs setting. J Clin Psychiatry 68:711–720
Gray MJ, Litz BT, Hsu JL, Lombardo TW (2004) Psychometric properties of the Life Events Checklist. Assessment 11:330–341. doi:10.1177/1073191104269954
Heger M (2013) Trial designs advance to overcome bitter pill of placebo effect. Nat Med 19:1353
Kaminski RM, Livingood MR, Rogawski MA (2004) Allopregnanolone Analogs That Positively Modulate GABAA Receptors Protect against Partial Seizures Induced by 6‐Hz Electrical Stimulation in Mice. Epilepsia 45:864–867
Kay T, Harrington DE, Adams R, Anderson T, Berrol S, Cicerone K et al (1993) Definition of mild traumatic brain injury. J Head Trauma Rehab 8:86–87
King LA, King DW, Vogt DS, Knight JA, Samper R (2006) Deployment Risk and Resilience Inventory: a collection of measures for studying deployment-related experiences of military personnel and veterans. Mil Psychol 18:89–120. doi:10.1207/s15327876mp1802_1
Kroenke K, Spitzer RL, Williams JB (2001) The PHQ-9: validity of a brief depression severity measure. J Gen Int Med 16:606–613. doi:10.1046/j.1525-1497.2001.016009606.x
Kubany ES, Leisen MB, Kaplan AS, Watson SB, Haynes SN, Owens JA et al (2000) Development and preliminary validation of a brief broad-spectrum measure of trauma exposure: the Traumatic Life Events Questionnaire. Psychol Assess 12:210–224. doi:10.1037/1040-3590.12.2.210
Lamba V, Yasuda K, Lamba JK, Assem M, Davila J, Strom S, Schuetz EG. PXR (NR1I2): splice variants in human tissues, including brain, and identification of neurosteroids and nicotine as PXR activators. Toxicol Applied Pharmacol 199:251–65
Lambert JJ, Belelli D, Peden DR, Vardy AW, Peters JA (2003) Neurosteroid modulation of GABA A receptors. Prog. Neurobiology 71:67–80
Laxer K, Blum D, Abou-Khalil BW, Morrell MJ, Lee DA, Data JL et al (2000) Assessment of ganaxolone’s anticonvulsant activity using a randomized, double-blind, presurgical trial design. Ganaxolone Presurgical Study Group. Epilepsia 41:1187–1194. doi:10.1111/j.1528-1157.2000.tb00324.x
Marshall RD, Beebe KL, Oldham M et al (2001) Efficacy and safety of paroxetine treatment for chronic PTSD: a fixed-dose, placebo-controlled study. Am J Psychiatry 158:1982–1988. doi:10.1176/appi.ajp.158.12.1982
Marx CE (2014) Neurosteroids as novel therapeutics and biomarker candidates in TBI, schizophrenia, and PTSD. Inaugural Neurosteroid Congress, Durham
McNair DM, Lorr M, Droppleman LF (1992) Revised manual for the profile of mood states. Educational and Industrial Testing Services, San Diego
Mody I, Pearce RA (2004) Diversity of inhibitory neurotransmission through GABA A receptors. Trends Neurosci 27:569–75
Monaghan EP, Navalta LA, Shum L, Ashbrook DW, Lee DA (1997) Initial human experience with ganaxolone, a neuroactive steroid with antiepileptic activity. Epilepsia. 38:1026–31
Nishith P, Nixon RD, Resick PA (2005) Resolution of trauma-related guilt following treatment of PTSD in female rape victims: A result of cognitive processing therapy targeting comorbid depression? J Affective Disorders 86:259–65
Nohria V, Giller E (2007) Ganaxolone. Neurotherapeutics 4:102–105. doi:10.1016/j.nurt.2006.11.003
Pepe MS, Anderson GL (1992) Two-stage experimental designs: early stopping with a negative result. Appl Stat 41:181–190. doi:10.2307/2347627
Pibiri F, Nelson M, Guidotti A, Costa E, Pinna G (2008) Decreased corticolimbic allopregnanolone expression during social isolation enhances contextual fear: a model relevant for posttraumatic stress disorder. Proc Natl Acad Sci 105:5567–5572. doi:10.1073/pnas.0801853105
Pinna G, Rasmusson AM (2014) Ganaxolone improves behavioral deficits in a mouse model of post-traumatic stress disorder. Front Cell Neurosci 8:1–11. doi:10.3389/fncel.2014.00256
Pinna G, Costa E, Guidotti A (2006) Fluoxetine and norfluoxetine stereospecifically and selectively increase brain neurosteroid content at doses inactive on 5-HT reuptake. Psychopharmacol 24:1–11. doi:10.1007/s00213-005-0213-2
Pitman RK, Rasmusson AM, Koenen KC, Shin LM, Orr SP, Gilbertson MW, Milad MR, Liberzon I (2012) Biological studies of post-traumatic stress disorder. Nature Rev Neurosci 13:769–87
Posner K, Brown GK, Stanley B, Brent DA, Yershova KV, Oquendo MA, Currier GW, Melvin GA, Greenhill L, Shen S, Mann JJ (2011) The Columbia–Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry 168:1266–1277
Rasmusson AM, Abdallah CG (2015) Biomarkers for PTSD treatment and diagnosis. PTSD Res Q 26:1–14
Rasmusson AM, Shalev AY (2014) Integrating the neuroendocrinology, neurochemistry, and neuroimmunology of PTSD to date and the challenges ahead. In Friedman M, Keane T, Resick P (eds) Handbook of PTSD, Second Edition, Guilford Publications, Inc, New York, NY, pp 166–189
Rasmusson AM, Pinna G, Paliwal P, Weisman D, Gottschalk C, Charney DS et al (2006) Decreased cerebrospinal fluid allopregnanolone levels in women with PTSD. Biol Psychiatry 60:704–713. doi:10.1016/j.biopsych.2006.03.026
Rasmusson A, King M, Gregor K, Scioli-Salter E, Pineles S, Valovski I, Hamouda M, Pinna G (2016) Sex differences in the enzyme site at which GABAergic neuroactive steroid synthesis is blocked in PTSD: implications for targeting of PTSD therapeutics. In symposium: sex specificity in posttraumatic stress disorder: from biological mechanisms to treatment response (Fellingham K: Chair; Jovanovich T: Discussant). 32nd Annual Meeting, International Society for Traumatic Stress Studies Dallas
Reddy DS, Castaneda DC, O'Malley BW, Rogawski MA (2004) Anticonvulsant activity of progesterone and neurosteroids in progesterone receptor knockout mice. J Pharmacol Exp Therapeutics. 310:230–9
Resick PA, Monson CM, Gutner CA, Maslej MM (2014) Psychosocial treatments for adults with PTSD. In: Friedman M, Keane T, Resick P (eds) Handbook of PTSD, 2nd edn. Guilford Publications, Inc., New York, pp 419–436
Romeo E, Strohle A, Spalletta G, di Michele F, Hermann B, Holsboer F et al (1998) Effects of antidepressant treatment on neuroactive steroids in major depression. Am J Psychiatry 155:910–913. doi:10.1176/ajp.155.7.910
Schüle C, Nothdurfter C, Rupprecht R (2014) The role of allopregnanolone in depression and anxiety. Prog Neurobiol. doi:10.1016/j.pneurobio.2013.09.003
Semyanov A, Walker MC, Kullmann DM, Silver RA (2004) Tonically active GABA a receptors: modulating gain and maintaining the tone. Trends Neurosci 27:262–269. doi:10.1016/j.tins.2004.03.005
Sheehan DV, Lecrubier Y, Sheehan KH, Amorim P, Janavs J, Weiller E et al (1998) The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry 59(Suppl 20):22–33 quiz 34-57
Sinz MW. Pregnane X receptor: prediction and attenuation of human CYP3A4 enzyme induction and drug–drug interactions (2008) Ann Reports Medicinal Chem 43:405–18
Sripada RK, Marx CE, King AP, Rampton JC, Ho SS, Liberzon I (2013) Allopregnanolone elevations following pregnenolone administration are associated with enhanced activation of emotion regulation neurocircuits. Biol Psychiatry. 73:1045–53
Timby E, Balgård M, Nyberg S, Spigset O, Andersson A, Porankiewicz-Asplund J et al (2006) Pharmacokinetic and behavioral effects of allopregnanolone in healthy women. Psychopharmacol 186:414–424. doi:10.1007/s00213-005-0148-7
Tucker P, Zaninelli R, Yehuda R (2001) Paroxetine in the treatment of chronic posttraumatic stress disorder: results of a placebo-controlled, flexible-dosage trial. J Clin Psychiatry 62:860–868
Uzunova V, Sheline Y, Davis J, Rasmusson A, Uzunova D, Costa E et al (1998) Increase in the cerebrospinal fluid content of neurosteroids in patients with unipolar major depression who are receiving fluoxetine or fluvoxamine. Proc Natl Acad Sci. 95:3239–3244.
van Broekhoven F, Bäckström T, van Luijtelaar G, Buitelaar JK, Smits P, Verkes RJ (2007) Effects of allopregnanolone on sedation in men, and in women on oral contraceptives. Psychoneuroendocrinol 32:555–564. doi:10.1016/j.psyneuen.2007.03.009
Zhang LM, Qiu ZK, Zhao N, Chen HX, Liu YQ, Xu JP, Zhang YZ, Yang RF, Li YF (2014) Anxiolytic-like effects of YL-IPA08, a potent ligand for the translocator protein (18 kDa) in animal models of post-traumatic stress disorder. Int J Neuropsychopharmacol 17:1659–1669. doi:10.1017/S1461145714000479
Acknowledgements
INTRuST Consortium lead site principal investigators
Duke University Medical Center: Gerald A. Grant, M.D., and Christine E. Marx, M.D., M.A.;
Spaulding Rehabilitation Hospital: Ross Zafonte, D.O.;
University of California, San Diego: Raul Coimbra, M.D., Ph.D.;
Dartmouth University: Thomas McAllister, M.D.;
Uniformed Services University of the Health Sciences: David Benedek, M.D.;
University of Cincinnati: Lori Shutter, M.D.;
Medical University of South Carolina: Mark S. George, M.D.
Clinical trial performance site principal investigators
VA Medical Center Cincinnati and University of Cincinnati College of Medicine: Thomas Geracioti, M.D.;
Ralph H. Johnson VA Medical Center: Mark Hamner, M.D.;
VA San Diego Healthcare System: James Lohr, M.D.;
Durham VA Medical Center: Christine E. Marx, M.D., M.A.;
VA Boston Healthcare System: Ann M. Rasmusson, M.D.;
Washington DC VA Medical Center: Richard Rosse, M.D.;
Manchester VA Medical Center: Lanier Summerall, M.D.;
White River Junction VA Medical Center: Lanier Summerall, M.D.
INTRuST Consortium clinical trialists and site monitors
Alice L. Mills, M.D., M.P.H., Wendy Ching, Kimberly Aguilar, Karen Stokes, Lisa Kallenberg, M.D., James Payamo, M.D.
Performance site clinical coordinators
Duke University Medical Center Durham VA: Susan O’Loughlin; Research Service, VA Boston Healthcare Service, and Department of Psychiatry, Boston University School of Medicine: Erica R. Scioli-Salter, Ph.D. and Kristin Gregor, Ph.D.; VA Medical Center Cincinnati and University of Cincinnati College of Medicine: Julie Baker-Nolan, MSW and Heather Dodge, MSW; Ralph H. Johnson VA Medical Center: Bridgette Heyward, B.S.; Washington DC VA Medical Center: Erika Roberge, B.S.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
U.S. Army Medical Research and Materiel Command (USAMRMC) Contract # W81XWH-08-2-0159 to: INTRuST Clinical Consortium Coordinating Center, 9500 Gilman Drive, # 0855, La Jolla, CA 92093-0855, Principal Investigator: Murray B. Stein, M.D., M.P.H.
Department of Veterans Affairs Rehabilitation Research and Development.
Career Development Award (1lK2RX000908) to Dr. Jennifer Naylor.
FDA IND# 106,104 & Source of Active and Placebo Medication for Trial
Marinus Pharmaceuticals, Inc.,
21 Business Park Drive,
Branford, Connecticut 06405.
Conflict of interest
Dr. Rasmusson has received compensation as a member of the scientific advisory board for Resilience Therapeutics, Inc., and as a consultant to Cohen Veterans Biosciences. Dr. Marx is an applicant on pending patent applications focused on neurosteroids and derivatives in CNS disorders (no patents issued; no licensing in place; VA 208 waiver issued). As noted in the author affiliations, Drs. Farfel and Tsai were employees of Marinus Pharmaceuticals during the clinical trial; Dr. Farfel is now an employee of Zogenix, Inc.; Dr. Tsai remains an employee of Marinus Pharmaceuticals. Dr. Gericioti was funded as the lead investigator for another DOD-INTRuST consortium clinical trial. He is also a member of the pharmaceutical development company, RxDino, LLC. Dr. Cusin has been a paid consultant to Janssen Pharmaceuticals, Inc. Dr. Stein has been paid in the past 3 years for consulting to Actelion, Janssen, Pfizer and Tonix pharmaceutical companies. He also has been paid for editorial work for UpToDate and the journal Biological Psychiatry, and he is a consultant to Resilience Therapeutics and Oxeia Biopharmaceuticals. Dr. Summerall receives no compensation outside of VA funding and has no conflict of interest. Drs. Hamner, Lohr, Rosse, and Lang have no disclosures.
Additional information
Ann M. Rasmusson and Christine E. Marx share first authorship and are the lead investigators responsible for the design and implementation of the study as submitted for funding and support to the INTRuST Clinical Consortium Coordinating Center.
Where the full trial protocol can be accessed
Marinus Pharmaceuticals, Inc.,
21 Business Park Drive,
Branford, Connecticut 06405
and
INTRuST Clinical Consortium Coordinating Center
9500 Gilman Drive, # 0855
La Jolla, CA 92093–0855
Rights and permissions
About this article

Cite this article
Rasmusson, A.M., Marx, C.E., Jain, S. et al. A randomized controlled trial of ganaxolone in posttraumatic stress disorder. Psychopharmacology 234, 2245–2257 (2017). https://doi.org/10.1007/s00213-017-4649-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-017-4649-y