
Abstract
Rationale
GABAA positive allosteric modulators (GABAA PAMs), such as diazepam and zolpidem, are used clinically for anxiety and insomnia, but abuse liability is a concern. Novel GABAA PAMS may have lower abuse liability while retaining clinical utility.
Objective
The present study compared abuse-related effects of the non-selective GABAA PAM diazepam, the α1-selective GABAA PAM zolpidem, and three novel GABAA PAMs (JY-XHe-053, XHe-II-053, and HZ-166) using intracranial self-stimulation (ICSS) in rats. These novel compounds have relatively low efficacy at α1-, α2-, and α3-containing GABAA receptors, putative in vivo selectivity at α2/α3-containing GABAA receptors, and produce anxiolytic-like effects with limited sedation in non-human primates.
Methods
Adult, male Sprague-Dawley rats (n = 17) were each implanted with a bipolar electrode in the medial forebrain bundle and trained to respond under a fixed-ratio 1 schedule of reinforcement for electrical brain stimulation. The potency and time course of effects were compared for diazepam (0.1–10 mg/kg), zolpidem (0.032–3.2 mg/kg), and the three novel compounds (JY-XHe-053, XHe-II-053, and HZ-166; all 3.2–32 mg/kg).
Results
Zolpidem and diazepam produced transient facilitation of ICSS at small doses and more sustained rate-decreasing effects at larger doses. JY-XHe-053 and HZ-166 produced weak and inconsistent ICSS facilitation, whereas XHe-II-053 had no effect on ICSS.
Conclusions
These results support a key role for α1-containing GABAA receptors in mediating GABAA PAM-induced ICSS facilitation. These results are concordant with drug self-administration studies in monkeys in suggesting that GABAA PAMs with low α1 efficacy and putative α2/α3 selectivity have lower abuse liability than high-efficacy non-selective or α1-selective GABAA PAMs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
GABAA positive allosteric modulators (GABAA PAMs) such as diazepam are used clinically for the treatment of anxiety (Bellantuono et al. 1980; Chouinard 2004; Shader and Greenblatt 1993), seizures (Dreifuss et al. 1998; Riss et al. 2008), alcohol withdrawal syndrome (Daeppen et al. 2002; Mayo-Smith 1997), and insomnia (McClusky et al. 1991; Pakes et al. 1981). Although GABAA PAMs are useful therapeutics and are frequently prescribed (O’Brien 2005), concerns exist regarding their abuse liability (Evans et al. 1990; Woods and Winger 1995). All clinically available GABAA PAMs in the USA are classified as schedule IV drugs by the Drug Enforcement Agency, and after opioids, they are the second most misused prescription drug class by people aged 12 or older in the USA (Hughes et al. 2016). Furthermore, GABAA PAMs carry a high risk of diversion and are often co-abused with other substances, which may result in fatalities (Inciardi et al. 2006; Jones et al. 2012; Pauly et al. 2011). GABAA receptors are pentameric ligand-gated ion channels that can be categorized into subtypes depending on their constituent subunit composition, and different subtypes may contribute to different behavioral and physiological effects. For example, much of the literature suggests that subtypes containing α1 subunits mediate the reinforcing and sedative effects of GABAA PAMs, whereas subtypes containing α2/α3 subunits have been implicated in anxiolytic effects (Ator 2005; Rudolph et al. 1999; Tan et al. 2010). For these reasons, novel GABAA PAM compounds selective for α2/α3-containing GABAA receptors might have lower abuse liability while still producing therapeutic effects such as anxiolysis (Ator 2005; Möhler et al. 2001; Skolnick 2012).
Drug self-administration procedures have played a key role in generating data on the abuse liability of GABAA PAMs and the role of GABAA receptor subtypes in mediating that abuse liability. For example, diazepam is a relatively non-selective and high-efficacy ligand across GABAA receptor subtypes, and it is self-administered across a wide range of reinforcement schedules in multiple species (Grant and Johanson 1987; Griffiths et al. 1979; Pilotto et al. 1984; Roache and Griffiths 1989; Stewart et al. 1994). Zolpidem is a high-efficacy α1-selective GABAA PAM clinically available for the treatment of insomnia (Dündar et al. 2004; Sanna et al. 2002; Siriwardena et al. 2008). Although zolpidem was originally thought to be a sleep aid devoid of abuse-related effects (Holm and Goa 2000; Victorri-Vigneau et al. 2007), it is also self-administered by non-human primates (Griffiths et al. 1992; Rowlett et al. 2005), and in humans, it produces subjective effects similar to benzodiazepines (Evans et al. 1990) and is abused (Griffiths and Johnson 2005; Hajak et al. 2003). Conversely, α2/α3-selective GABAA PAMs are self-administered at lower rates and across a narrower range of experimental conditions than diazepam or zolpidem (Ator et al. 2010; Rowlett et al. 2005; Shinday et al. 2013). For instance, TPA023B and TP003, compounds lacking efficacy at GABAA α1-containing receptors, failed to maintain self-administration in rhesus monkeys trained to self-administer cocaine, and they maintained relatively low rates of self-administration in monkeys trained to self-administer the non-selective GABAA PAM midazolam (Shinday et al. 2013). Similarly, L-838,417, an agonist at α2/α3/α5-containing GABAA receptors and an antagonist at α1-containing GABAA receptors, maintained low self-administration rates in rhesus monkeys trained to self-administer the barbiturate methohexital (Rowlett et al. 2005). GABAA PAMs that selectively activate α2/α3-containing GABAA receptors have not been approved for clinical use, and their actual abuse liability in humans remains to be determined; however, these data from drug self-administration studies have contributed to the impression that abuse-related effects of GABAA PAMs are mediated primarily by GABAA receptors containing α1 subunits.
Intracranial self-stimulation (ICSS) is another family of procedures that has been used to assess abuse liability of drugs (Carlezon and Chartoff 2007; Negus and Miller 2014). In ICSS procedures, subjects are trained to emit an operant response reinforced by pulses of brain stimulation delivered via an electrode to a brain reward area, and many drugs of abuse increase (or “facilitate”) ICSS. Consistent with its reinforcing effects in drug self-administration procedures and its abuse liability in humans, diazepam has been shown previously to facilitate ICSS in rodents (Caudarella et al. 1982; Reynolds et al. 2012; Straub et al. 2010; Tracy et al. 2014). However, zolpidem has been examined in only one study, where it failed to facilitate ICSS in mice (Reynolds et al. 2012), and effects of α2/α3-selective compounds have not been examined. Thus, the degree to which GABAA PAM effects in ICSS procedures parallel results from drug self-administration procedures has not been extensively investigated. Accordingly, the goal of this study was to compare the potency, efficacy, and time course of diazepam, zolpidem, and three novel GABAA PAMs in an ICSS procedure that has been used previously to examine effects of opioids, monoamine transporter ligands, and other classes of drugs (Negus and Miller 2014). The novel GABAA PAMs selected for study were JY-XHe-053, XHe-II-053, and HZ-166. In vitro data on receptor binding and functional activity suggest that, in comparison to diazepam, these compounds have similar non-selective binding profiles but progressively lower efficacies at α1-, α2-, and α3-containing GABAA receptor subtypes (diazepam > JY-XHe-053 > XHe-II-053 > HZ-166; Fischer et al. 2010; Rivas et al. 2009). This decline in efficacy also appears to result in α2/α3 selectivity of behavioral effects insofar as these compounds retain sufficient efficacy to produce effects mediated by GABAA receptors containing α2/α3 but not α1 subunits. For example, HZ-166 produced antinociception in mice that was eliminated with mutated α2 subunits (Ralvenius et al. 2015), and both HZ-166 and XHe-II-053 produced anxiolytic effects with reduced sedation compared to diazepam in rhesus monkeys (Fischer et al. 2010). We hypothesized that, in agreement with drug self-administration results, diazepam and zolpidem would produce greater abuse-related ICSS facilitation than JY-XHe-053, XHe-II-053, or HZ-166.
Methods
Subjects
Studies were conducted on a total of 17 adult Sprague-Dawley rats (Harlan, Frederick, MD), and studies were conducted in males to permit direct comparison to previous studies with other classes of drugs (Negus and Miller 2014). All rats had ad libitum access to water and rodent chow and were housed individually on a 12-h light-dark cycle (lights on from 0600 to 1800) in a facility accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care. At the time of surgery, rats weighed between 300 and 350 g. Experiments were performed with the approval of the Virginia Commonwealth University Institutional Animal Care and Use Committee in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals 8th edition (National Research Council 2011).
Surgery
Rats were anesthetized with 2.5–3% isoflurane (Webster Veterinary, Phoenix, AZ, USA) in oxygen until unresponsive to toe pinch before the stereotaxic implantation of a stainless steel, bipolar electrode (Plastics One, Roanoke, VA, USA). A cathode (0.25 mm in diameter, covered with polyamide insulation except at the tip) was implanted into the left medial forebrain bundle at the level of the hypothalamus (2.8 mm posterior to the bregma, 1.7 mm lateral to the midsagittal suture, 8.8 mm ventral to the skull). Three screws were placed into the skull, and the anode (0.125 mm in diameter, non-insulated) was wrapped around one screw to serve as the ground. Dental acrylic was used to secure the electrode to the screws and to the skull. An intraperitoneal (IP) injection of ketoprofen (5 mg/kg) served as postoperative analgesic immediately and 24 h after surgery. Rats recovered for 7 days prior to initiation of ICSS training.
Apparatus
Studies were conducted in sound-attenuating boxes containing modular acrylic and metal test chambers (29.2 × 30.5 × 24.1 cm; Med Associates, St. Albans, VT, USA). Each chamber contained a response lever (4.5 cm wide, 2.0 cm deep, 3.0 cm above the floor), three stimulus lights (red, yellow, and green) centered 7.6 cm above the lever, a 2-W house light, and an ICSS stimulator. Electrodes were connected to the stimulator via bipolar cables routed through a swivel commutator (Model SL2C, Plastics One, Roanoke, VA, USA). Programming software controlled all operant sessions and data collection (Med PC-IV, Med Associates).
Training
The behavioral procedure was similar to that described previously (Negus and Miller 2014). After initial shaping of lever pressing, rats were trained to respond under a fixed-ratio 1 (FR 1) schedule of reinforcement for electrical brain stimulation. Each lever press resulted in the delivery of a 0.5-s train of square wave cathodal pulses (0.1 ms per pulse) and illumination of all stimulus lights above the lever. Under the terminal schedule, daily sessions consisted of multiple 10-min components. Each component consisted of 10 60-s trials, and the available brain-stimulation frequency decreased in 0.05 log Hz increments from one trial to the next (158 to 56 Hz). In the first 10 s of each trial, five non-contingent priming stimulations were delivered at the stimulation frequency available during that trial, and responding had no scheduled consequences. The remaining 50 s of each trial consisted of a response period, during which responding produced electrical brain stimulation under an FR 1 schedule. Training continued until frequency-rate curves were not statistically different over three consecutive days as indicated by a lack of a significant effect of “day” in a two-way ANOVA with frequency and day as the two variables (see data analysis). Some rats were tested acutely with other drugs prior to initiation of studies reported here; however, all rats were drug-free for at least 1 week and had stable frequency-rate curves before transitioning to these studies.
Testing
Testing was conducted using dose-effect and time-course procedures. For dose-effect studies, test sessions consisted of three sequential “baseline” ICSS components followed first by a 15-min time-out period and then by two sequential “test” ICSS components. Drugs were administered IP at the beginning of the time out, and the drugs and doses tested were as follows: diazepam (0.1–10 mg/kg), zolpidem (0.032–3.2 mg/kg), HZ-166 (3.2–3.2 mg/kg), JY-XHe-053 (3.2–32 mg/kg), and XHe-II-053 (3.2–32 mg/kg). At the conclusion of each dose-effect study, one or two doses of each drug were selected for time-course studies (1 and 10 mg/kg diazepam, 0.32 and 3.2 mg/kg zolpidem, and 32 mg/kg for HZ-166, JY-XHe-053, and XHe-II-053). For time-course studies, test sessions consisted of three consecutive baseline ICSS components followed first by IP drug injection and then by pairs of test components that began 10, 30, 100, 180, and 300 min after injection. If drug effects persisted after 300 min, then an additional pair of test components was implemented 24 h after drug injection. Drug doses were administered in a counterbalanced order, dose-effect studies were completed before time-course studies, and all testing for one drug was completed before advancing to another drug. Additionally, vehicle was tested before and after dose-effect testing with each drug, and data from these vehicle tests were averaged. In the only exception to this general rule, one rat in the zolpidem group lost its electrode before testing with the largest dose and the second vehicle administration, so, for this rat, only the completed vehicle test contributed to data analysis. Test sessions were generally conducted twice a week with at least 48 h between drug doses, and three-component training sessions were conducted on all other weekdays. Each dose-effect or time-course study was conducted in a group of at least five rats, consistent with our previous studies (Negus and Miller 2014).
Data analysis
The first baseline component of each test session was considered to be a “warm up” component, and data were discarded. All remaining data were analyzed as previously described (Bauer et al. 2013; Negus and Miller 2014). The primary dependent measure was reinforcement rate in stimulations per minute during each frequency trial. Raw reinforcement rates for each rat from each trial were converted to percent maximum control rate (%MCR), with MCR determined daily and defined as the mean of the maximal rates observed at any trial during the second and third baseline components for that day. Thus, %MCR values for each trial were calculated as {(reinforcement rate during a frequency trial ÷ MCR) × 100}. For each test session, data from each pair of baseline ICSS components before drug injection and for each pair of test components after drug injection were averaged to yield group mean baseline and test frequency-rate curves, respectively. Test data were analyzed by repeated-measures two-way ANOVA, with ICSS frequency as one factor and drug dose or pretreatment time as the second factor. A significant ANOVA was followed by Holm-Sidak post hoc test. The criterion for statistical significance was P < 0.05.
Two additional dependent measures were calculated to summarize data from frequency-rate curves. First, the total number of stimulations across all 10 frequency trials was determined for each test component. Test data were normalized and expressed as a percentage of the average number of total stimulations per component during the second and third baseline components: % baseline total stimulations per component = (mean total stimulations per test component)/(mean total stimulations per baseline component) × 100. This approach includes all data from baseline and test frequency-rate curves and permits calculation of summary data even under conditions of robust ICSS facilitation or depression. ICSS facilitation is indicated by increases in % baseline total stimulations per component. As a second summary measure, the threshold frequency (θ 0) in log hertz required to maintain responding was calculated for each frequency-rate curve. Specifically, θ 0 values were determined where possible using linear regression through data on the linear portion of each frequency-rate curve, and θ 0 was defined as the x-intercept of this regression. Test data were normalized and expressed as the difference from the mean θ 0 during the second and third baseline components: ∆θ 0 = (mean θ 0 per test component) − (mean θ 0 per baseline component). Threshold calculations require application of data filtering and correction procedures and can be applied only to data sets that involve lateral shifts in frequency-rate curves without robust ICSS facilitation or depression (Carlezon and Chartoff 2007; Negus and Miller 2014). ICSS facilitation is indicated by negative ∆θ 0 values. Paired t tests were used to compare ∆θ 0 values for the dose of each drug producing maximal ICSS facilitation with ∆θ 0 values after vehicle administration.
Drugs
Diazepam (Hospira, Lake Forest, IL) and zolpidem (Sigma-Aldrich, St. Louis, MO) were purchased from commercial suppliers. HZ-166, JY-XHe-053, and XHe-II-053 were prepared by Dr. James Cook. Diazepam dilutions were made with a vehicle of 40% polyethylene glycol, 10% ethanol, and 50% sterile water. Zolpidem, HZ-166, JY-XHe-053, and XHe-II-053 were suspended in a vehicle of 60% polyethylene glycol, 20% ethanol, and 20% sterile water. All injections were administered IP in a volume of 1 mL/kg except 10 mg/kg diazepam, 32 mg/kg HZ-166, and the second vehicle determination for HZ-166, which were administered in volumes of 2 mL/kg.
Results
Baseline data
The mean ± SEM MCR for the 17 rats used in this study was 64.02 ± 1.39 stimulations per trial. The mean ± SEM number of total baseline stimulations per component was 301.14 ± 11.26 stimulations per component. The mean ± SEM θ 0 for all rats was 1.91 ± 0.01 log Hz.
Effects of diazepam
Figure 1a shows that diazepam produced biphasic effects on ICSS after a 15-min pretreatment time, and two-way ANOVA indicated a significant dose × frequency interaction [F(45,180) = 5.74, P < 0.0001]. Small doses (0.1–1.0 mg/kg) of diazepam dose-dependently increased rates of ICSS at intermediate brain-stimulation frequencies (1.85–1.95 log Hz). A larger dose (3.2 mg/kg) of diazepam also facilitated ICSS, though to a lesser degree than 1.0 mg/kg. Lastly, 10 mg/kg diazepam only decreased ICSS at the six highest frequencies (1.95–2.20 log Hz). Figure 1c, e shows the time courses of effects for 1.0 and 10 mg/kg diazepam, respectively. The smaller dose (1.0 mg/kg) of diazepam facilitated ICSS at five intermediate frequencies (1.85–2.05 log Hz) after 10 min, but this effect dissipated after 30 min (significant time × frequency interaction [F(27,108) = 1.67, P = 0.0337]). Conversely, the larger dose (10 mg/kg) of diazepam depressed ICSS from 10 to 100 min followed by weak facilitation at one frequency (1.9 log Hz) after 300 min; ICSS recovered to baseline levels after 24 h (significant time × frequency interaction [F(54,216) = 9.82, P < 0.0001]). Summary data for diazepam effects on the total number of stimulations per component across all 10 brain-stimulation frequencies are shown in Fig. 1b, d, f.

Effects of diazepam (0.1–10 mg/kg) on ICSS in rats. Left panels (a, c, e) show drug effects on frequency-rate curves. Abscissae Frequency of electrical brain stimulation in log hertz. Ordinates ICSS rate expressed as percent maximum control rate (%MCR). Filled symbols indicate frequencies at which ICSS rates after diazepam were different than those observed after vehicle (a) or at baseline (c, e) as determined by the Holm-Sidak post hoc test following a significant two-way ANOVA. Right panels (b, d, f) show summary data expressed as percent baseline total stimulations delivered across all frequencies of brain stimulation per test component. Abscissae Drug dose (mg/kg) or time (min or h) after drug administration. Ordinates % baseline total stimulations per component. Upward/downward arrows indicate significant drug-induced increases/decreases, respectively, in ICSS relative to vehicle or baseline for at least one brain-stimulation frequency as determined by analysis of full-frequency rate curves in the left panels. All points show mean ± SEM for five rats
Effects of zolpidem
Figure 2a shows that zolpidem produced biphasic effects on ICSS after a 15-min pretreatment time, and two-way ANOVA indicated a significant dose × frequency interaction [F(36,144) = 2.16, P = 0.0007]. Small doses (0.032–0.32 mg/kg) of zolpidem dose-dependently increased rates of ICSS at intermediate brain-stimulation frequencies (1.90–2.05 log Hz). A 1.0-mg/kg dose of zolpidem only decreased ICSS at the four highest frequencies (2.05–2.2 log Hz). The largest dose (3.2 mg/kg) of zolpidem was tested in four rats and thus was not included in statistical analysis, but it also only decreased ICSS. Figure 2c, e shows the time courses of effects for 0.32 and 3.2 mg/kg zolpidem, respectively. The smaller dose (0.32 mg/kg) of zolpidem produced a biphasic effect on rates of ICSS (significant time × frequency interaction [F(45,315) = 3.33, P < 0.0001]). Zolpidem produced the greatest ICSS facilitation at intermediate frequencies (1.95–2.05 log Hz) after 10 min, and weaker ICSS facilitation was still apparent after 30 min. From 100 to 300 min, zolpidem depressed ICSS at intermediate frequencies (2.0–2.1 log Hz). After 24 h, rates of responding returned to baseline. The larger dose (3.2 mg/kg) of zolpidem depressed ICSS from 10 to 300 min, and ICSS returned to baseline after 24 h (significant time × frequency interaction [F(45,315) = 2.65, P < 0.0001]). Summary data for zolpidem effects on the total number of stimulations per component across all 10 brain-stimulation frequencies are shown in Fig. 2b, d, f.

Effects of zolpidem (0.032–3.2 mg/kg) on ICSS in rats. Left panels (a, c, e) show drug effects on frequency-rate curves. Filled symbols indicate frequencies at which ICSS rates after zolpidem were different than those observed after vehicle (a) or at baseline (c, e) as determined by the Holm-Sidak post hoc test following a significant two-way ANOVA. Right panels (b, d, f) show summary data expressed as percent baseline total stimulations delivered across all frequencies of brain stimulation per test component. Upward/downward arrows indicate significant drug-induced increases/decreases, respectively, in ICSS relative to vehicle or baseline for at least one brain-stimulation frequency as determined by analysis of full-frequency rate curves in the left panels. All points show mean ± SEM for four to eight rats (dose-effect curve: 0.032–3.2 mg/kg n = 5, 3.2 mg/kg n = 4; time courses: n = 8). The asterisk in panel b indicates that results with 3.2 mg/kg zolpidem were not included in the statistical analysis because one rat lost its headcap before this dose was tested. Other details as in Fig. 1
Effects of JY-XHe-053, XHe-II-053, and HZ-166
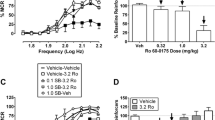
Figure 3a shows that JY-XHe-053 produced small effects on ICSS, and two-way ANOVA indicated a significant dose × frequency interaction [F(27,108) = 2.34, P = 0.0011]. A small dose (3.2 mg/kg) of JY-XHe-053 had no effects on ICSS, but larger doses (10 and 32 mg/kg) of JY-XHe-053 increased ICSS at one frequency (1.95 log Hz). Figure 3c shows that no dose of XHe-II-053 affected ICSS at any frequency of brain stimulation. Figure 3e shows that HZ-166 produced small effects on ICSS, and two-way ANOVA indicated a significant dose × frequency interaction [F(27,135) = 1.70, P = 0.0268]. The small dose (3.2 mg/kg) of HZ-166 had no effects on ICSS at any frequency of brain stimulation. The medium (10 mg/kg) and large (32 mg/kg) doses of HZ-166 increased ICSS at 2.0 and 1.95 log Hz, respectively. Summary data for effects of each drug on the total number of stimulations per component across all 10 brain-stimulation frequencies are shown in Fig. 3b, d, f. Larger doses for all three compounds were not able to be tested due to limited solubility. Time-course studies with these compounds are included in Online Resource 1. Supplemental Fig. 1a–d shows that 32 mg/kg of either JY-XHe-053 or XHe-II-053 had no effect on ICSS at any time point. Supplemental Fig. 1e–f shows that 32 mg/kg HZ-166 decreased ICSS rates only 10 min after injection at the three highest frequencies (2.1–2.2 log Hz) (significant time × frequency interaction [F(45,225) = 1.52, P = 0.0259]). Thus, none of these GABAA PAMs facilitated ICSS in time-course studies.

Effects of JY-XHe-053 (3.2–32 mg/kg), XHe-II-053 (3.2–32 mg/kg), and HZ-166 (3.2–32 mg/kg) on ICSS in rats. Left panels (a, c, e) show drug effects on frequency-rate curves. Filled symbols indicate frequencies at which ICSS rates after test drug were different than those observed after vehicle as determined by the Holm-Sidak post hoc test following a significant two-way ANOVA. Right panels (b, d, f) show summary data expressed as percent baseline total stimulations delivered across all frequencies of brain stimulation per test component. Upward/downward arrows indicate significant drug-induced increases/decreases, respectively, in ICSS relative to vehicle or baseline for at least one brain-stimulation frequency as determined by analysis of full-frequency rate curves in the left panels. All points show mean ± SEM for five to six rats (JY-XHe-053 and XHe-II-053: n = 5; HZ-166: n = 6). Other details as in Fig. 1
Drug effects on ICSS thresholds
Figure 4 shows the maximum effect of each drug on ICSS thresholds (∆θ 0 values). Only diazepam (0.32 mg/kg) [t(4) = 4.628, P = 0.0098] and zolpidem (0.32 mg/kg) [t(4) = 3.076, P = 0.0371] produced significant decreases in ∆θ 0 values relative to their respective drug vehicles.

Effects of GABAA PAMs on ICSS thresholds. Abscissa: compound and dose (mg/kg) that were most effective to facilitate ICSS. Ordinate: ∆θ 0 in log hertz. The dollar sign indicates a significant difference between effects of drug and vehicle as indicated by t test
Discussion
This study compared the effects of five GABAA PAMs in an ICSS procedure in rats. There were two main findings. First, both diazepam and zolpidem produced dose- and time-dependent ICSS facilitation consistent with the known abuse potential of these compounds. Second, three lower-efficacy PAMs thought to produce behavioral effects primarily by acting at α2/α3-containing GABAA receptors produced at best only weak and inconsistent ICSS facilitation across a range of doses. These results support evidence from drug self-administration studies in non-human primates that implicates α1-containing receptors as the principal GABAA receptors contributing to abuse-related effects of GABAA PAMs. These results also provide additional evidence for concordance between rewarding drug effects on ICSS in rats and reinforcing drug effects in drug self-administration procedures in non-human primates.
Rewarding effects of diazepam
The present results agree with those of previous reports showing facilitation of ICSS by diazepam in rodents under a variety of experimental conditions. For example, one of the earliest studies examining diazepam effects on ICSS showed that a single dose of diazepam (5 mg/kg) increased the rate at which rats pressed a pedal to receive electrical stimulation of the posterior hypothalamus (Olds 1966). Diazepam also produced an increase in lever pressing for brain stimulation in procedures that manipulated diazepam dose and stimulation intensity in rats (Caudarella et al. 1982; Gomita et al. 1983). Similarly, three previous studies in mice have shown that diazepam dose-dependently increases ICSS rates in frequency-rate procedures (Reynolds et al. 2012; Straub et al. 2010; Tracy et al. 2014) and ICSS break points in a progressive-ratio procedure (Tracy et al. 2014). The facilitation of ICSS by diazepam in rodents is also consistent with the reinforcing effects of diazepam in drug self-administration procedures in rats (Naruse and Asami 1987; Pilotto et al. 1984) and non-human primates (Grant and Johanson 1987; Griffiths and Weerts 1997; Rowlett et al. 2005; Stewart et al. 1994). For example, diazepam was self-administered via multiple routes of administration (Grant and Johanson 1987; Stewart et al. 1994) and across a range of schedule conditions (Griffiths and Weerts 1997; Rowlett et al. 2005) in rhesus monkeys and baboons. The rewarding effects of diazepam in ICSS procedures and reinforcing effects of diazepam in drug self-administration procedures are consistent with the well-established abuse liability of diazepam and other non-selective GABAA PAMs in humans (Evans et al. 1990; Griffiths et al. 1979; Woods and Winger 1995).
Rewarding effects of zolpidem
This is the first study to investigate zolpidem effects on ICSS in rats, but the facilitation of ICSS by zolpidem in this study is generally consistent with the evidence for the reinforcing effects of zolpidem in non-human primates (Ator 2002; Griffiths et al. 1992; Rowlett et al. 2005). For example, zolpidem maintained self-administration both in rhesus monkeys responding under a progressive-ratio schedule (Rowlett et al. 2005) and in baboons responding under a fixed-ratio schedule (Griffiths et al. 1992). These preclinical results also agree with the classification of zolpidem as a scheduled IV substance by the US Drug Enforcement Administration (DEA) and with evidence for zolpidem abuse by humans (Griffiths and Johnson 2005; Hajak et al. 2003). Taken together, these results support a role for α1-containing subunits in mediating abuse-related effects of GABAA PAMs.
One point of potential difference between the present ICSS results in rats and drug self-administration studies in non-human primates is the relative strength of abuse-related effects produced by zolpidem in comparison to diazepam and other non-selective GABAA PAMs. In the present study, the magnitude of ICSS facilitation by zolpidem was not larger than that produced by diazepam. In contrast, zolpidem has been shown by several metrics to have higher reinforcing effectiveness than diazepam and other non-selective GABAA PAMs in rhesus monkeys (Licata and Rowlett 2011; Rowlett and Lelas 2007; Rowlett et al. 2005). This difference in relative strength of abuse-related effects for diazepam and zolpidem in rat ICSS vs. monkey drug self-administration procedures contrasts with the highly correlated metrics for ICSS facilitation and drug self-administration produced by monoamine transporter substrates (e.g., amphetamine) in these procedures (Bauer et al. 2013). The reason for this discrepancy is currently unclear and could be related to either differences in species (rat vs. rhesus monkey) or procedure (ICSS vs. drug self-administration). Studies of zolpidem self-administration in rats might help to clarify this issue, but such studies have yet to be reported.
One finding to suggest that species might be an important factor is that the present results with zolpidem in rats disagree with results obtained in a similar ICSS procedure in mice (Reynolds et al. 2012). In that study, diazepam produced ICSS facilitation, but zolpidem failed to facilitate ICSS and produced only a dose-dependent decrease in responding. Moreover, genetic mutation of α1 subunits eliminated the rate-decreasing effects of zolpidem but failed to eliminate the ICSS-facilitating effects of diazepam; rather, ICSS facilitation by diazepam was blocked by mutations to α2 and α3 subunits. These results were interpreted to suggest that GABAA receptors containing α2/3 subunits, but not those containing α1 subunits, play a key role in mediating ICSS facilitation by GABAA PAMs in mice, a conclusion clearly at odds with the present results from rat ICSS studies or previous results from drug self-administration studies in rhesus monkeys. Overall, prevailing evidence suggests that abuse-related effects of zolpidem are absent in mice, present but roughly equivalent to those of diazepam in rats, and stronger than those of diazepam in non-human primates.
Effects of JY-XHe-053, XHe-II-053, and HZ-166
This is the first study to examine the effects of GABAA PAMs with putative α2/α3 selectivity on ICSS in any species, and relative to diazepam and zolpidem, JY-XHe-053, XHe-II-053, and HZ-166 produced weak and inconsistent changes in ICSS. Failure to observe more robust effects of these compounds on ICSS is not likely due to inadequate dosing for two reasons. First, a dose of 10 mg/kg HZ-166 reduced physiological effects of urinary bladder distension in rats (Kannampalli et al. 2017), suggesting that doses of HZ-166 tested in this study are pharmacologically relevant in rats. Second, JY-XHe-053, XHe-II-053, and HZ-166 were all as effective as diazepam, and no more than threefold less potent than diazepam, to increase punished responding in monkeys (Fischer et al. 2010); but in the present study, these compounds were tested at doses of up to 320-fold higher than the lowest effective dose of diazepam. Rather, the present results are consistent with evidence from studies in non-human primates to suggest that high efficacy at α1-containing GABAA receptors contributes to abuse-related effects (Ator et al. 2010; Rowlett et al. 2005; Shinday et al. 2013) and sedative effects (Fischer et al. 2010) of GABAA PAMs, whereas α2/α3-containing GABAA receptors play a lesser role in mediating these effects. Despite their failure to reliably facilitate ICSS in the present study, JY-XHe-053, XHe-II-053, and HZ-166 all produced anxiolytic effects in rhesus monkeys, and XHe-II-053 and HZ-166 produced anxiolysis with less sedation than diazepam (Fischer et al. 2010). Taken together, these results support the proposition that anxiolytic and abuse-related effects of GABAA PAMs can be dissociated.
Rate-decreasing effects of GABAA PAMs
In addition to facilitating ICSS at lower doses, both diazepam and zolpidem also reliably depressed ICSS at higher doses. Conversely, JY-XHe-053, XHe-II-053, and HZ-166 not only failed to reliably facilitate ICSS but also failed to reliably depress ICSS as well. These results are consistent with other evidence to suggest that α1-containing GABAA receptors mediate sedative as well as abuse-related effects of GABAA PAMS (Möhler et al. 2001) and suggest that lower levels of stimulation of α1-containing GABAA receptors are required to produce rewarding ICSS facilitation than ICSS depression.
Implications for use of ICSS for abuse liability testing
Drug self-administration is the most widely accepted procedure for preclinical abuse liability assessment (Carter and Griffiths 2009; O’Connor et al. 2011). ICSS is an alternative method for preclinical abuse liability assessment, and in general, there is a high degree of correlation between results from drug self-administration and ICSS procedures (Negus and Miller 2014). ICSS is especially advantageous for some applications, such as assessment of abuse-related effects in drug-naïve subjects, simultaneous assessment of both abuse-related ICSS facilitation and abuse-limiting rate-decreasing effects, assessment of the time course of drug effects, and evaluation of compounds that may be difficult to deliver via the intravenous route of administration commonly used for drug self-administration (Lazenka and Negus 2017; Negus and Miller 2014). Results of the present study provide new data that permit comparison of results from ICSS studies in rats and drug self-administration studies in non-human primates. In general, results across procedures are similar in showing greater abuse-related effects by high-efficacy non-selective and α1-selective GABAA PAMs than by lower-efficacy PAMs that produce behavioral effects mediated primarily by α2/α3-containing GABAA receptors. Similarly, both procedures support a key role for α1-containing GABAA receptors in mediating the abuse-related effects of GABAA PAMs. Overall, these results provide further pharmacological evidence to support use of ICSS as a complement to drug self-administration procedures for preclinical abuse liability testing.
References
Ator NA (2002) Relation between discriminative and reinforcing effects of midazolam, pentobarbital, chlordiazepoxide, zolpidem, and imidazenil in baboons. Psychopharmacology 163(3–4):477–487
Ator NA (2005) Contributions of GABA A receptor subtype selectivity to abuse liability and dependence potential of pharmacological treatments for anxiety and sleep disorders. CNS Spectr 10(1):31–39
Ator NA, Atack JR, Hargreaves RJ, Burns HD, Dawson GR (2010) Reducing abuse liability of GABAA/benzodiazepine ligands via selective partial agonist efficacy at α1 and α2/3 subtypes. J Pharmacol Exp Ther 332(1):4–16
Bauer C, Banks M, Blough B, Negus S (2013) Use of intracranial self-stimulation to evaluate abuse-related and abuse-limiting effects of monoamine releasers in rats. Br J Pharmacol 168(4):850–862
Bellantuono C, Reggi V, Tognoni G, Garattini S (1980) Benzodiazepines: clinical pharmacology and therapeutic use. Drugs 19(3):195–219
Carlezon WA, Chartoff EH (2007) Intracranial self-stimulation (ICSS) in rodents to study the neurobiology of motivation. Nature Protoc 2(11):2987–2995
Carter LP, Griffiths RR (2009) Principles of laboratory assessment of drug abuse liability and implications for clinical development. Drug Alcohol Depend 105(Suppl 1):S14–S25
Caudarella M, Campbell K, Milgram N (1982) Differential effects of diazepam (valium) on brain stimulation reward sites. Pharmacol Biochem Behav 16(1):17–21
Chouinard G (2004) Issues in the clinical use of benzodiazepines: potency, withdrawal, and rebound. J Clin Psychiatry 65(Suppl 5):7–12
Daeppen JB, Gache P, Landry U, Sekera E, Schweizer V, Gloor S, Yersin B (2002) Symptom-triggered vs fixed-schedule doses of benzodiazepine for alcohol withdrawal: a randomized treatment trial. Arch Intern Med 162(10):1117–1121
Dreifuss FE, Rosman NP, Cloyd JC, Pellock JM, Kuzniecky RI, Lo WD, Matsuo F, Sharp GB, Conry JA, Bergen DC (1998) A comparison of rectal diazepam gel and placebo for acute repetitive seizures. N Engl J Med 338(26):1869–1875
Dündar Y, Boland A, Strobl J, Dodd S, Haycox A, Bagust A, Bogg J, Dickson R, Walley T (2004) Newer hypnotic drugs for the short-term management of insomnia: a systematic review and economic evaluation. Health Technol Assess 8(24): iii-x, 1–125
Evans SM, Funderburk FR, Griffiths RR (1990) Zolpidem and triazolam in humans: behavioral and subjective effects and abuse liability. J Pharmacol Exp Ther 255(3):1246–1255
Fischer BD, Licata SC, Edwankar RV, Wang ZJ, Huang S, He X, Yu J, Zhou H, Johnson EM, Cook JM et al (2010) Anxiolytic-like effects of 8-acetylene imidazobenzodiazepines in a rhesus monkey conflict procedure. Neuropharmacology 59(7–8):612–618
Gomita Y, Ichimaru Y, Moriyama M (1983) Effects of benzodiazepines on low rate responding for low current brain stimulation rewards. Jpn J Pharmacol 33(2):498
Grant KA, Johanson CE (1987) Diazepam self-administration and resistance to extinction. Pharmacol Biochem Behav 28(1):81–86
Griffiths RR, Johnson MW (2005) Relative abuse liability of hypnotic drugs: a conceptual framework and algorithm for differentiating among compounds. J Clin Psychiatry 66(Suppl 9):31–41
Griffiths RR, Weerts EM (1997) Benzodiazepine self-administration in humans and laboratory animals—implications for problems of long-term use and abuse. Psychopharmacology 134(1):1–37
Griffiths RR, Bigelow G, Liebson I (1979) Human drug self-administration: double-blind comparison of pentobarbital, diazepam, chlorpromazine and placebo. J Pharmacol Exp Ther 210(2):301–310
Griffiths RR, Sannerud CA, Ator NA, Brady JV (1992) Zolpidem behavioral pharmacology in baboons: self-injection, discrimination, tolerance and withdrawal. J Pharmacol Exp Ther 260(3):1199–1208
Hajak G, Müller WE, Wittchen HU, Pittrow D, Kirch W (2003) Abuse and dependence potential for the non-benzodiazepine hypnotics zolpidem and zopiclone: a review of case reports and epidemiological data. Addiction 98(10):1371–1378
Holm KJ, Goa KL (2000) Zolpidem: an update of its pharmacology, therapeutic efficacy and tolerability in the treatment of insomnia. Drugs 59(4):865–889
Hughes A, Williams MR, Lipari RN, Bose J, Copello EAP, Kroutil LA (2016) Prescription drug use and misuse in the United States: results from the 2015 National Survey on Drug Use and Health. NSDUH Data Review
Inciardi JA, Surratt HL, Kurtz SP, Burke JJ (2006) The diversion of prescription drugs by health care workers in Cincinnati, Ohio. Subst Use Misuse 41(2):255–264
Jones JD, Mogali S, Comer SD (2012) Polydrug abuse: a review of opioid and benzodiazepine combination use. Drug Alcohol Depend 125(1–2):8–18
Kannampalli P, Babygirija R, Zhang J, Poe MM, Li G, Cook JM, Shaker R, Banerjee B, Sengupta JN (2017) Neonatal bladder inflammation induces long-term visceral pain and altered responses of spinal neurons in adult rats. Neuroscience 346:349–364
Lazenka MF, Negus SS (2017) Oral modafinil facilitates intracranial self-stimulation in rats: comparison with methylphenidate. Behav Pharmacol
Licata SC, Rowlett JK (2011) Self-administration of bretazenil under progressive-ratio schedules: behavioral economic analysis of the role intrinsic efficacy plays in the reinforcing effects of benzodiazepines. Drug Alcohol Depend 113(2–3):157–164
Mayo-Smith MF (1997) Pharmacological management of alcohol withdrawal: a meta-analysis and evidence-based practice guideline. JAMA 278(2):144–151
McClusky HY, Milby JB, Switzer PK, Williams V, Wooten V (1991) Efficacy of behavioral versus triazolam treatment in persistent sleep-onset insomnia. Am J Psychiatry 148(1):121–126
Möhler H, Crestani F, Rudolph U (2001) GABA A-receptor subtypes: a new pharmacology. Curr Opin Pharmacol 1(1):22–25
Naruse T, Asami T (1987) Intravenous self-administration of diazepam in rats. Eur J Pharmacol 135(3):365–373
National Research Council (2011) Guide for the care and use of laboratory animals. National Academies Press, Washington
Negus SS, Miller LL (2014) Intracranial self-stimulation to evaluate abuse potential of drugs. Pharmacol Rev 66(3):869–917
O’Brien CP (2005) Benzodiazepine use, abuse, and dependence. J Clin Psychiatry 66(Suppl 2):28–33
O’Connor EC, Chapman K, Butler P, Mead AN (2011) The predictive validity of the rat self-administration model for abuse liability. Neurosci Biobehav Rev 35(3):912–938
Olds ME (1966) Facilitatory action of diazepam and chlordiazepoxide on hypothalamic reward behavior. J Comp Physiol Psychol 62(1):136
Pakes GE, Brogden RN, Heel RC, Speight TM, Avery GS (1981) Triazolam: a review of its pharmacological properties and therapeutic efficacy in patients with insomnia. Drugs 22(2):81–110
Pauly V, Frauger E, Pradel V, Nordmann S, Pourcel L, Natali F, Sciortino V, Lapeyre-Mestre M, Micallef J, Thirion X (2011) Monitoring of benzodiazepine diversion using a multi-indicator approach. Int Clin Psychopharmacol 26(5):268–277
Pilotto R, Singer G, Overstreet D (1984) Self-injection of diazepam in naive rats: effects of dose, schedule and blockade of different receptors. Psychopharmacology 84(2):174–177
Ralvenius WT, Benke D, Acuña MA, Rudolph U, Zeilhofer HU (2015) Analgesia and unwanted benzodiazepine effects in point-mutated mice expressing only one benzodiazepine-sensitive GABAA receptor subtype. Nat commun 6:6803
Reynolds LM, Engin E, Tantillo G, Lau HM, Muschamp JW, Carlezon WA, Rudolph U (2012) Differential roles of GABAA receptor subtypes in benzodiazepine-induced enhancement of brain-stimulation reward. Neuropsychopharmacology 37(11):2531–2540
Riss J, Cloyd J, Gates J, Collins S (2008) Benzodiazepines in epilepsy: pharmacology and pharmacokinetics. Acta Neurol Scand 118(2):69–86
Rivas FM, Stables JP, Murphree L, Edwankar RV, Edwankar CR, Huang S, Jain HD, Zhou H, Majumder S, Sankar S, Roth BL, Ramerstorfer J, Furtmüller R, Sieghart W, Cook JM (2009) Antiseizure activity of novel ɣ-aminobutyric acid (A) receptor subtype-selective benzodiazepine analogues in mice and rat models. J Med Chem 52(7):1795–1798
Roache JD, Griffiths RR (1989) Diazepam and triazolam self-administration in sedative abusers: concordance of subject ratings, performance and drug self-administration. Psychopharmacology 99(3):309–315
Rowlett JK, Lelas S (2007) Comparison of zolpidem and midazolam self-administration under progressive-ratio schedules: consumer demand and labor supply analyses. Exp Clin Psychopharmacol 15(4):328–337
Rowlett JK, Platt DM, Lelas S, Atack JR, Dawson GR (2005) Different GABAA receptor subtypes mediate the anxiolytic, abuse-related, and motor effects of benzodiazepine-like drugs in primates. Proc Natl Acad Sci U S A 102(3):915–920
Rudolph U, Crestani F, Benke D, Brünig I, Benson JA, Fritschy J-M, Martin JR, Bluethmann H, Möhler H (1999) Benzodiazepine actions mediated by specific γ-aminobutyric acid(A) receptor subtypes. Nature 401(6755):796–800
Sanna E, Busonero F, Talani G, Carta M, Massa F, Peis M, Maciocco E, Biggio G (2002) Comparison of the effects of zaleplon, zolpidem, and triazolam at various GABA(A) receptor subtypes. Eur J Pharmacol 451(2):103–110
Shader RI, Greenblatt DJ (1993) Use of benzodiazepines in anxiety disorders. N Engl J Med 328(19):1398–1405
Shinday NM, Sawyer EK, Fischer BD, Platt DM, Licata SC, Atack JR, Dawson GR, Reynolds DS, Rowlett JK (2013) Reinforcing effects of compounds lacking intrinsic efficacy at α1 subunit-containing GABAA receptor subtypes in midazolam-but not cocaine-experienced rhesus monkeys. Neuropsychopharmacology 38(6):1006–1014
Siriwardena AN, Qureshi MZ, Dyas JV, Middleton H, Orner R (2008) Magic bullets for insomnia? Patients’ use and experiences of newer (Z drugs) versus older (benzodiazepine) hypnotics for sleep problems in primary care. Br J Gen Pract 58(551):417–422
Skolnick P (2012) Anxioselective anxiolytics: on a quest for the Holy Grail. Trends Pharmacol Sci 33(11):611–620
Stewart RB, Lemaire GA, Roache JD, Meisch RA (1994) Establishing benzodiazepines as oral reinforcers: midazolam and diazepam self-administration in rhesus monkeys. J Pharmacol Exp Ther 271(1):200–211
Straub CJ, Carlezon WA, Rudolph U (2010) Diazepam and cocaine potentiate brain stimulation reward in C57BL/6J mice. Behav Brain Res 206(1):17–20
Tan KR, Brown M, Labouèbe G, Yvon C, Creton C, Fritschy JM, Rudolph U, Lüscher C (2010) Neural bases for addictive properties of benzodiazepines. Nature 463(7282):769–774
Tracy ME, Slavova-Hernandez GG, Shelton KL (2014) Assessment of reinforcement enhancing effects of toluene vapor and nitrous oxide in intracranial self-stimulation. Psychopharmacology 231(7):1339–1350
Victorri-Vigneau C, Dailly E, Veyrac G, Jolliet P (2007) Evidence of zolpidem abuse and dependence: results of the French Centre for Evaluation and Information on Pharmacodependence (CEIP) network survey. Br J Clin Pharmacol 64(2):198–209
Woods JH, Winger G (1995) Current benzodiazepine issues. Psychopharmacology 118(2):107–115
Acknowledgements
The research was supported by the National Institutes of Health grants R01 NS070715 (SSN) and NS076517 and MH096463 (JMC). Additional support was provided by the Shimadzu Analytical Facility of Southeastern Wisconsin. All authors have made significant contributions to the research and manuscript, and all authors have read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Experiments were performed with the approval of the Virginia Commonwealth University Institutional Animal Care and Use Committee in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals 8th edition (National Research Council 2011).
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(DOCX 151 kb)
Rights and permissions
About this article

Cite this article
Schwienteck, K.L., Li, G., Poe, M.M. et al. Abuse-related effects of subtype-selective GABAA receptor positive allosteric modulators in an assay of intracranial self-stimulation in rats. Psychopharmacology 234, 2091–2101 (2017). https://doi.org/10.1007/s00213-017-4615-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-017-4615-8