
Abstract
Rationale
Noradrenergic system plays a critical role in the hypothalamic-pituitary-adrenal (HPA) axis regulation and the stress response. A dysregulated HPA axis may be indicative of an increased biological vulnerability for depression. In addition, a variety of studies have focused on specific alterations of α2-adrenoceptors as a mechanism involved in the pathogenesis of mood disorders and antidepressant response.
Objectives
This study aimed to evaluate the effect of subchronic corticosterone administration on rat brain α2-adrenoceptor functionality by in vitro [35S]GTPγS binding stimulation assays and in vivo dual-probe microdialysis determination of extracellular noradrenaline concentrations.
Results
Implantation of a time release corticosterone pellet during 14 days induced sustained changes in endocrine function. However, there were no differences in α2-adrenoceptor agonist UK14304-induced stimulation of [35S]GTPγS binding in prefrontal cortex (PFC) between corticosterone-treated and control rats. In the same way, the in vivo evaluation of α2-adrenoceptor-mediated noradrenaline release responses to the α2-adrenoceptor agonist clonidine local administration into the locus coeruleus (LC), and the PFC did not show differences between the groups.
Conclusions
The present results show that subchronic corticosterone administration does not induce changes on functionality of α2-adrenoceptors neither in the LC nor in noradrenergic cortical terminal areas.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Noradrenergic system and hypothalamic-pituitary-adrenal (HPA) axis are both activated in response to stress. On the one hand, HPA axis activation leads to corticosterone release which promotes the mobilization of resources, among others, peripheral monoamine release or central nervous system regulation. It is released to compensate for adverse effects of stressful events and become especially relevant when stress is maintained for a long time. On the other hand, stress results in an increased synthesis, release and turnover of noradrenaline (NA) in several brain regions involved in the stress response, including cerebral cortex, hippocampus, hypothalamus and amygdala (Stanford 1995). The main source of noradrenergic innervation in the central nervous system (CNS) remains in the locus coeruleus. Furthermore, inhibitory α2-adrenoceptors modulate noradrenergic neurone activity and NA release both in locus coeruleus (LC) and noradrenergic terminal areas (Mateo and Meana 1999). Alterations in these α2-adrenoceptors have been associated to psychiatric disorders, such as anxiety or depression, and also to the effect of antidepressants drugs. In this sense, an upregulation of α2-adrenoceptors has been reported in the brain of depressed subjects and/or suicide victims (Callado et al. 1998; De Paermentier et al. 1997; Valdizan et al. 2010), as well as in platelets of depressed untreated patients (Garcia-Sevilla et al. 2004). In contrast, decreased α2-adrenoceptor binding sites have been described in platelets from patients with anxiety disorders with respect to the control subjects (Bremner et al. 1996). Moreover, chronic administration of antidepressants reduces the activity of α2-adrenoceptors in rat brain (Invernizzi et al. 2001; Mateo et al. 2001; Parini et al. 2005) and in human platelets (Garcia-Sevilla et al. 1990; Garcia-Sevilla et al. 2004). However, the role of noradrenergic system in chronic stress conditions remains poorly understood. The notion that chronic stress exposure could be related to affective disorders is supported by findings of altered HPA axis function in depressed and anxious subjects. Thus, altered 24-h urinary cortisol excretion, blunted adenocorticotrophin hormone (ACTH), responses to exogenous corticotrophin releasing factor (CRF) administration, and increased concentration of CRF in the cerebrospinal fluid (CSF) has been reported in patients suffering from depression (Charmandari et al. 2005).
Numerous animal models have been developed for the purpose of studying mood and anxiety disorders. In this sense, exogenous corticosterone administration has been described as a useful method to study the relationship between stress, glucocorticoids, and depression/anxiety. Exogenous corticosterone administration in animals provides a valid and reliable paradigm to study the possible influence of stress system dysregulation on the aetiology, development, and treatment of anxiety and depression (Sterner and Kalynchuk 2010). In this context, the aim of the present study was to evaluate the effect of subchronic corticosterone administration for 14 days in rat brain α2-adrenoceptor in vitro and in vivo functionality.
Materials and methods
Animals and treatments
Male Sprague-Dawley rats (256 ± 4 g; n = 33) were obtained from Harlan Interfauna Ibérica, S.A. (Barcelona, Spain) and were treated subchronically with corticosterone or vehicle. Commercially available corticosterone pellets (Innovative Research of America, USA) designed to release 56 mg of corticosterone during 14 days (16 mg kg−1 day−1) were used. Control pellets (Innovative Research of America, USA) were exactly the same formulation but in the absence of corticosterone. Subcutaneous pellets were implanted in the back of the neck by surgical procedure. Animal care and all experimental protocols were performed in agreement with the European Ethical Standards (European Union Directive 2010/63/EU) and translation into Spanish legislation (RD 53/2013) and approved by the UPV/EHU Ethical Board for Animal Welfare (CEEA).
Plasma corticosterone determination
Fourteen days after surgery, animals were killed by decapitation and trunk blood was collected in EDTA tubes. Blood was centrifuged at 3000g for 20 min at 4 °C, and plasma was removed and stored at −80 °C until use. Plasma samples were analysed using a commercially available radioimmunoassay kit for corticosterone (sensitivity 5.7 ng ml−1; Coat-a-count® rat corticosterone, DPS DIPESA S.A., Madrid, Spain). The adrenal glands were also dissected and weighed.
[35S]GTPγS binding assays in membrane preparation
Rat brain cortex samples were immediately dissected after animal decapitation and stored at −80 °C until assay was performed. Membranes from whole cortex were prepared as previously described (Gonzalez-Maeso et al. 2000). The α2-adrenoceptor-mediated functional G-protein activity was assessed by [35S]GTPγS binding assay as previously described (Gonzalez-Maeso et al. 2000) with slight modifications to adapt the incubation conditions to 96-well microplates. The α2-adrenoceptor full agonist UK14304 (10−9 to 10−3 mol l−1) was used to determine receptor-stimulated [35S]GTPγS binding concentration-response curves. The specific binding was determined as the difference between total and nonspecific binding defined as the remaining [35S]GTPγS binding in the presence of 10 μM GTPγS.
Surgery, microdialysis procedure, drug administration and chromatography
At day 13 after pellet implantation, animals were anesthetised and two microdialysis probes were implanted in the proximity of the right LC and in the ipsilateral PFC as previously described (Ortega et al. 2012). In these conditions, in vitro recovery for NA was in the 10–15 % range. After 24 h for recovery, experiments were performed. In order to evaluate α2-adrenoceptor functionality, the agonist clonidine was dissolved in artificial CSF (0.1–100 μM, four concentrations) and administered locally by reverse dialysis in the LC or PFC as previously described (Fernandez-Pastor et al. 2013). Clonidine was selected as α2-adrenoceptor agonist because of the existence of previous validated data of its activity in vivo (Mateo and Meana 1999; Mateo et al. 2001). NA concentrations were measured immediately after collection of the samples by HPLC coupled to an electrochemical detector as previously described (Ortega et al. 2012).
Data analyses
Data from [35S]GTPγS binding stimulations are expressed as percentage of maximal stimulation (E max) over specific basal binding. Nonlinear regression analysis was performed on individual concentration-response curves of the agonist. The concentration of drug that elicited half-maximal effect (EC50) was obtained and normalized as −log EC50 value before statistical analysis. All data are expressed as mean values ± SEM. Differences of [35S]GTPγS binding parameters between different experimental groups were assessed by unpaired Student’s t test. The level of significance was chosen at p < 0.05.
In microdialysis experiments, statistical analyses were performed on normalized values as percentages of the basal concentrations. One-way ANOVA followed by Dunnett’s post-hoc test was used to assess the effect of the drug over time. The effect of clonidine on corticosterone-treated animals was compared with the effect on the control group by two-way ANOVA of repeated measures followed by Bonferroni’s post-hoc test. In these analyses, all the experimental points, including basal values, were considered. F values were expressed as F tr (treatment; between the groups), F t (time; within groups) or F i (treatment × time; interaction). Maximal effects (E max) were expressed as percentage over basal values. The area under the curve (AUC) values of each curve was also calculated by the addition of the percent change from basal value. Differences of AUC between different groups were assessed by unpaired Student’s t test. Results are expressed as mean values ± SEM. All statistical procedures were performed using GrahPad Prism™ (GraphPad Software, San Diego, CA, USA).
Results
Physiological parameters
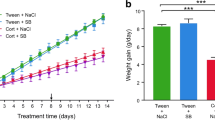
At day 14, after pellet implantation, plasma corticosterone concentration was higher in the group treated subchronically with corticosterone (141 ± 19 ng ml−1) than in the control group (72 ± 18 ng ml−1) (p = 0.02) (Fig. 1a). Body weight was also measured and the weight gain was lower (2 ± 4 g) than in control group (34 ± 7 g) (p < 0.001) (Fig. 1b). Thus, adrenal gland weight was corrected for body weight before comparison between the groups, and as expected, a significant reduction of adrenal gland size in corticosterone-treated group was found (p < 0.001) (Fig. 1c).

a Plasma corticosterone concentration, b body weight gain and c adrenal: body weight ratio obtained for each experimental condition. Rats received corticosterone (16 mg kg−1 day−1) or vehicle during 14 days from subcutaneous pellet. Data are shown as mean ± SEM (n = 8 for each group; *p < 0.05; ***p < 0.0001 compared with control)
[35S]GTPγS binding assays
The [35S]GTPγS basal binding was similar in the group treated subchronically with corticosterone (1075 ± 135 fmol mg prot.−1, n = 12) and in the control group (813 ± 165 fmol mg prot.−1, n = 8). The α2-adrenoceptor agonist UK14304 stimulated [35S]GTPγS binding in a concentration-dependent manner (10−9–10−3 M, ten concentrations) in both groups (Fig. 2a). The potency of UK14304 to stimulate [35S]GTPγS binding in corticosterone-treated group (−log EC50 = 7.0 ± 0.3) was not different to that in the control group (−log EC50 = 6.7 ± 0.3). UK14304-induced maximal stimulation of [35S]GTPγS binding did not differ between the groups (corticosterone E max = 14 ± 2 % vs control E max = 12 ± 2 %) either.

a Concentration-response curves of [35S]GTPγS binding stimulation by α2-adrenoceptor agonist UK14304 (10−9 to 10−3 M, ten concentrations) in rat brain cortex membrane homogenates of the control and corticosterone-treated animals. Points represent mean ± SEM values from eight to 12 experiments carried out in triplicates and are expressed as percentages over basal binding (BB) values. b Effect of local administration of the α2-adrenoceptor agonist clonidine (0.1–100 μM) in PFC by reverse dialysis over extracellular NA concentrations in PFC. Rats received corticosterone (16 mg kg−1 day−1) or vehicle along 14 days from subcutaneous pellet. Data are shown as mean ± SEM values from seven experiments and are expressed as percentages of the corresponding basal values. Bar graphs represent mean ± SEM area under the curve expressed in arbitrary units
Microdialysis
Basal extracellular NA concentrations in LC and PFC
Basal extracellular NA concentrations were obtained as average of three consecutive basal samples per animal. There were no statistical changes of basal extracellular NA concentration in LC in corticosterone-treated group (2.74 ± 0.49 nM; n = 36) when compared to the control group (2.53 ± 0.93 nM; n = 23). In the same way, no alterations of basal extracellular NA concentrations in PFC were observed in corticosterone-treated group (3.30 ± 0.74 nM; n = 36) in comparison with controls (3.60 ± 0.70 nM; n = 28).
Effect of local administration of clonidine into PFC on extracellular NA concentration evaluated in PFC
Local administration of the α2-adrenoceptor agonist clonidine (0.1–100 μM, four concentrations) into the PFC induced a significant decrease of extracellular NA concentration in PFC of corticosterone-treated (E max = −56 ± 11 %, p < 0.0001) (F[10,66] = 12.79; p < 0.0001; n = 7) and control (E max = −48 ± 16 %, p < 0.001) (F[10,55] = 4.55; p = 0.0001; n = 6) groups (Fig. 2b). There were not significant differences in the effect induced by clonidine between the groups (F tr[1,11] = 1.50; p = 0.25; F t[10,110] = 14.10; p < 0.0001; F i[10,110] = 1.18; p = 0.31). The analysis of the AUC values revealed a decrease (14 %) in the corticosterone-treated group that did not reach statistical significance (t = 1.17; p = 0.27).
Effect of local administration of clonidine into LC on extracellular NA concentrations evaluated in LC and PFC
Local administration of clonidine (0.1–100 μM, four concentrations) into the LC significantly decreased extracellular NA concentration in LC in a concentration-dependent manner (Fig. 3a) in control (E max = −41 ± 1 %, p < 0.001) (F[10,66] = 14.57; p < 0.0001; n = 7) and in corticosterone-treated (E max = −42 ± 7 %, p < 0.001) (F[10,88] = 12.93; p < 0.0001; n = 9) groups. There were not significant differences in the effect induced by local administration of clonidine in LC between experimental groups (F tr[1,14] = 0.08; p = 0.77; F t[10,140] = 24.17; p < 0.0001; F i[10,140] = 1.42; p = 0.18). As expected, the percentage of decrease measured as AUC was not different between the groups (t = 0.11; p = 0.91).

Effect of local administration of the α2-adrenoceptor agonist clonidine (0.1–100 μM) into the LC by reverse dialysis on extracellular NA concentrations in LC (a) and PFC (b). Data are shown as mean ± SEM values from 7 to 9 experiments and are expressed as percentages of the corresponding basal values. Rats received corticosterone (16 mg kg−1 day−1) or vehicle along 14 days from subcutaneous pellet. Bar graphs represent mean ± SEM area under the curve (AUC) in arbitrary units
Local administration of clonidine into the LC also induced a significant decrease in the extracellular NA concentration in PFC both in control (E max = −46 ± 12 %, p < 0.001) (F[10,77] = 14.26; p < 0.0001; n = 8) and in the corticosterone-treated groups (E max = −49 ± 16 %, p < 0.001) (F[10,88] = 13.93; p < 0.0001; n = 9) (Fig. 3b). The observed effect was not different between the groups (F tr[1,15] = 1.34; p = 0.26; F t[10,150] = 27.23; p < 0.0001; F i[10,150] = 0.50; p = 0.88). When the effect induced by clonidine was measured as AUC, there were not significant differences between the groups (t = 0.88; p = 0.39).
Discussion
A wide variety of studies have linked dysfunctions in central noradrenergic system and HPA axis with stress and psychiatric disorders, particularly anxiety and depression. In the present study, we observed that subchronic corticosterone administration for 14 days, even though producing profound changes in the HPA axis related physiological parameters, did not induce alterations in the functionality of brain α2-adrenoceptors, analysed by both in vitro and in vivo methods.
Several animal models have been developed for the purpose of studying depressive symptomatology and response to antidepressant drugs. Animal models of chronic mild stress have been described as useful paradigms for studying the neurobiological bases of mood disorders (Hill et al. 2012). In that respect, chronic stress exposure induces functional changes of α2-adrenoceptors in LC neurones. These changes are region-specific and time-dependent leading to some contradictory results in the literature. A single cortisol injection, which induces similar effects to those triggered by stress, downregulates α2adrenoceptor density in several brain regions of adult male tree shrews, whereas the oral administration of cortisol for 5 days induces regional receptor protein upregulation (Flugge 1999). In the same way, chronic psychosocial stress in male tree shrews during 10 days downregulates brain α2-adrenoceptors but an upregulation after 28 days is also observed (Flugge 1996). On the other hand, chronic exposure to cold during 14 days, another paradigm of stress, induces the desensitization of α2-adrenoceptors in LC (Jedema et al. 2008).
The possible role of glucocorticoids in the stress-induced abnormalities on noradrenergic system remains unclear. In this sense, exogenous corticosterone administration in animals has also been described as a useful model of mood disorders (Sterner and Kalynchuk 2010). Thus, functional desensitization of somatodendritic 5-HT1A autoreceptors has been described as a consequence of subchronic corticosterone administration (Leitch et al. 2003) with a similar protocol to the one applied in the present work. The 5-HT1A receptors are the main inhibitory serotonin receptors that regulate the firing of serotonergic raphe neurones, affecting serotonin levels at projection sites throughout the brain. Thus, these receptors play a similar role for serotonergic system as α2-adrenoceptors do in the noradrenergic function.
In the present work, α2-adrenoceptor functionality was studied in rats treated with time release corticosterone pellets during 14 days. This treatment induces a flattened diurnal blood corticosterone rhythm without raising the total daily amount of circulating corticosterone. As described in previous works (Gartside et al. 2003; Leitch et al. 2003), corticosterone-induced HPA axis dysfunction was observed in physiological parameters such as adrenal and body weight and plasma corticosterone concentration.
Interestingly, basal concentrations of NA did not differ between corticosterone-treated and control rats, indicating that corticosterone treatment does not affect the basal release of NA from noradrenergic neurones in LC or PFC. The α2-adrenoceptors located in these areas regulate noradrenergic neurone activity and NA release via inhibitory G-proteins (Gi/o). Results obtained in [35S]GTPγS binding assays showed that in corticosterone-treated rats, the functional coupling to G-proteins of α2-adrenoceptors located in PFC is not altered. However, α2-adrenoceptors functionality evaluated by in vitro [35S]GTPγS binding assays does not discriminate between auto and heteroreceptors. Therefore, microdialysis was used to evaluate the functionality of the subpopulation of α2-adrenoceptor located on noradrenergic neurones acting as inhibitory feedback input that controls NA release to the synaptic cleft in both LC and PFC areas. In order to evaluate this α2adrenoceptor sensitivity in vivo, we used the well-characterized α2-adrenoceptors selective agonist clonidine (Mateo and Meana 1999). It has been previously described that local administration of clonidine in the PFC, a noradrenergic terminal area, induces a decrease of local NA release by acting on presynaptic α2-adrenoceptors (Mateo et al. 2001). Our results confirm that clonidine-induced NA decrease in PFC was similar in both experimental groups and demonstrate that no differences in the functionality of these receptors are induced by the present paradigm of corticosterone treatment.
The firing activity of LC noradrenergic neurones is controlled by somatodendritic α2-adrenoceptors located in this area exerting a tonic inhibitory effect on NA release by noradrenergic terminal areas (Fernandez-Pastor and Meana 2002). Therefore, NA concentration in PFC is tightly controlled by α2-adrenoceptors-mediated inhibitory modulation of LC firing rate (Mateo et al. 1998). In the present study, we measured NA concentrations in the PFC after local administration of clonidine into the LC in order to evaluate the role of this somatodendritic α2-adrenoceptor subpopulation expressed in the LC area. There were no differences of clonidine response when NA concentration was measured in PFC. In the other hand, it has been described another subpopulation of α2-adrenoceptors located in LC that regulates somatodendritic NA release in this area (Callado and Stamford 1999; Fernandez-Pastor and Meana 2002). The evaluation of functionality of this α2-adrenoceptor population in the LC did not show changes associated to the corticosterone treatment.
Although there are evidences of corticosterone-induced effects on rat physiology and in contrast to previously observed evidences in somatodendritic 5-HT1A receptors, α2-adrenoceptors show resistance in the present paradigm. However, this fact does not exclude a putative role of corticosterone as inductor of α2-adrenoceptor alterations on aetiology of mood disorders after longer modulations of corticosterone levels. Here, we cannot completely exclude the risk of making a type II or β error (failing to reject the false null hypothesis), because of the size of the sample. Indeed, taking into account that clonidine administration induced a 14-% reduction in NA release in PFC of corticosterone-treated rats (Fig. 2), the sample size needed to ensure a 0.80 statistical power would be as high as 40 rats per group. Therefore, potential differences, if existing, would be of low biological relevance.
Despite there is not any alteration in α2-adrenoceptor functionality, we cannot preclude definitive alterations in noradrenergic system. In this regard, here α2-adrenoceptor activity has been evaluated in LC and PFC, but we cannot discard changes in other noradrenergic projection areas such as hippocampus. On the other hand, we have measured extracellular NA concentration in basal conditions and, therefore, the possibility of an altered noradrenergic system in response to acute stress conditions in corticosterone-treated animals cannot be discarded.
It is important to remark that alterations induced by stressors on α2-adrenoceptor functionality seem to be time-dependent. In fact, the density of α2-adrenoceptors and probably also the sensitivity of the receptor system are dependent on the duration of the stress period. Similarly, the treatment length and/or dose could also be important in the pharmacological response to corticosterone (Sterner and Kalynchuk 2010). In this line, there are evidences suggesting that long-term treatments are necessary to alter adrenoceptor functionality, as it has been demonstrated for administration of different antidepressant treatments (Invernizzi et al. 2001; Mateo et al. 2001). In this context, evaluation of α2-adrenoceptors functionality on a longer corticosterone treatment paradigm could be of interest.
The present data demonstrate that corticosterone administration during 14 days by time release subcutaneous pellet did not induce alterations on α2-adrenoceptor functionality in noradrenergic neurones in the CNS. Larger treatments would be necessary to confirm or discard that α2-adrenoceptors are sensitive to long-term corticosterone effects.
References
Bremner JD, Krystal JH, Southwick SM, Charney DS (1996) Noradrenergic mechanisms in stress and anxiety: II. Clinical studies. Synapse 23:39–51
Callado LF, Stamford JA (1999) α2A- but not α2B/C-adrenoceptors modulate noradrenaline release in rat locus coeruleus: voltammetric data. Eur J Pharmacol 366:35–39
Callado LF, Meana JJ, Grijalba B, Pazos A, Sastre M, Garcia-Sevilla JA (1998) Selective increase of α2A-adrenoceptor agonist binding sites in brains of depressed suicide victims. J Neurochem 70:1114–1123
Charmandari E, Tsigos C, Chrousos G (2005) Endocrinology of the stress response. Annu Rev Physiol 67:259–284
De Paermentier F, Mauger JM, Lowther S, Crompton MR, Katona CL, Horton RW (1997) Brain α-adrenoceptors in depressed suicides. Brain Res 757:60–68
Fernandez-Pastor B, Meana JJ (2002) In vivo tonic modulation of the noradrenaline release in the rat cortex by locus coeruleus somatodendritic α2-adrenoceptors. Eur J Pharmacol 442:225–229
Fernandez-Pastor B, Ortega JE, Meana JJ (2013) Involvement of serotonin 5-HT3 receptors in the modulation of noradrenergic transmission by serotonin reuptake inhibitors: a microdialysis study in rat brain. Psychopharmacology 229:331–344
Flugge G (1996) Alterations in the central nervous α2-adrenoceptor system under chronic psychosocial stress. Neuroscience 75:187–196
Flugge G (1999) Effects of cortisol on brain α2-adrenoceptors: potential role in stress. Neurosci Biobehav Rev 23:949–956
Garcia-Sevilla JA, Padro D, Giralt MT, Guimon J, Areso P (1990) α2-adrenoceptor-mediated inhibition of platelet adenylate cyclase and induction of aggregation in major depression. Effect of long-term cyclic antidepressant drug treatment. Arch Gen Psychiatry 47:125–132
Garcia-Sevilla JA, Ventayol P, Perez V, Rubovszky G, Puigdemont D, Ferrer-Alcon M, Andreoli A, Guimon J, Alvarez E (2004) Regulation of platelet α2A-adrenoceptors, Gi proteins and receptor kinases in major depression: effects of mirtazapine treatment. Neuropsychopharmacology 29:580–588
Gartside SE, Leitch MM, Young AH (2003) Altered glucocorticoid rhythm attenuates the ability of a chronic SSRI to elevate forebrain 5-HT: implications for the treatment of depression. Neuropsychopharmacology 28:1572–1578
Gonzalez-Maeso J, Rodriguez-Puertas R, Gabilondo AM, Meana JJ (2000) Characterization of receptor-mediated [35S]GTPgammaS binding to cortical membranes from postmortem human brain. Eur J Pharmacol 390:25–36
Hill MN, Hellemans KG, Verma P, Gorzalka BB, Weinberg J (2012) Neurobiology of chronic mild stress: parallels to major depression. Neurosci Biobehav Rev 36:2085–2117
Invernizzi RW, Parini S, Sacchetti G, Fracasso C, Caccia S, Annoni K, Samanin R (2001) Chronic treatment with reboxetine by osmotic pumps facilitates its effect on extracellular noradrenaline and may desensitize α2-adrenoceptors in the prefrontal cortex. Br J Pharmacol 132:183–188
Jedema HP, Gold SJ, Gonzalez-Burgos G, Sved AF, Tobe BJ, Wensel T, Grace AA (2008) Chronic cold exposure increases RGS7 expression and decreases α2-autoreceptor-mediated inhibition of noradrenergic locus coeruleus neurons. Eur J Neurosci 27:2433–2443
Leitch MM, Ingram CD, Young AH, McQuade R, Gartside SE (2003) Flattening the corticosterone rhythm attenuates 5-HT1A autoreceptor function in the rat: relevance for depression. Neuropsychopharmacology 28:119–125
Mateo Y, Meana JJ (1999) Determination of the somatodendritic α2-adrenoceptor subtype located in rat locus coeruleus that modulates cortical noradrenaline release in vivo. Eur J Pharmacol 379:53–57
Mateo Y, Pineda J, Meana JJ (1998) Somatodendritic α2-adrenoceptors in the locus coeruleus are involved in the in vivo modulation of cortical noradrenaline release by the antidepressant desipramine. J Neurochem 71:790–798
Mateo Y, Fernandez-Pastor B, Meana JJ (2001) Acute and chronic effects of desipramine and clorgyline on α2-adrenoceptors regulating noradrenergic transmission in the rat brain: a dual-probe microdialysis study. Br J Pharmacol 133:1362–1370
Ortega JE, Mendiguren A, Pineda J, Meana JJ (2012) Regulation of central noradrenergic activity by 5-HT3 receptors located in the locus coeruleus of the rat. Neuropharmacology 62:2472–2479
Parini S, Renoldi G, Battaglia A, Invernizzi RW (2005) Chronic reboxetine desensitizes terminal but not somatodendritic α2-adrenoceptors controlling noradrenaline release in the rat dorsal hippocampus. Neuropsychopharmacology 30:1048–1055
Stanford SC (1995) Central noradrenergic neurones and stress. Pharmacol Ther 68:297–242
Sterner EY, Kalynchuk LE (2010) Behavioral and neurobiological consequences of prolonged glucocorticoid exposure in rats: relevance to depression. Prog Neuro-Psychopharmacol Biol Psychiatry 34:777–790
Valdizan EM, Diez-Alarcia R, Gonzalez-Maeso J, Pilar-Cuellar F, Garcia-Sevilla JA, Meana JJ, Pazos A (2010) α2-adrenoceptor functionality in postmortem frontal cortex of depressed suicide victims. Biol Psychiatry 68:869–872
Acknowledgments
This work was supported by Spanish MICINN (SAF 04/02784), MINECO (SAF2009-08460, SAF2013-48586-R), ERDF Funds and the Basque Government (IT-616-13).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Animal care and all experimental protocols were performed in agreement with the European Ethical Standards (European Union Directive 2010/63/EU) and translation into Spanish legislation (RD 53/2013) and approved by the UPV/EHU Ethical Board for Animal Welfare (CEEA)
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Horrillo, I., Ortega, J.E., Diez-Alarcia, R. et al. Effect of subchronic corticosterone administration on α2-adrenoceptor functionality in rat brain: an in vivo and in vitro study. Psychopharmacology 233, 3861–3867 (2016). https://doi.org/10.1007/s00213-016-4418-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-016-4418-3