
Abstract
Rationale
Brain-derived neurotrophic factor (BDNF) and signaling at its receptor, tropomyosin-related kinase B (TrkB), are implicated in the rapid and long-lasting antidepressant effects of ketamine. Moreover, a TrkB agonist, 7,8-dihydroxyflavone (7,8-DHF), and/or TrkB antagonist, ANA-12, shows antidepressant effects in animal models of depression.
Objective
The objective of this study is to compare the influence of ketamine, 7,8-DHF, and ANA-12 on antidepressant activity in the social defeat stress model.
Results
In the tail suspension and forced swimming tests, ketamine, 7,8-DHF, or ANA-12 markedly attenuated the increased immobility time in depressed mice compared with the vehicle-treated group. In the sucrose preference test, all drugs significantly improved the reduced preference in depressed mice at both 1 and 3 days after a single dose. Antidepressant effect of ketamine, but not 7,8-DHF or ANA-12, was still detectable 7 days after a single dose. Western blot analyses showed that ketamine, but not 7,8-DHF or ANA-12, markedly attenuated reduced levels of BDNF and postsynaptic density protein 95 (PSD-95) in the prefrontal cortex (PFC), dentate gyrus (DG), and CA3 of the hippocampus in depressed mice 8 days after a single dose. Furthermore, ketamine markedly increased reduced levels of GluA1 in the PFC and DG of depressed mice. In contrast, ketamine showed no effect against increased levels of BDNF, PSD-95, and GluA1 observed in the nucleus accumbens of depressed mice.
Conclusions
Compared with 7,8-DHF and ANA-12, ketamine is a longer-lasting antidepressant in the social defeat stress model, and synaptogenesis may be required for the mechanisms that promote sustained antidepressant effects of ketamine.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Depression is one of the most common psychiatric disorders and the leading cause of disability worldwide. According to the World Health Organization (WHO), more than 350 million individuals of all ages suffer from depression (World Health Organization WHO 2012). Antidepressant therapy is generally effective in the treatment of depression. However, complete therapeutic benefits of antidepressant therapy may require several weeks before influencing patients’ mood and/or behavior. In addition, despite efficacious standard treatments, approximately two thirds of patients with depression fail to respond to pharmacotherapy. Therefore, to provide therapy to patients with treatment-resistant depression, development of new drugs capable of inducing rapid and robust antidepressant effects is critical.
Accumulating evidence suggests a key role for glutamate neurotransmission via the N-methyl-d-aspartate (NMDA) receptor in the pathophysiology of depression (Hashimoto et al. 2007, 2013; Sanacora et al. 2008; Hashimoto 2009, 2011; Skolnick et al. 2009; Zarate et al. 2010; Tokita et al. 2012). Ketamine, a NMDA receptor antagonist, is one of the most attractive antidepressants for treatment-resistant depression (Aan Het Rot et al. 2012; Krystal et al. 2013; Hashimoto 2014; Hashimoto 2015; Yang and Hashimoto 2014; Sanacora and Schatzberg 2015). A single subanesthetic dose (0.5 mg/kg) of ketamine was shown to produce a rapid and robust antidepressant response in two thirds of patients with treatment-resistant depression, which could last for over 1 week (Aan Het Rot et al. 2012; Berman et al. 2000; Zarate et al. 2006, 2012; Diazgranados et al. 2010). As such, several studies suggest involvement of brain-derived neurotrophic factor (BDNF) in the mechanistic action of ketamine in promoting antidepressant effects (Autry et al. 2011; Yang et al. 2013; Lepack et al. 2014; Zhou et al. 2014).
In this context, BDNF and signaling at its receptor, tropomyosin-related kinase B (TrkB), represent other therapeutic targets for treating depression (Nestler et al. 2002; Hashimoto et al. 2004; Duman and Monteggia 2006; Nestler and Carlezon 2006; Hashimoto 2010, 2013; Castrén 2014; Homberg et al. 2014). Systemic administration of the commercially available TrkB agonist 7,8-dihydroxyflavone (7,8-DHF) (Jang et al. 2010) and/or TrkB antagonist ANA-12 (Cazorla et al. 2011) induces antidepressant effects in the inflammation model of depression (Zhang et al. 2015). However, to our knowledge, the antidepressive effects of ketamine, 7,8-DHF, and ANA-12 have not been compared in the animal model of depression.
The purpose of this study was to compare antidepressant effects of ketamine, 7,8-DHF, and ANA-12 in the social defeat stress model of depression. In addition, BDNF and synaptogenesis are implicated in the mechanisms of antidepressant action. Thus, using western blot analysis, we studied potential mechanisms of antidepressant action for BDNF, along with postsynaptic density protein 95 (PSD-95) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor (GluA1), both of which are required for synaptic plasticity in selected brain regions (Nestler and Carlezon 2006; Hashimoto 2010; Duman and Aghajanian 2012; Dwyer and Duman 2013; Ohgi et al. 2015).
Materials and methods
Animals
Male adult C57BL/6 mice, aged 8 weeks (body weight 20–25 g, Japan SLC, Inc., Hamamatsu, Japan), and male adult CD1 mice, aged 13–15 weeks (body weight >40 g, Japan SLC, Inc., Hamamatsu, Japan), were used in experiments. Animals were housed under controlled temperatures and 12-h light/dark cycles (lights on between 07:00 and 19:00 h), with ad libitum food and water. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Chiba University Institutional Animal Care and Use Committee (Permit Number: 26–31). All efforts were made to minimize suffering.
Drug administration
On the day of injection, ketamine (ketamine hydrochloride, Daiichi Sankyo Pharmaceutical Ltd., Tokyo, Japan, 10 mg/kg), 7,8-dihydroxyflavone (7,8-DHF; Catalog number: D1916, Tokyo Chemical Co., Ltd., Tokyo, Japan, 10 mg/kg), and ANA-12, N2-(2-{[(2-oxoazepan-3-yl) amino]carbonyl}phenyl)benzo[b]thiophene-2-carboxamide (Catalog number: BTB06525SC, Maybridge, UK, 0.5 mg/kg) were prepared in vehicle of 17 % dimethyl sulfoxide (DMSO) in phosphate-buffered saline. The doses of ketamine (10 mg/kg), 7,8-DHF (10 mg/kg), and ANA-12 (0.5 mg/kg) were selected as reported previously (Cazorla et al. 2011; Zhang et al. 2015; Ren et al. 2013, 2014; Li et al. 2014; Berton et al. 2006). All compounds were administered intraperitoneally (i.p.) to mice.
Social defeat procedure
The social defeat procedure was performed as previously reported (Berton et al. 2006; Tsankova et al. 2006; Golden et al. 2011; Zhao et al. 2013). Every day, the C57BL/6 mice were exposed to a different CD1 aggressor mouse for 10 min, total for 10 days. When the social defeat session ended, the resident CD1 mouse and the intruder mouse were housed in one half of the cage separated by a perforated Plexiglas divider to allow visual, olfactory, and auditory contact for the remainder of the 24-h period. At 24 h after the last session, all mice were housed individually. On day 11, a social avoidance test was performed to identify subgroups of mice that were susceptible and unsusceptible to social defeat stress. This was accomplished by placing mice in an interaction test box (42 × 42 cm) with an empty wire mesh cage (10 × 4.5 cm) located at one end. The movement of the mice was tracked for 2.5 min, followed by 2.5 min in the presence of an unfamiliar aggressor confined in the wire mesh cage. The duration of the subject’s presence in the “interaction zone” (defined as the 8-cm-wide area surrounding the wire mesh cage) was recorded by a stopwatch. The interaction ratio was calculated as time spent in an interaction zone with an aggressor/time spent in an interaction zone without an aggressor. An interaction ratio of 1 was set as the cutoff: mice with scores <1 were defined as “susceptible” to social defeat stress, and those with scores ≥1 were defined as “unsusceptible” (Zhao et al. 2013). Approximately 70 % of mice were susceptible in this study. Only susceptible (depressed) mice were used in the experiments.
Behavioral tests
Behavioral tests were performed as reported previously (Zhang et al. 2014, 2015; Li et al. 2014; Ma et al. 2014; Yao et al. 2015). Locomotion: The locomotor activity was measured by an animal movement analysis system SCANETMV-40 (MELQUEST Co., Ltd., Toyama, Japan), and the mice were placed in experimental cages (length × width × height: 560 × 560 × 330 mm). The cumulative exercise was recorded for 60 min. Cages were cleaned between testing session.
Tail suspension test
A small piece of adhesive tape was placed approximately 2 cm from the tip of the tail for mouse. A single hole was punched in the tape, and mice were hung individually, on a hook. The immobility time was recorded for 10 min. Mice were considered immobile only when they hung passively and completely motionless.
Forced swimming test
The forced swimming test (FST) was tested by an automated forced swim apparatus SCANETMV-40 (MELQUEST Co., Ltd., Toyama, Japan). The mice were placed individually in a cylinder (diameter, 23 cm; height, 31 cm) containing 15 cm of water, maintained at 23 ± 1 °C. Immobility time from activity time as (total) − (active) time was calculated by the apparatus analysis software. The immobility time for mouse was recorded for 6 min.
Sucrose preference test
Mice were exposed to water and 1 % sucrose solution for 48 h, followed by 4 h of water and food deprivation and a 1-h exposure to two identical bottles: one is water and another is 1 % sucrose solution. The bottles containing water and sucrose were weighed before and at the end of this period and the sucrose preference was determined.
Western blot analysis of proBDNF, BDNF, PSD-95, and GluA1
The brain samples of prefrontal cortex (PFC), nucleus accumbens (NAc), CA1, CA3, and dentate gyrus (DG) of the hippocampus were prepared since these regions are implicated in depression-like phenotype, as described previously (Zhang et al. 2015). Western blot analysis was performed as reported previously (Zhang et al. 2015; Yang et al. 2015). Basically, tissue samples were homogenized in Laemmli lysis buffer. Aliquots (10 μg) of protein were measured using the DC protein assay kit (Bio-Rad, Hercules, CA) and incubated for 5 min at 95 °C, with an equal volume of 125 mM Tris/HCl, pH 6.8, 20 % glycerol, 0.1 % bromophenol blue, 10 % β-mercaptoethanol, and 4 % sodium dodecyl sulfate, and subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis, using 10 % mini-gels (Mini-PROTEAN® TGX™ Precast Gel; Bio-Rad, CA, USA). Proteins were transferred onto polyvinylidene difluoride (PVDF) membranes using a Trans Blot Mini Cell (Bio-Rad). For immunodetection, the blots were blocked with 2 % BSA in TBS + 0.1 % Tween 20 (TBST) for 1 h at room temperature (RT) and kept with primary antibodies overnight at 4 °C. The following primary antibodies were used: BDNF (1: 1000, Santa Cruz Biotechnology, Inc., CA, USA), proBDNF (1:400, Alomone Labs, Jerusalem, Israel), postsynaptic density protein 95 (PSD-95) (1 μg/ml, Invitrogen, Carlsbad, CA, USA), and AMPA glutamate receptor 1 (GluA1) (1 μg/ml, Abcam, Cambridge, MA, USA). The next day, the blots were washed three times in TBST and incubated with horseradish peroxidase-conjugated anti-rabbit antibody (1:5000) for 1 h at RT. After final three washes with TBST, the bands were detected using enhanced chemiluminescence (ECL) plus the Western Blotting Detection system (GE Healthcare Bioscience). The blots then were washed three times in TBST and incubated with the primary antibody directed against β-actin. Images were captured with a Fuji LAS3000-mini imaging system (Fujifilm, Tokyo, Japan), and immunoreactive bands were quantified.
Statistical analysis
The data show as the mean ± standard error of the mean (SEM). Analysis was performed using PASW Statistics 20 (formerly SPSS Statistics; SPSS). Comparisons between groups were performed using the one-way analysis of variance (ANOVA), followed by post hoc Fisher’s least significant difference (LSD) test. The P values of less than 0.05 were considered statistically significant.
Results
Antidepressant effects of ketamine, 7,8-DHF, and ANA-12 in the social defeat stress model of depression
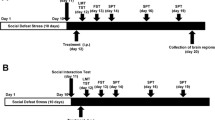
Ketamine produces rapid and long-lasting antidepressant effects in the chronic mild stress model of depression (Li et al. 2011; Ma et al. 2013). Recently, we demonstrated that both 7,8-DHF (a TrkB agonist; 10 mg/kg) and ANA-12 (a TrkB antagonist; 0.5 mg/kg) showed antidepressant effects in lipopolysaccharide (LPS)-induced depression-like behavior (Zhang et al. 2015). In this study, we compared the antidepressant effects of ketamine, 7,8-DHF, and ANA-12 in the social defeat stress model of depression (Fig. 1a).

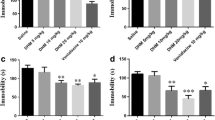
Ketamine showed greater potency and long-lasting antidepressant effects in social defeat mice. a The schedule of social defeat stress (10 days), drug treatment, and Western blot analyses. LMT, TST, and FST were performed 1, 3, and 5 h after a single injection, respectively. SPT was performed 1 day (day 13), 3 days (day 15), and 7 days (day 19) after injection. b Locomotion (LMT). c TST. d FST. e–g SPT. The values represent the mean ± SEM (n = 10–13). *P < 0.05, **P < 0.01, ***P < 0.001 compared with the vehicle + stress group. Con control, Veh vehicle, DHF 7,8-dihydroxyflavone, ANA ANA-12, Ket ketamine, LMT locomotion test, TST tail suspension test, FST forced swimming test, SIT social interaction test, SPT 1 % sucrose preference test
In the tail suspension test (TST) and forced swimming test (FST), ketamine (10 mg/kg), 7,8-DHF (10 mg/kg), and ANA-12 (0.5 mg/kg) significantly attenuated an increased immobility time in depressed mice (Fig. 1c, d). One-way ANOVA detected statistical significance in both the TST and FST (TST: F 4,53 = 3.363, P = 0.016; FST: F 4,52 = 4.474, P = 0.004) among the five groups (Fig. 1c, d). Locomotion showed no difference (F 4,53 = 0.229, P = 0.921) among the five groups (Fig. 1b). In the sucrose preference test (SPT), preference of mice after an injection of ketamine, 7,8-DHF, or ANA-12 was significantly higher (E (day 13): F 4,53 = 9.791, P < 0.001, F (day 15): F 4,53 = 5.236, P = 0.001) than that of the vehicle-treated group (Fig. 1e, f). Ketamine, but not 7,8-DHF or ANA-12, significantly (F 4,53 = 5.056, P = 0.002) improved the decreased preference in the SPT 7 days after a single dose (Fig. 1g). These behavioral data suggest that ketamine, 7,8-DHF, and ANA-12 promote rapid antidepressant effects in the social defeat stress model of depression and that ketamine produces longer-lasting antidepressant effects compared to 7,8-DHF and ANA-12.
In the TST, FST, and SPT, a single dose of ketamine (10 mg/kg), 7,8-DHF (10 mg/kg), or ANA-12 (0.5 mg/kg) did not show antidepressant-like effects in the control mice (Supplemental Fig. 1).
Levels of proBDNF and BDNF protein in mouse brain regions 4 days after a single administration of ketamine, 7,8-DHF, or ANA-12
Accumulation evidence suggests that PFC, NAc, and hippocampus play a key role in the pathophysiology of depression (Li et al. 2011; Nestler and Carlezon 2006; Zhang et al. 2015; Yang et al. 2015). Therefore, we performed Western blot analysis of BDNF and its precursor, proBDNF, in the selected brain regions (PFC, NAc, DG, CA1, and CA3 of the hippocampus) 4 days after a single dose (Fig. 1a). One-way ANOVA of proBDNF data showed a significant change in PFC [PFC: F 4,23 = 3.473, P = 0.023; NAc: F 4,23 = 0.146, P = 0.963; DG: F 4,23 = 0.060, P = 0.993; CA1: F 4,23 = 0.414, P = 0.797; CA3: F 4,23 = 0.643, P = 0.637] (Fig. 2a). Social defeat stress significantly increased the protein levels of proBDNF in the PFC. Furthermore, ketamine significantly attenuated an increased level of proBDNF in the PFC. In contrast, we did not find any change of proBDNF in other brain regions.

The levels of proBDNF and BDNF in the brain regions 4 days after ketamine, 7,8-DHF, or ANA-12 treatment. a Western blot analysis of proBDNF in PFC, NAc, CA1, CA3, and DG of the hippocampus. The value was expressed as a percentage of that of control mice. The values represent the mean ± SEM (n = 5–6). b Western blot analysis of BDNF (mature form) in PFC, NAc, CA1, CA3, and DG of the hippocampus. The value was expressed as a percentage of that of control mice. The values represent the mean ± SEM (n = 5–6). *P < 0.05, **P < 0.01 compared with the vehicle + stress group. Con control, Veh vehicle, DHF 7,8-dihydroxyflavone, ANA ANA-12, Ket ketamine
One-way ANOVA of BDNF data showed statistical significances in all regions, except CA1 [PFC: F 4,23 = 3.120, P = 0.035; NAc: F 4,23 = 3.989, P = 0.013; DG: F 4,23 = 7.319, P = 0.001; CA1: F 4,23 = 0.824, P = 0.523; CA3: F 4,23 = 7.498, P = 0.001] (Fig. 2b). Social defeat stress significantly caused the marked reduction of BDNF in the PFC, DG, and CA3, but not in CA1, while the stress significantly increased the levels of BDNF in the NAc (Fig. 2b), consistent with previous data from the inflammation model (Zhang et al. 2015) and the rat learned helplessness model (Yang et al. 2015) of depression. Interestingly, ketamine significantly attenuated the reduced levels of BDNF in the PFC, CA3, and DG after social defeat stress, although ketamine did not affect the increased levels of BDNF in the NAc (Fig. 2b). In this model, 7,8-DHF and ANA-12 did not affect BDNF levels in any region (Fig. 2b).
Levels of PSD-95 and GluA1 in the brain regions 4 days after a single administration of ketamine, 7,8-DHF, or ANA-12
We performed Western blot analysis of the synaptogenesis markers, PSD-95 and GluA1, in the selected brain regions. One-way ANOVA of PSD-95 data showed significant differences [PFC: F 4,23 = 4.670, P = 0.007; NAc: F 4,23 = 3.045, P = 0.038; DG: F 4,23 = 7.455, P = 0.001; CA1: F 4,23 = 0.282, P = 0.887; CA3: F 4,23 = 4.139, P = 0.011] (Fig. 3a). Four days after a single dose, social defeat stress significantly decreased the protein levels of PSD-95 in the PFC, DG, and CA3 of the hippocampus, but not CA1, whereas the stress significantly increased the protein levels of PSD-95 in the NAc (Fig. 3a). Interestingly, ketamine and 7,8-DHF significantly attenuated the reduction of PSD-95 in the PFC, DG, and CA3 after social defeat stress, although these compounds had no effect in the NAc (Fig. 3a). Furthermore, ANA-12 significantly attenuated the increased levels of PSD-95 in the NAc after social defeat stress, although it had no effect in the PFC, DG, and CA3 (Fig. 3a).

The levels of postsynaptic density protein 95 (PSD-95) and AMPA receptor 1 (GluA1) in the brain regions 4 days after ketamine, 7,8-DHF, or ANA-12 treatment. a Western blot analysis of PSD-95 in PFC. NAc, CA1, CA3, and DG of the hippocampus. The value was expressed as a percentage of that of control mice. The values represent the mean ± SEM (n = 5–6). b Western blot analysis of GluA1 in PFC, NAc, CA1, CA3, and DG of the hippocampus. The value was expressed as a percentage of that of control mice. Values represent the mean ± SEM (n = 5–6). *P < 0.05, **P < 0.01, ***P < 0.001 compared with the vehicle + stress group. Con control, Veh vehicle, DHF 7,8-dihydroxyflavone, ANA ANA-12, Ket ketamine
One-way ANOVA of GluA1 data showed significant changes in different brain regions, except CA1 [PFC: F 4,23 = 4.305, P = 0.010; NAc: F 4,23 = 5.628, P = 0.003; DG: F 4,22 = 6.600, P = 0.001; CA1: F 4,23 = 1214, P = 0.332; CA3: F 4,23 = 5.177, P = 0.004]. Social defeat stress significantly decreased the protein levels of GluA1 in PFC, DG, and CA3, but not CA1 (Fig. 3b). In contrast, the stress significantly increased the protein levels of GluA1 in the NAc (Fig. 3b). Ketamine and 7,8-DHF significantly attenuated the reduction of GluA1 protein in PFC, DG, and CA3. In contrast, ANA-12 significantly attenuated the increased levels of GluA1 in the NAc after social defeat stress, although it had no effect in the PFC, DG, and CA3 (Fig. 3b).
Levels of proBDNF and BDNF protein in mouse brain regions 8 days after a single administration of ketamine, 7,8-DHF, or ANA-12
To explore the mechanism underlying long-lasting antidepressant effects of ketamine, we performed Western blot analysis of BDNF and its precursor, proBDNF, in selected brain regions 8 days after a single dose (Fig. 1a). One-way ANOVA of proBDNF data showed no significant changes in any test group, for any brain region (Fig. 4a). One-way ANOVA of BDNF data detected statistical significances in all regions, except CA1 [PFC: F 4,24 = 4.639, P = 0.006; NAc: F 4,24 = 5.256, P = 0.003; DG: F 4,23 = 4.661, P = 0.007; CA1: F 4,24 = 0.554, P = 0.698; CA3: F 4,24 = 4.527, P = 0.007] (Fig. 4b). We found that social defeat stress significantly decreased protein levels of BDNF in the PFC, DG, and CA3, but not in CA1, while this significantly increased BDNF protein in the NAc (Fig. 4b). Ketamine significantly attenuated reduced levels of BDNF protein in the PFC, CA3, and DG after social defeat stress, although ketamine did not affect the increased levels of BDNF protein in the NAc in this model (Fig. 4b). 7,8-DHF and ANA-12 did not affect BDNF levels in any region (Fig. 4b).

The levels of proBDNF and BDNF in the brain regions 8 days after ketamine, 7,8-DHF, or ANA-12 treatment. a Western blot analysis of proBDNF in PFC, NAc, CA1, CA3, and DG of the hippocampus. The value was expressed as a percentage of that of control mice. Values represent the mean ± SEM (n = 5–6) b Western blot analysis of BDNF in PFC, NAc, CA1, CA3, and DG of the hippocampus. The value was expressed as a percentage of that of control mice. Values represent the mean ± SEM (n = 5–6). *P < 0.05, **P < 0.01 compared with the vehicle + stress group. Con control, Veh vehicle, DHF 7,8-dihydroxyflavone, ANA ANA-12, Ket ketamine
Levels of PSD-95 and GluA1 in the mouse brain regions 8 days after a single administration of ketamine, 7,8-DHF, or ANA-12
We performed Western blot analysis of PSD-95 and GluA1, in selected brain regions 8 days after a single dose (Fig. 1a). One-way ANOVA of PSD-95 data showed statistically significant differences [PFC: F 4,24 = 14.508, P < 0.001; NAc: F 4,24 = 3.090, P = 0.035; DG: F 4,25 = 3.374, P = 0.024; CA1: F 4,25 = 1.806, P = 0.159; CA3: F 4,24 = 4.501, P = 0.007] (Fig. 4a). Eight days after a single dose of test drug, social defeat stress significantly decreased levels of PSD-95 in the PFC, DG, and CA3 of the hippocampus, but not CA1, whereas it significantly increased the levels of PSD-95 in the NAc (Fig. 5a). Interestingly, ketamine significantly attenuated the reduction of PSD-95 protein in the PFC, DG, and CA3 after social defeat stress, although it had no effect on the increased PSD-95 in the NAc in this stress model (Fig. 5a). Furthermore, neither 7,8-DHF nor ANA-12 affected alterations in PSD-95 levels following social defeat stress (Fig. 5a).

The levels of markers for synaptogenesis, postsynaptic density protein 95 (PSD-95), and AMPA receptor 1 (GluA1) in the brain regions 8 days after ketamine, 7,8-DHF, or ANA-12 treatment. a Western blot analysis of PSD-95 in PFC, NAc, CA1, CA3, and DG of the hippocampus. The value was expressed as a percentage of that of control mice. Values represent the mean ± SEM (n = 5–6). b Western blot analysis of GluA1 in PFC, NAc, CA1, CA3, and DG of the hippocampus. The value was expressed as a percentage of that of control mice. Values represent the mean ± SEM (n = 5–6). *P < 0.05, **P < 0.01, ***P < 0.001 compared with the vehicle + stress group. Con control, Veh vehicle, DHF 7,8-dihydroxyflavone, ANA ANA-12, Ket ketamine
Next, we performed Western blot analysis of GluA1 in selected mouse brain regions. One-way ANOVA of GluA1 levels showed statistically significant changes in different brain regions [PFC: F 4,24 = 8.194, P < 0.001; NAc: F 4,25 = 3.638, P = 0.019; DG: F 4,24 = 20.229, P < 0.001; CA1: F 4,24 = 0.391, P = 0.813; CA3: F 4,25 = 21.158, P < 0.001]. Social defeat stress decreased levels of GluA1 in PFC and DG, but not CA1 and CA3 (Fig. 5b). However, this stress significantly increased levels of GluA1 in the NAc, consistent with the PSD-95 data. Unexpectedly, ketamine significantly increased the levels of GluA1 in PFC, DG, and CA3. In contrast, neither 7,8-DHF nor ANA-12 affected GluA1 protein levels in any brain region (Fig. 5b).
Discussion
The major findings of this study suggest that (1) a single dose of ketamine, 7,8-DHF, or ANA-12 promotes rapid antidepressant responses in the social defeat stress model of depression, (2) ketamine produces longer-lasting antidepressant effects compared with both 7,8-DHF and ANA-12, and (3) the antidepressant effects of a single dose of ketamine, but not that of 7,8-DHF and ANA-12, are still detectable 7 days after administration in the social defeat stress model of depression. As such, these data clearly show that the dose of ketamine administered in this study provides more favorable and sustained antidepressant effects compared with 7,8-DHF and ANA-12.
Previously, Li et al. (2011) reported that in the SPT, ketamine ameliorates anhedonia in rats under the chronic mild stress (CMS) model (21 days), while also influencing long-lasting (up to 7 days) increases in sucrose preference compared with untreated CMS rats. Therefore, it is notable that the rapid and sustained antidepressant effects of ketamine in the CMS and social defeat stress models (present study) are similar in time course to the therapeutic effects of ketamine in patients with treatment-resistant depression or bipolar depression (Aan Het Rot et al. 2012; Zarate et al. 2006, 2012; Diazgranados et al. 2010). Accordingly, because of the potential therapeutic benefits of ketamine and TrkB ligands (7,8-DHF and ANA-12) in patients with treatment-resistant depression, our novel comparison of the antidepressive effects of these therapies in the social defeat stress model of depression may have important clinical implications.
We have previously reported marked reduction in levels of BDNF in the PFC, DG, and CA3, but not CA1, in inflammation-induced depressed mice (Zhang et al. 2015) and learned helplessness rats (Yang et al. 2015; Shirayama et al. 2015). In the present study, we showed that there is a marked reduction in levels of BDNF in the PFC, DG, CA3, but not CA1, in depressed mice after social defeat stress. Shirayama et al. (2002) reported that direct infusion of BDNF into the DG and CA3, but not CA1, promoted rapid and sustained antidepressant effects in the rat learned helplessness model of depression, which implicates potential causative pathways in the DG and CA3, but not CA1, in the antidepressant action of BDNF.
In contrast to our observations of a marked reduction in levels of BDNF in the PFC, DG, and CA3, we have previously also shown that inflammation and learned helplessness induce a marked increase in levels of BDNF in the NAc (Zhang et al. 2015; Shirayama et al. 2015; Yang et al. 2015), which is consistent with high BDNF levels in the NAc of depressed mice following social defeat stress. Notably, in this study, we showed that social defeat stress produces an opposing effect on levels of BDNF in the PFC, hippocampus, and NAc. Thus, we suggest that the ventral tegmental area (VTA)–NAc pathway plays a critical integrative role in the depression phenotype (Nestler and Carlezon 2006; Shirayama et al. 2015; Zhang et al. 2015; Yang et al. 2015).
Previously, it was reported that intra-VTA BDNF injections cause depression-like behavior, whereas a blockade of BDNF activity in the NAc produces antidepressant-like effects (Nestler and Carlezon 2006). Accordingly, social defeat stress probably causes reduction in levels of BDNF in the hippocampus and PFC but increases those in the NAc, resulting in depression-like behavior in mice. Consistent with this hypothesis, in this study, we demonstrated that there are long-lasting increases in levels of BDNF in the PFC, CA3, and DG, but not NAc, after a single dose of ketamine, suggesting that long-lasting activation at BDNF–TrkB signaling might be involved in the sustained antidepressant effects of ketamine. In contrast, the TrkB ligands (7,8-DHF and ANA-12) compared with ketamine did not show similar sustained antidepressant effects in the social defeat stress model, likely because these drugs cannot increase levels of BDNF protein. Therefore, we suggest that the antidepressant effects of TrkB ligands may be dependent upon the drug concentration in the brain following a single dose because these compounds can bind to the TrkB receptor.
Studies using postmortem brain samples have shown that there are reduced levels of BDNF in the hippocampus and PFC of psychiatric disorder patients who had committed suicide compared with non-psychiatric controls (Dwivedi et al. 2003; Karege et al. 2005). Similarly, another study using postmortem brain samples showed markedly higher levels of BDNF (40 % increase) in the NAc of patients with depression compared with controls (Krishnan et al. 2007). Thus, these observations of postmortem brain studies are consistent with our current social defeat stress model data. Abnormal levels of BDNF in the hippocampus, PFC, and NAc likely play a causative role in the pathophysiology of depression.
BDNF–TrkB signaling is involved in the antidepressant effects of ketamine (Autry et al. 2011; Lepack et al. 2014; Zhou et al. 2014). A study using BDNF conditional knockout mice has confirmed that ketamine’s antidepressant effects are dependent on BDNF (Autry et al. 2011). Subsequent studies have shown that increased levels of BDNF in the PFC and hippocampus may also be required for fast-acting and long-lasting antidepressant-like actions of ketamine (Autry et al. 2011; Zhou et al. 2014). In contrast, ketamine does not appear to alter levels of BDNF in the NAc (Autry et al. 2011). The aforementioned findings are consistent with the present results, suggesting that ketamine can induce rapid and log-lasting antidepressant effects in the social defeat stress model via increasing levels of BDNF in the PFC and hippocampus. We did not observe any alteration of levels of proBDNF and BDNF in the brain regions resulting from administration of 7,8-DHF or ANA-12 in the social defeat stress model, indicating that antidepressant effects of 7,8-DHF and ANA-12 are not related to levels of BDNF.
We have recently showed that 7,8-DHF and ANA-12 influence antidepressant activity on inflammation-induced depressive behavior by normalizing altered dendritic spines in the PFC and hippocampus and NAc, respectively (Zhang et al. 2015). Similarly, direct infusions of 7,8-DHF, but not ANA-12, into the hippocampus (CA3 and DG) and PFC and ANA-12, but not 7,8-DHF, into the NAc promote antidepressant effects in the rat learned helplessness model (Shirayama et al. 2015), suggesting that stimulation at TrkB in the PFC, CA3, and DG as well as blockade of TrkB in the NAc confers antidepressant effects. Therefore, it is likely that 7,8-DHF and ANA-12 affect depression by normalizing altered BDNF–TrkB signaling in the PFC and hippocampus and NAc, respectively. Consistent with this, here, we showed that ketamine increased altered levels of BDNF protein in the PFC, CA3, and DG, but not NAc, at 4 and 8 days after a single dose. Thus, it is unlikely that BDNF–TrkB signaling in the NAc is necessary to mediate antidepressant effects of ketamine, although further studies are needed to confirm this.
Accumulating evidence indicates that AMPA receptors are crucial for the antidepressant effects of ketamine because these effects are blocked by the AMPA receptor antagonist, NBQX (Li et al. 2010, 2011; Maeng et al. 2008; Koike et al. 2011; Koike and Chaki 2014; Freudenberg et al. 2015). In this study, a single dose of ketamine resulted in a marked increase in levels of GluA1 protein in the PFC, DG, and CA3 but influenced no change in the NAc. Therefore, considering the role of synaptogenesis in the sustained antidepressant effects of ketamine (Duman and Aghajanian 2012; Dwyer and Duman 2013; Ohgi et al. 2015), long-lasting increases in levels of GluA1 in the PFC, DG, and CA3 may underlie ketamine’s long-lasting antidepressive effects. Further detailed studies on the role of GluA1 in the sustained antidepressant response to ketamine are needed.
The half-life of ketamine in mouse (C57/B6) plasma is approximately 30 min (Sato et al. 2004), indicating a possible rapid clearance of ketamine from the body. Thus far, the pharmacokinetic profile of 7,8-DHF and ANA-12 is unknown. In this study, we detected a sustained antidepressant response of 7 days in response to a single dose of ketamine. Taken together, it is unlikely that the differential antidepressant effects between ketamine and the tested TrkB ligands are due to differences in pharmacokinetic profiles.
A limitation of the current study may be related to the use of only a single dose of ketamine (10 mg/kg), 7,8-DHF (10 mg/kg), and ANA-12 (0.5 mg/kg) to assess the effects of these compounds on antidepressive effects in the social defeat stress model of depression. Nevertheless, drug dosages used are consistent with those used for the same drugs previously (Berton et al. 2006; Cazorla et al. 2011; Zhang et al. 2015; Ren et al. 2013, 2014; Li et al. 2014). However, because threshold doses of ketamine and other drugs vary depending on the behavioral task, we need to examine the dose–response curves of these compounds in other animal models of depression.
In conclusion, this study shows that single doses of ketamine, 7,8-DHF, or ANA-12 can produce rapid antidepressant effects in the social defeat stress model of depression. In addition, unlike 7,8-DHF and ANA-12, ketamine elicits sustained antidepressant effects in the rodent model of depression. Furthermore, it is likely that increased synaptogenesis in the PFC, DG, and CA3 of the hippocampus may be involved in this sustained antidepressant response.
References
Aan Het Rot M, Zarate CA Jr, Charney DS, Mathew SJ (2012) Ketamine for depression: where do we go from here? Biol Psychiatry 72:537–547
Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, Kavalali ET, Monteggia LM (2011) NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475:91–95
Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH (2000) Antidepressant effects of ketamine in depressed patients. Biol Psychiatry 47:351–354
Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM, Bolanos CA, Rios M, Monteggia LM, Self DW, Nestler EJ (2006) Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science 311:864–868
Castrén E (2014) Neurotrophins and psychiatric disorders. Handb Exp Pharmacol 220:461–479
Cazorla M, Prémont J, Mann A, Girard N, Kellendonk C, Rognan D (2011) Identification of a low-molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J Clin Invest 121:1846–1857
Diazgranados N, Ibrahim L, Brutsche NE, Newberg A, Kronstein P, Khalife S, Kammerer WA, Quezado Z, Luckenbaugh DA, Salvadore G, Machado-Vieira R, Manji HK, Zarate CA Jr (2010) A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Arch Gen Psychiatry 67:793–802
Duman RS, Aghajanian GK (2012) Synaptic dysfunction in depression: potential therapeutic targets. Science 338:68–72
Duman RS, Monteggia LM (2006) A neutrotrophic model for stress-related mood disorders. Biol Psychiatry 59:1116–1127
Dwivedi Y, Rizavi HS, Conley RR, Roberts RC, Tamminga CA, Pandey GN (2003) Altered gene expression of brain-derived neurotrophic factor and receptor tyrosin kinase B in postmortem brain of suicide subjects. Arch Gen Psychiatry 60:804–815
Dwyer JM, Duman RS (2013) Activation of mammalian target of rapamycin and synaptogenesis: role in the actions of rapid-acting antidepressants. Biol Psychiatry 73:1189–1198
Freudenberg F, Celikel T, Reif A (2015) The role of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors in depression: central mediators of pathophysiology and antidepressant activity? Neurosci Biobehav Rev 52:193–206
Golden SA, Covington HE III, Berton O, Russo SJ (2011) A standard protocol for repeated social defeat stress in mice. Nat Protoc 6:1183–1191
Hashimoto K (2009) Emerging role of glutamate in the pathophysiology of major depressive disorder. Brain Res Rev 61:105–123
Hashimoto K (2010) Brain-derived neurotrophic factor as a biomarker for mood disorders: an historical overview and future directions. Psychiatry Clin Neurosci 64:341–357
Hashimoto K (2011) The role of glutamate on the action of antidepressants. Prog Neuropsychopharmacol Biol Psychiatry 35:1558–15568
Hashimoto K (2013) Sigma-1 receptor chaperone and brain-derived neurotrophic factor: emerging links between cardiovascular disease and depression. Prog Neurobiol 100:15–29
Hashimoto K (2014) The R-stereoisomer of ketamine as an alternative for ketamine for treatment-resistant major depression. Clin Psychopharmacol Neurosci 12:72–73
Hashimoto K (2015) Inflammatory biomarkers as differential predictors of antidepressant response. Int J Mol Sci 16:7796–7801
Hashimoto K, Shimizu E, Iyo M (2004) Critical role of brain-derived neurotrophic factor in mood disorders. Brain Res Rev 45:104–114
Hashimoto K, Sawa A, Iyo M (2007) Increased levels of glutamate in brains from patients with mood disorders. Biol Psychiatry 62:1310–1316
Hashimoto K, Malchow B, Falkai P, Schmitt A (2013) Glutamate modulators as potential therapeutic drugs in schizophrenia and affective disorders. Eur Arch Psychiatry Neurosci 263:367–377
Homberg JR, Molteni R, Calabrese F, Riva MA (2014) The serotonin-BDNF duo: developmental implications for the vulnerability to psychopathology. Neurosci Biobehav Rev 43:35–47
Jang SW, Liu X, Yepes M, Shepherd KR, Miller GW, Liu Y, Wilson WD, Xiao G, Blanchi B, Sun YE, Ye K (2010) A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc Natl Acad Sci U S A 107:2687–2692
Karege F, Vaudan G, Schwald M, Perround N, La Harpe R (2005) Neurotrophin levels in postmortem brains of suicide victims and the effects of antemortem diagnosis and psychotropic drugs. Mol Brain Res 136:29–37
Koike H, Chaki S (2014) Requirement of AMPA receptor stimulation for the sustained antidepressant activity of ketamine and LY341495 during the forced swim test in rats. Behav Brain Res 271:111–115
Koike H, Iijima M, Chaki S (2011) Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behav Brain Res 224:107–111
Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, Laplant Q, Graham A, Lutter M, Lagace DC, Ghose S, Reister R, Tannous P, Green TA, Neve RL, Chakravarty S, Kumar A, Eisch AJ, Self DW, Lee FS, Tamminga CA, Cooper DC, Gershenfeld HK, Nestler EJ (2007) Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 131:391–404
Krystal JH, Sanacora G, Duman RS (2013) Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry 73:1133–1141
Lepack AE, Fuchikami M, Dwyer JM, Banasr M, Duman RS (2014) BDNF release is required for the behavioral actions of ketamine. Int J Neuropsychopharmacol Oct 31;18(1) doi: 10.1093/ijnp/pyu033
Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS (2010) mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329:959–964
Li N, Liu RJ, Dwyer JM, Banasr M, Lee B, Son H, Li XY, Aghajanian G, Duman RS (2011) Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry 69:754–761
Li SX, Zhang JC, Wu J, Hashimoto K (2014) Antidepressant effects of ketamine on depression-like behavior in juvenile mice after neonatal dexamethasone exposure. Clin Psychopharmacol Neurosci 12:124–127
Ma XC, Dang YH, Jia M, Ma R, Wang F, Wu J, Gao CG, Hashimoto K (2013) Long-lasting antidepressant action of ketamine, but not glycogen synthase kinase-3 inhibitor SB216763, in the chronic mild stress model of mice. PLoS One 8:e56053
Ma M, Ren Q, Zhang JC, Hashimoto K (2014) Effects of brilliant blue G on serum tumor necrosis factor-α levels and depression-like behavior in mice after lipopolysaccharide administration. Clin Psychopharmacol Neurosci 12:31–36
Maeng S, Zarate CA Jr, Du J, Schloesser RJ, McCammon J, Chen G, Manji HK (2008) Cellular mechanisms underlying the antidepressant effects of ketamine: role of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry 63:349–352
Nestler EJ, Carlezon WA Jr (2006) The mesolimbic dopamine reward circuit in depression. Biol Psychiatry 59:1151–1159
Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM (2002) Neurobiology of depression. Neuron 34:13–25
Ohgi Y, Futamura T, Hashimoto K (2015) Glutamate signaling in synaptogenesis and NMDA receptors as potential therapeutic targets for psychiatric disorders. Curr Mol Med 15:206–221
Ren Q, Zhang JC, Fujita Y, Ma M, Wu J, Hashimoto K (2013) Effects of TrkB agonist 7,8-dihydroxyflavone on sensory gating deficits in mice after administration of methamphetamine. Pharmacol Biochem Behav 106:124–127
Ren Q, Zhang JC, Ma M, Fujita Y, Wu J, Hashimoto K (2014) Protective effects of TrkB agonist 7,8-dihydroxyflavone on the behavioral changes and neurotoxicity in mice after administration of methamphetamine. Psychopharmacology (Berlin) 231:159–166
Sanacora G, Schatzberg AF (2015) Ketamine: promising path or false prophecy in the development of novel therapeutics for mood disorders? Neuropsychopharmacology 40:259–267
Sanacora G, Zarate CA, Krystal JH, Manji HK (2008) Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov 7:426–437
Sato Y, Kobayashi E, Hakamata Y, Kobahashi M, Wainai T, Murayama T, Mishina M, Seo N (2004) Chronopharmacological studies of ketamine in normal and NMDA epsilon1 receptor knockout mice. Br J Anaesth 92:859–864
Shirayama Y, Chen AC-H, Nakagawa S, Russell DS, Duman RS (2002) Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci 22:3251–3261
Shirayama Y, Yang C, Zhang JC, Ren Q, Yao W, Hashimoto K (2015) Alterations in brain-derived neurotrophic factor (BDNF) and its precursor proBDNF in the brain regions of a learned helplessness rat model and the antidepressant effects of a TrkB agonist and antagonist. Eur Neuropsychopharmacol in press.
Skolnick P, Popik P, Trullas R (2009) Glutamate-based antidepressants: 20 years on. Trends Pharmacol Sci 30:563–569
Tokita K, Yamaji T, Hashimoto K (2012) Role of glutamate signaling in preclinical and/or mechanistic models of depression. Pharmacol Biochem Behav 100:688–704
Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ (2006) Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci 9:519–525
World Health Organization (WHO) (2012) Depression fact sheet No. 369/October 2012. Available at http://www.who.int/mediacentre/factsheets/fs369/en/index.html
Yang C, Hashimoto K (2014) Rapid antidepressant effects and abuse liability of ketamine. Psychopharmacology (Berlin) 231:2041–2042
Yang C, Hu YM, Zhou ZQ, Zhang GF, Yang JJ (2013) Acute administration of ketamine in rats increases hippocampal BDNF and mTOR levels during forced swimming test. Ups J Med Sci 118:3–8
Yang C, Shirayama Y, Zhang JC, Ren Q, Hashimoto K (2015) Regional differences in brain-derived neurotrophic factor levels and dendritic spine density confer Resilience to inescapable stress. Int J Neuropsychopharmacol 18:pyu121
Yao W, Zhang JC, Dong C, Zhuang C, Hirota S, Inanaga K, Hashimoto K (2015) Effects of amycenone on serum levels of tumor necrosis factor-α and depression-like behaviors in mice after administration of lipopolysaccharide. Pharmacol Biochem Behav 136:7–12
Zarate CA Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK (2006) A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 63:856–864
Zarate C Jr, Machado-Vieira R, Henter I, Ibrahim L, Diazgranados N, Salvadore G (2010) Glutamatergic modulators: the future of treating mood disorders? Harv Rev Psychiatry 18:293–303
Zarate CA Jr, Brutsche NE, Ibrahim L, Franco-Chaves J, Diazgranados N, Cravchik A, Selter J, Marquardt CA, Liberty V, Luckenbaugh DA (2012) Replication of ketamine’s antidepressant efficacy in bipolar depression: a randomized controlled add-on trial. Biol Psychiatry 71:939–946
Zhang JC, Li SX, Hashimoto KJ (2014) R(−)-ketamine shows greater potency and longer lasting antidepressant effects than S(+)-ketamine. Pharmacol Biochem Behav 116:137–141
Zhang JC, Wu J, Fujita Y, Yao W, Ren Q, Yang C, Li SX, Shirayama Y, Hashimoto K (2015) Antidepressant effects of TrkB ligands on depression-like behavior and dendritic changes in mice after inflammation. Int J Neuropsychopharmacol 18:pyu077
Zhao T, Huang GB, Muna SS, Bagalkot TR, Jin HM, Chae HJ, Chung YC (2013) Effects of chronic social defeat stress on behavior and choline acetyltransferase, 78-kDa glucose-regulated protein, and CCAAT/enhancer-binding protein (C/EBP) homologous protein in adult mice. Psychopharmacology (Berlin) 228:217–230
Zhou W, Wang N, Yang C, Li XM, Zhou ZQ, Yang JJ (2014) Ketamine-induced antidepressant effects are associated with AMPA receptors-mediated upregulation of mTOR and BDNF in rat hippocampus and prefrontal cortex. Eur Psychiatry 29:419–423
Acknowledgments
This study was supported by a Grant-in-Aid for Scientific Research on Innovative Areas of the Ministry of Education, Culture, Sports, Science and Technology, Japan (to K.H., #24116006).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplemental Fig. 1
Ketamine, 7,8-DHF and ANA-12 did not show antidepressant effects in control mice. A: Treatment schedule and behavioral tests. B: LMT, C: TST and D: FST were performed 1 hour, 3 hours, and 5 hours after injection of ketamine (10 mg/kg), 7,8-DHF (10 mg/kg) or ANA-12 (0.5 mg/kg), respectively. E-G: SPT was performed 1 day (day 2), 3 days (day 4) and 7 days (day 8) after injection of ketamine, 7,8-DHF or ANA-12. The values represent the mean ± S.E.M. (n=8-10). Con: control; Veh: vehicle; DHF: 7,8-dihydroxyflavone; ANA: ANA-12; Ket: ketamine; LMT: locomotion test; TST: tail suspension test; FST: forced swimming test; SPT: 1% sucrose preference test.(GIF 109 kb)
Rights and permissions
About this article

Cite this article
Zhang, Jc., Yao, W., Dong, C. et al. Comparison of ketamine, 7,8-dihydroxyflavone, and ANA-12 antidepressant effects in the social defeat stress model of depression. Psychopharmacology 232, 4325–4335 (2015). https://doi.org/10.1007/s00213-015-4062-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-015-4062-3