
Abstract
Licochalcone A (Lico A), a flavonoid found in licorice, possesses multiple pharmacological activities in modulating oxidative stress, glycemia, inflammation, and lipid metabolism. This study aimed to explore the potential mechanism of Lico A in mitigating ferroptosis associated with doxorubicin-induced cardiotoxicity (DIC). Initially, network pharmacology analysis was applied to identify the active components present in licorice and their targeted genes associated with DIC. Subsequently, to assess the role of Lico A in a DIC mouse model, electrocardiograms, myocardial injury markers, and myocardial histopathological changes were measured. Additionally, cell viability, reactive oxygen species (ROS), ferrous iron, glutathione/glutathione disulfide (GSH/GSSG), and malondialdehyde (MDA) were measured in the cell model as hallmarks of ferroptosis. Finally, the PI3K/AKT/MDM2/p53 signaling pathway and ferroptosis-related proteins were measured in vitro and in vivo. Bioinformatics results revealed that 8 major compounds of licorice, including Lico A, primarily regulated targets such as p53 and the PI3K/AKT signaling pathways in DIC. In the mouse model of DIC, Lico A significantly ameliorated serum biomarkers, histopathology, and electrocardiogram abnormalities. Pretreatment with Lico A enhanced the viability of H9C2 cells treated with doxorubicin. Furthermore, Lico A administration resulted in decreased levels of ROS, ferrous iron, and MDA and increased levels of GSH/GSSG. At the protein level, Lico A increased the phosphorylation of PI3K/AKT/MDM2, reduced p53 accumulation, and induced the upregulation of SLC7A11 and GPX4 expression. However, selective inhibition of PI3K/AKT and plasmid-based overexpression of p53 significantly abolished the anti-ferroptosis functions of Lico A. In conclusion, Lico A attenuates DIC by suppressing p53-mediated ferroptosis through activating PI3K/AKT/MDM2 signaling.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Doxorubicin (DOX), an effective antineoplastic drug widely used in chemotherapy, is limited due to its cardiovascular toxicity in cancer patients. Although the underlying mechanisms of DOX-induced cardiotoxicity (DIC) remain unclear, extensive studies have suggested that distinct modalities of cell death including apoptosis, autophagy, necroptosis, and pyroptosis are involved in its pathogenesis (Renu et al. 2018). Ferroptosis, a newly recognized regulated cell death process, is characterized by the accumulation of iron and lipid reactive oxygen species (ROS), leading to cellular antioxidant depletion and lipid peroxidation of the cell membrane (Kitakata et al. 2022). Over the past decade, several studies have implicated ferroptosis in various cardiovascular diseases, including the development of DIC. In fact, inhibition of ferroptosis using ferrostatin-1 (Fer-1) significantly reduced DOX-induced mortality in mouse models (Fang et al. 2019). Mechanistically, DOX treatment induces iron overload and the formation of DOX-iron complexes, triggering excessive ROS production through a Fenton-type reaction (Christidi and Brunham 2021). Normally, cystine/glutamate antiporter system Xc- (SLC7A11) promotes cystine uptake and intracellular glutathione (GSH) synthesis to guarantee the anti-lipid peroxidization function of glutathione peroxidase 4 (GPX4) (Fratta Pasini et al. 2023). However, DOX downregulates these key antiferroptosis proteins, such as GPX4 and SLC7A11, ultimately leading to ferroptosis (Li et al. 2022b). In addition, activation of the p53 protein has been demonstrated to reduce SLC7A11 levels, rendering normal cells more susceptible to ferroptosis (Jiang et al. 2015). The phosphatidylinositol 3-kinase/AKT (PI3K/AKT) pathway has been extensively studied to determine its roles in protecting against heart ischemia/reperfusion (IR) injury and DIC and mitigating cardiomyocyte death (Ghafouri-Fard et al. 2022; Lu et al. 2023). Moreover, AKT promotes phosphorylating murine double minute 2 (MDM2) and thus leads to p53 degradation, regulating the cell cycle. A recent study showed that propofol inhibits ferroptosis in cardiac IR injury through the AKT/p53 signaling pathway (Li et al. 2022a). Given the essential roles of the above signaling pathway and cell death phenotypes, we hypothesized that the PI3K/AKT/MDM2/p53 pathway and ferroptosis might be involved in DIC.
Licorice, an herbaceous perennial legume, has been utilized in clinical practice for centuries due to its antibacterial, anti-inflammatory, cardioprotective, neuroprotective, and hepatoprotective activities. The ethanolic extract of licorice possesses potent antioxidant effects and attenuates lipoprotein oxidation (Visavadiya et al. 2009). A recent study also demonstrated the regulatory effects of licorice on mitochondrial function, lipid homeostasis, cardiac metabolism, and cardiac hypertrophy, thereby mitigating DIC (Upadhyay et al. 2020). However, the major active component responsible for these effects and its modulation of transcription factors and signaling pathways remain unclear. Therefore, in this study, bioinformatics analysis was applied to explore the practical chemical components and therapeutic targets of licorice in treating DIC. Furthermore, the effect and mechanism of the active component on DIC were evaluated by in vitro and in vivo tests.
Materials and methods
Construction and analysis of the active component-gene target-disease network
Major components of licorice and their putative targets were identified from the Traditional Chinese Medicine Systematic Pharmacology Online Database (TCMSP). The initial screening criteria for major components were oral bioavailability (OB) ≥ 30.00% and drug-likeness (DL) ≥ 0.18 (Ru et al. 2014). DIC-related genes were obtained by searching “doxorubicin-induced cardiotoxicity” in the GeneCards online database. Possible gene targets of major components in licorice were compared with DIC-related genes to screen out the overlapping ones, which were regarded as the potential therapeutic targets of licorice in DIC. Active components were defined as compounds associated with these therapeutic targets.
Construction and analysis of the protein‒protein interaction network
A protein‒protein interaction (PPI) network illustrating the intersection of licorice and DIC-related targets was constructed by using the String online platform. Next, the network was visualized and analyzed using Cytoscape software, with nodes representing the genes and edges representing the interactions between the nodes (Shannon et al. 2003; Szklarczyk et al. 2017).
Pathway enrichment analysis
KEGG enrichment analysis of the potential therapeutic targets was performed, and the pathways were obtained and visualized by using the ClusterProfiler and Bioconductor packages in R studio (Robinson et al. 2010; Yu et al. 2012).
Molecular docking
According to the PPI network, core targets were selected to analyze their interactions with core compounds of licorice by using molecular docking technology. First, the 2D chemical structures of the obtained active components (isorhamnetin, MOL5281654; kaempferol, MOL5280863; licochalcone A (Lico A), MOL5318998; licochalcone B, MOL5318999; licochalcone G, MOL49856081; medicarpin, MOL336327; naringenin, MOL439246; quercetin, MOL5280343) were downloaded from the PubChem online database and then transmitted into Chem3D software to generate 3D structures with minimal energy states for molecular docking (Kim et al. 2019). We acquired the 3D chemical structures of the target proteins (AKT1, 7nh52; p53, 5olc2) from the PDB database (Burley et al. 2017). PyMOL and AutoDockTools v1.5.6 were used for the preparation of the protein and ligand structures, including the removal of water molecules and unwanted ligands, hydrogenation, and determination of the active pocket of the protein (Trott and Olson 2010; Eberhardt et al. 2021). Finally, the binding energies between the target protein and major active components in licorice were calculated by AutoDock Vina, and the protein‒ligand binding affinities were determined by the lowest binding energy value. The three-dimensional structures of the protein‒ligand binding complexes were visualized using PyMOL.
Animals
Five-week-old male ICR mice weighing 25 ± 2 g were purchased from the National Laboratory Animal Centre, Fujian Medical University, Fujian Province, PR China. Specific-pathogen-free (SPF) grade mice were fed in a barrier SPF environment at a constant temperature with alternating 12-hour light/dark cycles. The animals were maintained on a standard pellet diet and water ad libitum.
Animal groupings
To investigate the role of Lico A in vivo, 30 ICR mice were randomly divided into five groups (n = 6/group): (1) Control group: mice received intraperitoneal injections of solvent for 5 weeks; (2) Lico A group: mice received intraperitoneal injections of 20 mg/kg/day Lico A throughout all experiments (5 weeks); (3) DOX group: mice received intraperitoneal injections of equal amounts of solvent throughout all experiments and 5 mg/kg/week DOX from weeks 4 to 6; (4) DOX + Lico A1 group: mice received intraperitoneal injections of 10 mg/kg/day Lico A throughout all experiments and 5 mg/kg/week DOX from weeks 4 to 6; and (5) DOX + Lico A2 group: mice received intraperitoneal injections of 20 mg/kg/day Lico A throughout all experiments and 5 mg/kg/week DOX from weeks 4 to 6. Lico A and DOX were both dissolved in the vehicle consisting of 10% DMSO, 40% PEG-300, 5% Tween-80, and 45% physiological saline in vitro experiments.
Surface electrocardiogram
Before the measurements, the mice were lightly anesthetized with 1.5% isoflurane, and their body temperatures were kept constant at 37 °C using a custom-made heating pad. The limbs of the mice were attached to electrocardiogram (ECG) machines for recording ECG data. The electrocardiographic analysis included RR interval (ms), PR interval (ms), QRS duration (ms), QT interval (ms), and QTc (QT/square root of the RR interval).
Examination of myocardial injury markers and antioxidant enzyme activities
The whole blood of ICR mice was collected by extracting eyeball blood under anesthesia and then centrifuged at 3000 rpm (4 °C) for 15 min to isolate serum. The serum samples were stored at -80 °C until assayed. The level of creatine kinase MB isoenzyme (CK-MB) was determined by using a mouse CK-MB ELISA kit (MU30025, Bioswamp, China). Creatine kinase (CK) and lactate dehydrogenase (LDH) activities were detected by applying CK and LDH activity kits (A020-2&A032-1, Nanjing Jiancheng, China). The total superoxide dismutase (SOD) activity was determined using a total superoxide dismutase assay kit with NBT (S0109, Beyotime, China).
Histopathological analysis
Mouse heart tissue samples for histopathological evaluation were fixed in 10% formalin solution for 48 h. After dehydration in graded ethanol, the samples were embedded in fresh paraffin. The blocks containing mouse hearts were serially sectioned at a thickness of 5 μm, deparaffinized, and rehydrated. Hematoxylin-eosin (HE) staining was performed followed by standard HE staining procedures (C0105S, Beyotime, China). For Masson staining, sections were stained strictly according to the instructions provided for the Masson Stain Kit (G1340, Solarbio, China). In addition, the heart slices were incubated with antibodies against p53 (1:200, 60283-2-Ig, Proteintech, China), SLC7A11 (1:200, 26864-1-AP, Proteintech, China), and GPX4 (1:200, 67763-1-Ig, Proteintech, China) overnight at 4 °C and then stained using an immunohistochemical and DAB substrate kit at room temperature (PV-9001, ZSGB-BIO, China). Histopathological changes in mouse hearts were examined by light microscopy.

Network pharmacology and molecular docking analyses to identify the active pharmaceutical components in licorice for treating DIC. a Venn diagram illustrating the shared gene targets associated with DIC and licorice. Among the 140 targets associated with DIC and 777 targets of the major components of licorice, 83 targets were common to both. b PPI network depicting the 83 common target genes associated with licorice and DIC. Nodes represent proteins, and edges represent protein‒protein interactions. Larger and darker nodes indicate higher connectivity. The top two targets with the highest degree values were AKT1 and P53. c Heatmap plot illustrating the binding energies of molecular docking between eight active components and AKT1 and p53. d Molecular docking diagram showing the interaction between p53 protein and Lico A. e Molecular docking diagram showing the interaction between AKT1 protein and Lico A. f Scatter plot of the enriched KEGG pathways for the 83 hub genes. GeneRatio indicates the ratio of target genes enriched in the certain KEGG pathway to the total hub genes found in the KEGG database. The color and size of the dots in the scatterplot represent the range of the adjusted p value and the number of genes mapped to the distinct pathways, respectively. The top twelve enriched pathways are presented. DIC Doxorubicin-induced cardiotoxicity
H9C2 cell culture
H9C2 cells were obtained from the Type Culture Collection at Wuhan University and maintained in Dulbecco’s Modified Eagle Medium (DMEM, 31,600,034, Thermo, United States) supplemented with 10% fetal bovine serum (FBS, ST30-3302, PAN Seratech, United States) and 1% penicillin/streptomycin (15,140,122, Thermo, United States) at 37 °C in a 5% CO2 incubator.
H9C2 cell viability assay
H9C2 cells were seeded into a 96-well culture plate for 24 h. Next, the cells were treated with different concentrations of DOX (D1515, Sigma‒Aldrich, United States, in water), Lico A (HY-N0372, MedChem Express, United States, in DMSO), and Fer-1 (HY-100,579, MedChem Express, United States, in DMSO). Cell viability was evaluated using Cell Counting Kit-8 (CCK8) according to the kit instructions (C0037, Beyotime, China).
H9C2 cell experimental grouping and treatment
To investigate the role of Lico A in a DOX-treated cell model, H9C2 cells were divided into four groups: the DOX group (treated with 1 µm DOX for 24 h), DOX + Lico A group (pretreated with 10 µm Lico A for 2 h and then cotreated with 1 µm DOX for 24 h), Lico A group (treated with 10 µm Lico A for 26 h), and control group (treated with an equal volume of solvent for 26 h).
To validate the essentiality of the PI3K/AKT/MDM2/p53 pathways, H9C2 cells were pretreated using the specific PI3K inhibitor LY294002 (20 µm, HY-10,108, MedChem Express, United States) and the specific AKT inhibitor Mk2206 (5 µm, HY-10,358, MedChem Express, United States) for 1 h and then treated with Lico A for 2 h prior to DOX stimulation for 24 h (DOX + Lico A + LY294002 group and DOX + Lico A + MK2206 group, respectively). In addition, H9C2 cells were transfected with a p53 overexpression plasmid for 24 h and then treated with Lico A for 2 h prior to DOX stimulation for 24 h (DOX + Lico A + pcDNA-p53 groups).

Impact of Lico A and DOX on H9C2 cell viability. a Molecular structure of DOX. b Molecular structure of Lico A. c-h Effects of DOX at concentrations of 0.1–2.0 µm (c-e) and Lico A at concentrations of 1.0–20 µm (f-h) for 24, 48, and 72 h on H9C2 cell viability. g-h Effects of Lico A on the viability of DOX-treated H9C2 cells. Cells were pretreated with Lico A at concentrations of 1.0–15 µm for 2 h, followed by cotreatment with 1 µm DOX for 24, 48, and 72 h. Cell viability was assessed using the CCK8. n = 3, *p < 0.05 compared with the control group; #p < 0.05 compared with the DOX group. DOX Doxorubicin, Lico A Licochalcone A
H9C2 cell transfection
The p53 overexpression plasmid was constructed using a pcDNA3.1 vector. H9C2 cells were transfected using Lipo8000 Transfection Reagent (C0533, Beyotime, China). Specifically, when H9C2 cells reached 80% confluency, a mixture of Lipofectamine 8000 transfection reagent (0.15 µl) and pcDNA3.1-p53 plasmid (0.2 µg) in serum-free DMEM (150 µl) was added to each well. The transfected cells were incubated at 37 °C for 24 h. Equal amounts of empty pcDNA3.1 vector were used as a negative control following the aforementioned methods.
Measurement of ROS production
DCFH-DA in serum-free DMEM (S0033S, Beyotime, China) was added to one well of a six-well plate, and the plates were incubated for 20 min in the dark. Fluorescence and bright-field pictures of cells were captured through a fluorescence microscope (Nikon, Japan). The DCFH fluorescence intensity of each group was measured by a flow cytometer (FACSCanto™ II, BD Biosciences, United States) at excitation and emission wavelengths of 488 and 525 nm, respectively. Fluorescence images and flow cytometry data were analyzed using ImageJ and FlowJo software, respectively.

Protective effect of Lico A pretreatment against DIC in ICR mice. a Flowchart depicting the animal groupings and experimental procedures. b-e Effects of Lico A on DOX-induced alterations in serum levels of LDH, CK, CKMB, and SOD activity in the mouse model. f Representative ECG recordings of mice in different groups. g Impact of Lico A on DOX-induced changes in ECG parameters, including heart rate, PR interval, QRS duration, and QTc. h Representative histological images of HE-stained myocardium from mice. i Representative histological images of Masson-stained myocardium samples from mice. n = 6, *p < 0.05 compared with the control group; #p < 0.05 compared with the DOX group. DOX Doxorubicin, Lico A Licochalcone A
Determination of ferrous iron levels, malondialdehyde (MDA), and the glutathione/oxidized glutathione (GSH/GSSG) ratio
H9c2 cells were collected and lysed in RIPA lysis buffer (P0013B, Beyotime, China), and their protein concentrations were determined using a BCA protein assay kit (23,227, Thermo, United States). Next, the MDA levels were determined according to the kit instructions (S0131S, Beyotime, China). Ferrous iron levels in H9C2 cells were measured using a colorimetric assay kit (E-BC-K881-M, Elabscience Biotechnology, China). The concentrations of GSH and GSSG were determined using a GSH/GSSG test kit (S0053, Beyotime, China).

Effect of Lico A pretreatment on DOX-induced ferroptosis in H9C2 cells. a-c Assessment of ROS levels in H9C2 cells using DCFH-DA staining by light microscopy (a), fluorescence microscopy (b), and flow cytometry (c). d Quantitative analysis of the fluorescence intensity in DAFH-stained H9C2 cells. e Quantitative analysis of DCFH-positive cells measured by flow cytometry. f Effects of ferrostatin on cell viability in DOX-treated H9C2 cells. g-k GSH levels (g), GSSG levels (h), GSH/GSSG ratio (i), ferrous iron levels (j), and MDA levels (k) in H9C2 cells. n = 3, *p < 0.05 compared with the control group; #p < 0.05 compared with the DOX group. DOX Doxorubicin, Lico A Licochalcone A
Western blot analysis
H9c2 cells and mouse heart tissue samples were lysed in Western Blot RIPA lysis buffer. After the protein concentrations were measured using the BCA protein assay, the cell lysates were separated by 10–15% SDS‒PAGE and transferred onto activated polyvinylidene difluoride membranes (PVDF membranes, IPVH00010, Millipore, United States). Each membrane was blocked with 1% BSA in TBS and then incubated with specific primary antibodies for 12 h, including anti-PI3K (1:1000, A11177, Abclonal, China), anti-phospho-PI3K (Y467/Y199/Y464) (1:1000, AP0427, Abclonal, China), anti-AKT1 (1:1000, 60203-2-Ig, Proteintech, China), anti-phospho-AKT1 (S473) (1:1000, 80455-1-rr, Proteintech, China), anti-MDM2 (1:1000, 66511-1-Ig, Proteintech, China), anti-phospho-MDM2 (S166) (1:1000, ab170880, Abcam, Japan), anti-p53 (1:1000, 60283-2-Ig, Proteintech, China), anti-phospho-p53 (S392) (1:1000, T40061S, Abmart, China), anti-SLC7A11 (1:1000, A13685, Abclonal, China), anti-GPX4 (1:1000, A1933, Abclonal, China), anti-β-actin (1:10000, 20536-1-AP, Proteintech, China) and anti-GAPDH (1:10000, 60004-1-Ig, Proteintech, China). After being washed, the bound antibodies were detected with 1:4000 diluted goat anti-rabbit and goat anti-mouse IgG-HRP (1:10000, SA00001-1/ SA00001-2, Proteintech, China). Finally, the bands were visualized using an ECL chemiluminescence kit (K12045D50, Advansta, United States). The relative expression levels of each detected protein to β-actin/GAPDH were quantified using ImageJ software.
Immunofluorescence staining
After being fixed with paraformaldehyde (AR1069, Boster, China), permeabilized with Triton X-100 (P0096, Beyotime, China), and blocked with immunostaining blocking solution (P0260, Beyotime, China) for 30 min, H9c2 cells were incubated with anti-p53 (1:500, 60283-2-Ig, Proteintech, China), anti-SLC7A11 (1:500, 26864-1-AP, Proteintech, China), and anti-GPX4 (1:500, 67763-1-Ig, Proteintech, China) at 4 °C overnight, incubated with Alexa Fluor 555-labeled donkey anti-rabbit/mouse IgG for 1 h at room temperature, and then stained with DAPI (Beyotime, C100, China) for 10 min. Images were acquired using a fluorescence microscope (Nikon, Japan).

Effect of Lico A pretreatment on ferroptosis-related protein expression in DIC mice. a-b Impact of Lico A on ferroptosis-related protein expression (p53, SLC7A11, and GPX4) in the myocardia of DIC mice. The expression of the protein was standardized by GAPDH. n = 3, *p < 0.05 compared with the control group; #p < 0.05 compared with the DOX group. c Representative images for p53, SLC7A11, and GPX4 immunohistochemical staining in mice myocardial tissue (400 ×, Scale bar = 50 μm, n = 6). DOX Doxorubicin, Lico A Licochalcone A
Statistical analysis
In the study, all continuous variables are presented as the mean ± S.E.M., and categorical variables are presented as proportions of 100%. Student’s t test and one-way ANOVA were implemented to demonstrate differences between two and more than two groups separately. Statistical analysis was performed by using GraphPad Prism ver.08 software.
Results
PPI network of licorice in the treatment of DIC
Utilizing the criteria of OB and DL, 92 major components of licorice and 777 target genes were retrieved from the TCMSP database. Furthermore, a comprehensive collection of 140 DIC-related targets was obtained from the GeneCards database. By performing comparative analysis, 83 overlapping genes associated with licorice and DIC were identified and are visually presented in Fig. 1a. Subsequently, the PPI network for these 83 genes was constructed using the STRING online database. To evaluate the interactive relationships, PPI network analysis was conducted using Cytoscape, revealing two prominent hub targets, p53 and AKT1 (Fig. 1b).

Effect of Lico A pretreatment on the PI3K/AKT/MDM2/p53 pathway and ferroptosis-associated proteins in H9C2 cells. a-b Impact of Lico A on the PI3K/AKT/MDM2/p53 pathway and ferroptosis-associated proteins in H9C2 cells. Protein expression was standardized to β-actin or GAPDH. n = 3, *p < 0.05 compared with the control group; #p < 0.05 compared with the DOX group. c Representative images for p53, SLC7A11, and GPX4 immunofluorescence staining in H9c2 cells (400 ×, Scale bar = 50 μm, n = 3). DOX Doxorubicin, Lico A Licochalcone A
Identification of the active components
To explore the active components of licorice involved in DIC treatment, an active component-gene target-disease network was established. Notably, the active components of licorice, including quercetin, naringenin, kaempferol, isorhamnetin, licochalcone A, licochalcone B, licochalcone G, and medicarpin, were identified.
Molecular docking analysis
We performed molecular docking simulations to investigate the binding affinities of the key active components of licorice with the protein structures of AKT1 and p53. The docking results demonstrated favorable interactions between the eight compounds and their respective target proteins. The binding energies, indicating the strength of the interactions, are presented in Fig. 1c. Furthermore, the molecular dockings between Lico A and AKT1 as well as p53 were visualized using PyMOL (Fig. 1d,e).

Effects of Lico A pretreatment on DOX-induced H9C2 cell ferroptosis through p53 protein downregulation. a-b Expression of p53 in H9C2 cells transfected with pcDNA3.1-p53 or an empty vector. c-d Effect of p53 overexpression on the PI3K/AKT/p53 pathway and ferroptosis-associated proteins in H9C2 cells cotreated with Lico A and DOX. Protein expression was standardized to β-actin or GAPDH. e Assessment of the ROS levels in H9C2 cells using DCFH-DA staining by flow cytometry. f Quantitative analysis of DCFH-positive cells measured by flow cytometry. g Evaluation of the MDA levels in H9C2 cells. n = 3, *p < 0.05 compared with the control group; #p < 0.05 compared with the DOX group; †p < 0.05 compared with the DOX + Lico A group. DOX Doxorubicin, Lico A Licochalcone A
KEGG pathway enrichment analyses
To gain insights into the underlying mechanisms, KEGG pathway analysis was conducted for the genes associated with licorice and DIC. A total of 157 significantly enriched KEGG pathways were identified (p < 0.05). The top 12 signaling pathways were shown in Fig. 1f and included lipid and atherosclerosis, PI3K-AKT, Kaposi sarcoma herpesvirus infection, fluid shear stress and atherosclerosis pathway, hepatitis B, human papillomavirus infection, hepatitis C, proteoglycans in cancer, FoxO signaling pathway, apoptosis, platinum drug resistance, HIF-1 signaling pathway, and prostate cancer.

Effects of Lico A pretreatment on DOX-induced H9C2 cell ferroptosis through activation of the PI3K/AKT/MDM2 pathway. a-d The effects of the PI3K inhibitor LY294002 and the AKT inhibitor MK2206 on the PI3K/AKT/MDM2/p53 pathway and ferroptosis-associated proteins in H9C2 cells cotreated with Lico A and DOX. Protein expression was standardized to β-actin or GAPDH. e Assessment of ROS levels in H9C2 cells using DCFH-DA staining by flow cytometry. f Quantitative analysis of DCFH-positive cells measured by flow cytometry. g Evaluation of the MDA levels in H9C2 cells. n = 3, *p < 0.05 compared with the control group; #p < 0.05 compared with the DOX group; †p < 0.05 compared with the DOX + Lico A group. DOX Doxorubicin, Lico A Licochalcone A
Effect of Lico A on an H9C2 cell model of DIC
Previous studies have reported the cardioprotective effects of quercetin, naringenin, kaempferol, isorhamnetin, and licochalcone B in mitigating myocardial tissue injury (Han et al. 2008, 2014; Xiao et al. 2012; Sun et al. 2013; Chen et al. 2019), but there have been no previous reports about the cardioprotective effects of Lico A. Therefore, we selected Lico A, as the new active compound involved in DIC, for further investigation. The chemical structures of DOX and Lico A are depicted in Fig. 2a,b.
To assess whether Lico A could attenuate DIC in vitro, we evaluated H9C2 cell viability using the CCK8 assay. The viability of H9c2 cells significantly decreased with increasing concentrations of DOX (p < 0.05). As illustrated in Fig. 2c-e, when the DOX concentration reached 1 µm, the cell viability was 48.52 ± 1.561% at 24 h, 43.05 ± 0.39% at 48 h, and 29.02 ± 1.20% at 72 h. To assess the potential treatment toxicity of Lico A, we treated H9C2 cells with various concentrations of Lico A for 24, 48, and 72 h. The results indicated that Lico A at doses lower than 15 µm did not induce significant cytotoxic effects in H9C2 cells (Fig. 2f-h). Consequently, we investigated whether Lico A, within the concentration range of 1 to 15 µm, could alleviate DOX-induced cytotoxicity. Subsequently, H9C2 cells were treated with different concentrations of Lico A for 2 h, and then cotreated with 1 µm DOX for 24, 48, and 72 h. As demonstrated in Fig. 2i-k, cell viability significantly decreased after DOX induction, but it was significantly restored following pretreatment with Lico A (p < 0.05). Notably, when the concentration of Lico A reached 10 µm, cell viability was restored to the highest value.
Effect of Lico A on an ICR mouse model of DIC
The specific procedure of the in vivo experiment is described in Fig. 3a. Regarding the biochemical indicators of myocardial injury, DOX treatment significantly increased the levels of LDH, CK, and CK-MB in mouse serum (p < 0.05). Lico A treatment notably decreased the levels of these biochemical indicators of myocardial injury induced by DOX (p < 0.05, Fig. 3b-d). Moreover, serum SOD levels were measured in each group to determine whether Lico A protects against oxidative damage in DIC. As depicted in Fig. 3e, serum SOD activity significantly decreased following exposure to DOX, but Lico A treatment significantly restored SOD activity (p < 0.05). In the ECG parameter measurements, the DOX group showed significant prolongments in the RR interval, QT interval, QTc, and QRS duration (p < 0.05), which indicated cardiac electrical signal conduction block in mice. However, Lico A could partially restore abnormal ECG parameters in mice (p < 0.05, Fig. 3f,g).
Histopathological analysis of myocardial tissues from mice treated with DOX revealed notable interstitial edema, inflammatory cellular infiltration around blood vessels, perinuclear vacuolation, and disrupted and disordered myocardial fibers. Conversely, as shown in Fig. 3h, myocardial tissues from mice pretreated with Lico A exhibited notable reductions in all pathological features. Additionally, Masson’s trichrome staining revealed a significant increase in interstitial fibrosis in the myocardial tissues of mice treated with DOX, while Lico A significantly decreased the cardiac fibrotic area (Fig. 3i).
Effect of Lico A on DOX-induced H9C2 cell ferroptosis
Subsequently, we attempted to examine the potential of Lico A in alleviating DOX-induced ferroptosis in vitro. According to the CCK-8 assay and previous studies (Liu et al. 2022), DOX at 1 μm and Lico A at 10 µm were selected for subsequent cell experiments. Considering the crucial role of ROS accumulation in ferroptosis, we assessed the extent of ROS generation in DOX-treated H9C2 cells. The level of ROS expression was determined by measuring the intensity of DCFH fluorescence. Both fluorescence microscopy and flow cytometric analysis demonstrated a significant increase in the ROS level of the DOX-treated group compared to the control group. However, pretreatment with Lico A substantially reduced the level of ROS in H9C2 cells (p < 0.05, Fig. 4a-e). These findings indicate that Lico A effectively mitigated DOX-induced excessive oxidative stress.
To investigate the involvement of ferroptosis in DIC cell death, we examined the impact of Fer-1 (0.5, 1.0, 2.0, and 4.0 µm) on DOX (1.0 µm)-treated H9C2 cells for 24, 48, and 72 h using the CCK-8 assay and observed a considerable enhancement in cell viability (p < 0.05, Fig. 4f). Next, we evaluated critical markers of ferroptosis, including the levels of GSH, GSSG, and ferrous iron. The DOX group exhibited increased GSSG accumulation along with corresponding decreases in GSH levels and the GSH/GSSG ratio. However, intervention with Lico A restored GSH levels and reversed the decrease in the GSH/GSSG ratio (p < 0.05, Fig. 4g-i). In addition, DOX treatment led to an elevation in intracellular ferrous iron, which was attenuated by Lico A pretreatment (p < 0.05, Fig. 4j). Another hallmark of ferroptosis, MDA, as the end-product of polyunsaturated fatty acid peroxidation, exhibited increased levels in the DOX group (p < 0.05). However, pretreatment with Lico A significantly decreased MDA levels compared with the DOX group (p < 0.05, Fig. 4k).
Lico A-mediated activation of the PI3K/AKT/MDM2/p53 pathway
For further validation, Western blotting and immunohistochemistry were performed to determine the expression levels of ferroptosis-related proteins, including p53, SLC7A1, and GPX4 in mouse heart tissues. By Western blot analysis, we found that p53 expression was significantly increased in DOX-treated mice, but the increase was significantly attenuated in Lico A-treated mice. Conversely, DOX led to anti-ferroptosis (SLC7A11 and GPX4) protein depletion, but this effect could be attenuated by Lico A pretreatment (Fig. 5a,b). Immunohistochemical staining of mouse heart sections also yielded similar results (Fig. 5c).
To elucidate the mechanisms through which Lico A inhibits DOX-induced ferroptosis, the PI3K/AKT/MDM2/p53 signaling pathway was selected for additional investigation based on the PPI network and KEGG results obtained from network pharmacology analyses. Western blot results revealed that DOX treatment significantly suppressed the phosphorylation of PI3K, AKT, and MDM2 (p-PI3K, p-AKT, and p-MDM2), promoted p53 and p-p53 accumulation, and inhibited the expression of SLC7A11 and GPX4 in H9C2 cells (p < 0.05). However, pretreatment with Lico A restored the phosphorylation levels of PI3K/AKT/MDM2, decreased the p53 and p-p53 protein levels, and protected cells from SLC7A11/GPX4 depletion (p < 0.05, Fig. 6a,b). The expression levels of p53, SLC7A11, and GPX4 in H9c2 cells (Fig. 6c) were similar to the results of immunofluorescence staining. The above results demonstrated the potential involvement of the PI3K/AKT/MDM2 and p53 pathways in Lico A-mediated ferroptosis inhibition.
Lico A-mediated relief of DOX-induced ferroptosis via the PI3K/AKT/MDM2/p53 pathway
To validate the significance of p53 in the Lico A-mediated relief of ferroptosis, we constructed a p53 overexpression plasmid and transfected it into cells. After 24 h of transfection, H9C2 cells were harvested for Western blot analysis (Fig. 7a,b). A comparison between the DOX + Lico A and DOX + Lico A + pcDNA-p53 groups revealed a significant decrease in the protein levels of SLC7A11/GPX4 in the latter (p < 0.05, Fig. 7c,d). Conceivably, Lico A treatment decreased excessive levels of ROS and MDA induced by DOX, whereas overexpression of p53 reversed these effects of Lico A (p < 0.05, Fig. 7e-g).
The results obtained from Western blot analysis indicated that pretreatment with LY294002 and MK2206 both inhibited PI3K/AKT/MDM2 phosphorylation and abolished the Lico A-induced inhibition of p53 and the upregulation of SLC7A11/GPX4 (p < 0.05, Fig. 8a-d). Similarly, in comparison with the DOX + Lico A group, the levels of both ROS and MDA in LY294002/MK2206-pre-treated H9C2 cells significantly increased again (p < 0.05, Fig. 8e-g). The above experiments further demonstrated that the PI3K/AKT/MDM2/p53 pathway was indispensable for the anti-ferroptosis effect of Lico A.

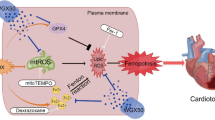
Protective mechanism of Lico A against DIC Lico A activates the PI3K/AKT/MDM2 pathway, leading to the downregulation of p53 protein. The depletion of p53 activates SLC7A11/GSH/GPX4, effectively attenuating ferroptosis in DIC
Discussion
DOX, the most commonly utilized chemotherapeutic agent, has been extensively employed for the treatment of various hematological and solid tumors. However, its clinical applications are greatly limited owing to its wide range of side effects, including cardiotoxicity, gastrointestinal symptoms, hepatotoxicity, and immunosuppression. Among these, cardiotoxicity is considered the most prevalent and severe complication in patients receiving DOX, often leading to myocardial injury and cardiac failure (Armenian et al. 2017). The etiology and pathogenesis of DIC are complex and not yet fully understood, which has hindered the development of specific treatment approaches for a long time. Dexrazoxane, the sole cardioprotective drug approved for DIC, has been found to chelate iron, attenuate the formation of iron-anthracycline complexes, and subsequently reduce ROS production (Padegimas et al. 2020). It is important to note that dexrazoxane may diminish chemotherapeutic efficacy, and there are concerns regarding its potential adverse effects, such as secondary tumor development (Shaikh et al. 2016). Nevertheless, the efficacy of the iron chelator dexrazoxane highlights the significance of myocardial iron overload in DIC. Previous studies have demonstrated that ferroptosis, characterized by the iron-dependent accumulation of lipid peroxides, is implicated in the pathogenesis of cardiotoxicity. DOX has been shown to increase ROS-induced lipid peroxidation and affect iron homeostasis by regulating transferrin receptor/ferritin (Kotamraju et al. 2002; Maccarinelli et al. 2014). Therefore, it is conceivable that inhibiting ferroptosis could serve as a possible treatment strategy for DIC. Currently, several potential drugs with antiferroptosis properties, including dexrazoxane, deferoxamine, ferrostatin-1, empagliflozin, and metformin, are being explored to determine their efficacy in DIC (Zhang et al. 2022).
Flavonoids, including flavonols, flavones, flavanes, flavanones, anthocyanins, proanthocyanidins, and chalcones, are considered the most abundant plant secondary metabolites and have been proven to possess various biological activities, such as antioxidant, inflammation elimination, cardioprotective, hepatoprotective, and neuroprotective effects (Wang et al. 2021). Due to the presence of specific metal binding sites, flavonoids are considered to be able to chelate iron and reduce iron accumulation and lipid peroxidation (Pietta 2000). Regarding their pharmacological characteristics, it is speculated that flavonoids may play a role in anti-ferroptosis in DIC. In this study, using network pharmacology methods, we hypothesized that a flavonoid in licorice, Lico A, could exert a cardioprotective effect via the PI3K/AKT and p53 pathways. While Lico A has been found to possess antioxidant activity, its antiferroptosis capacity has not been investigated in detail (Su et al. 2018). Indeed, ferroptosis, iron overload, and oxidative stress are interrelated and influence each other. Thus, we experimentally demonstrated that Lico A could activate the PI3K/AKT signaling pathway to alleviate p53-mediated ferroptosis.
The concept of ferroptosis initially emerged during the screening of the small molecular compounds erastin and RSL3 for their ability to inhibit tumor growth (Dolma et al. 2003; Yang and Stockwell 2008). Subsequent studies determined that erastin and RSL3 mediate SLC7A11 and GPX4, respectively, to induce a new form of cell death driven by iron-dependent lipid peroxidation (Dixon et al. 2012). Mechanistically, SLC7A11 exports intracellular glutamate while importing cystine, which is rapidly reduced to cysteine, the limiting amino acid for glutathione (GSH) synthesis. GSH is a co-factor of GPX4 and directly enhances GPX4 activity to eliminate lipid peroxides. Therefore, the SLC7A11/GSH/GPX4 pathway has been established as one of the crucial mechanisms protecting cells against ferroptosis and has also been shown to be inhibited in DIC (Li et al. 2022b).
p53, a well-established tumor suppressor gene, is involved in cell cycle inhibition, aging, and apoptosis and plays important roles in tumorigenesis and progression. p53 can downregulate the expression of SLC7A11, which inhibits cystine uptake, thereby affecting GPX4 activity and leading to lipid ROS accumulation and ferroptosis (Lei et al. 2021). Additionally, overexpression of the oncoprotein MDM2 leads to p53 protein degradation and suppression of p53 activity. It has been reported that AKT phosphorylation promotes MDM2-p53 association, enhancing p53 degradation (Freedman et al. 1999). A recent study on cardiovascular medication has revealed that propofol protects the heart against IR injury and inhibits ferroptosis through the AKT/p53 signaling pathway (Li et al. 2022a).
AKT is a key downstream effector of PI3K, and the PI3K/AKT pathway contributes to a wide spectrum of biological processes, such as cell metabolism, cell cycle progression, and cell survival (Shi et al. 2019). In addition to its role in regulating the survival of cardiomyocytes, angiogenic processes, and inflammatory responses, the PI3K/AKT pathway is implicated in myocardial damage induced by toxic substances such as H2O2, lipopolysaccharides, and anthracyclines (Ghafouri-Fard et al. 2022). Since ferroptosis was first described as a new form of nonapoptotic cancer cell death, emerging evidence has proven that the PI3K/AKT signaling pathway is involved in ferroptosis in multiple myeloma and colorectal cancer (Li et al. 2023; Yin et al. 2023). Previous findings have indicated that DOX treatment decreases the phosphorylation of PI3K/AKT, while treatment with kirenol, for instance, activates the PI3K/AKT pathway and prevents DIC-related cardiac hypertrophy, oxidative stress, and cell apoptosis (Alzahrani et al. 2021).
Given the essential roles of related pathways and programmed cell death in DIC, we investigated whether small-molecule activators of the PI3K/AKT/MDM2 pathway from licorice could alleviate p53-mediated ferroptosis. Our results demonstrated that Lico A, identified through bioinformatics methods, might be a candidate drug for DIC. Furthermore, in vitro and in vivo DIC models were employed to evaluate the cardioprotective effects of Lico A. DOX treatment triggered GSH depletion, ROS production, lipid peroxidation, and iron overload while decreasing the expression of SLC7A11 and GPX4 in H9C2 cells. However, these characteristic changes in ferroptosis were mitigated after Lico A administration. Mechanistically, we identified that the PI3K/AKT/MDM2 pathway was inhibited by DOX, but Lico A pretreatment mitigated this inhibition and attenuated p53-mediated ferroptosis in H9C2 cells. To further verify the specific signaling pathways involved in the protective effects of Lico A, we employed LY294002, Mk2206, and p53 plasmid interventions and found that PI3K/AKT inhibition and p53 overexpression could partly abolish the antiferroptotic effects of Lico A.
Conclusion
In conclusion, this study is the first to identify Lico A, an active compound found in licorice, as a therapeutic agent for DIC. Its cardioprotective function is mediated by the reduction of p53-mediated ferroptosis through activation of the PI3K/AKT/MDM2 pathway (Fig. 9).
Data Availability
All data in this study are available from the corresponding authors upon reasonable request.
References
Alzahrani AM, Rajendran P, Veeraraghavan VP, Hanieh H (2021) Cardiac protective effect of Kirenol against Doxorubicin-Induced Cardiac Hypertrophy in H9c2 cells through Nrf2 Signaling via PI3K/AKT pathways. Int J Mol Sci 22(6):3269
Armenian SH, Lacchetti C, Lenihan D (2017) Prevention and Monitoring of Cardiac Dysfunction in survivors of adult cancers: American Society of Clinical Oncology Clinical Practice Guideline Summary. J Oncol Pract 13:270–275
Burley SK, Berman HM, Kleywegt GJ, Markley JL, Nakamura H, Velankar S (2017) Protein Data Bank (PDB): the single global Macromolecular structure archive. Methods Mol Biol 1607:627–641
Chen X, Peng X, Luo Y, You J, Yin D, Xu Q, He H, He M (2019) Quercetin protects cardiomyocytes against doxorubicin-induced toxicity by suppressing oxidative stress and improving mitochondrial function via 14-3-3gamma. Toxicol Mech Methods 29:344–354
Christidi E, Brunham LR (2021) Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis 12:339
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149:1060–1072
Dolma S, Lessnick SL, Hahn WC, Stockwell BR (2003) Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human Tumor cells - ScienceDirect. Cancer Cell 3:285–296
Eberhardt J, Santos-Martins D, Tillack AF, Forli S (2021) AutoDock Vina 1.2.0: new docking methods, expanded force field, and Python Bindings. J Chem Inf Model 61:3891–3898
Fang X, Wang H, Han D, Xie E, Yang X, Wei J, Gu S, Gao F, Zhu N, Yin X, Cheng Q, Zhang P, Dai W, Chen J, Yang F, Yang HT, Linkermann A, Gu W, Min J, Wang F (2019) Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A 116:2672–2680
Fratta Pasini AM, Stranieri C, Busti F, Di Leo EG, Girelli D, Cominacini L (2023) New insights into the role of ferroptosis in Cardiovascular Diseases. Cells 12(6):867
Freedman DA, Wu L, Levine AJ (1999) Functions of the MDM2 oncoprotein. Cell Mol Life Sci 55:96–107
Ghafouri-Fard S, Khanbabapour Sasi A, Hussen BM, Shoorei H, Siddiq A, Taheri M, Ayatollahi SA (2022) Interplay between PI3K/AKT pathway and heart disorders. Mol Biol Rep 49:9767–9781
Han X, Pan J, Ren D, Cheng Y, Fan P, Lou H (2008) Naringenin-7-O-glucoside protects against doxorubicin-induced toxicity in H9c2 cardiomyocytes by induction of endogenous antioxidant enzymes. Food Chem Toxicol 46:3140–3146
Han J, Wang D, Yu B, Wang Y, Ren H, Zhang B, Wang Y, Zheng Q (2014) Cardioprotection against ischemia/reperfusion by licochalcone B in isolated rat hearts. Oxid Med Cell Longev 2014: 134862
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W (2015) Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520:57–62
Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, Li Q, Shoemaker BA, Thiessen PA, Yu B, Zaslavsky L, Zhang J, Bolton EE (2019) PubChem 2019 update: improved access to chemical data. Nucleic Acids Res 47:D1102–D1109
Kitakata H, Endo J, Ikura H, Moriyama H, Shirakawa K, Katsumata Y, Sano M (2022) Therapeutic targets for DOX-Induced Cardiomyopathy: role of apoptosis vs. Ferroptosis. Int J Mol Sci 23(3):1414
Kotamraju S, Chitambar CR, Kalivendi SV, Joseph J, Kalyanaraman B (2002) Transferrin receptor-dependent iron uptake is responsible for doxorubicin-mediated apoptosis in endothelial cells: role of oxidant-induced iron signaling in apoptosis. J Biol Chem 277:17179–17187
Lei G, Zhang Y, Hong T, Zhang X, Liu X, Mao C, Yan Y, Koppula P, Cheng W, Sood AK, Liu J, Gan B (2021) Ferroptosis as a mechanism to mediate p53 function in Tumor radiosensitivity. Oncogene 40:3533–3547
Li S, Lei Z, Yang X, Zhao M, Hou Y, Wang D, Tang S, Li J, Yu J (2022a) Propofol protects Myocardium from Ischemia/Reperfusion Injury by inhibiting ferroptosis through the AKT/p53 signaling pathway. Front Pharmacol 13:841410
Li X, Liang J, Qu L, Liu S, Qin A, Liu H, Wang T, Li W, Zou W (2022b) Exploring the role of ferroptosis in the doxorubicin-induced chronic cardiotoxicity using a murine model. Chem Biol Interact 363:110008
Li J, Jiang JL, Chen YM, Lu WQ (2023) KLF2 inhibits Colorectal cancer progression and Metastasis by inducing ferroptosis via the PI3K/AKT signaling pathway. J Pathol Clin Res
Liu X, Li D, Pi W, Wang B, Xu S, Yu L, Yao L, Sun Z, Jiang J, Mi Y (2022) LCZ696 protects against doxorubicin-induced cardiotoxicity by inhibiting ferroptosis via AKT/SIRT3/SOD2 signaling pathway activation. Int Immunopharmacol 113:109379
Lu Y, Min Q, Zhao X, Li L, Zhao G, Dong J (2023) Eupatilin attenuates doxorubicin-induced cardiotoxicity by activating the PI3K-AKT signaling pathway in mice. Mol Cell Biochem. https://doi.org/10.1007/s11010-023-04769-1
Maccarinelli F, Gammella E, Asperti M, Regoni M, Biasiotto G, Turco E, Altruda F, Lonardi S, Cornaghi L, Donetti E, Recalcati S, Poli M, Finazzi D, Arosio P, Cairo G (2014) Mice lacking mitochondrial ferritin are more sensitive to doxorubicin-mediated cardiotoxicity. J Mol Med (Berl) 92:859–869
Padegimas A, Clasen S, Ky B (2020) Cardioprotective strategies to prevent Breast cancer therapy-induced cardiotoxicity. Trends Cardiovasc Med 30:22–28
Pietta PG (2000) Flavonoids as antioxidants. J Nat Prod 63:1035–1042
Renu K, V GA, Arunachalam PBT S (2018) Molecular mechanism of doxorubicin-induced cardiomyopathy - an update. Eur J Pharmacol 818:241–253
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140
Ru J, Li P, Wang J, Zhou W, Li B, Huang C, Li P, Guo Z, Tao W, Yang Y, Xu X, Li Y, Wang Y, Yang L (2014) TCMSP: a database of systems pharmacology for drug discovery from herbal medicines. J Cheminform 6:13
Shaikh F, Dupuis LL, Alexander S, Gupta A, Mertens L, Nathan PC (2016) Cardioprotection and Second Malignant Neoplasms Associated with Dexrazoxane in Children receiving Anthracycline Chemotherapy: a systematic review and Meta-analysis. J Natl Cancer Inst 108(4):djv357
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504
Shi X, Wang J, Lei Y, Cong C, Tan D, Zhou X (2019) Research progress on the PI3K/AKT signaling pathway in gynecological cancer (review). Mol Med Rep 19:4529–4535
Su X, Li T, Liu Z, Huang Q, Liao K, Ren R, Lu L, Qi X, Wang M, Chen J, Zhou H, Leung EL, Pan H, Liu J, Wang H, Huang L, Liu L (2018) Licochalcone A activates Keap1-Nrf2 signaling to suppress arthritis via phosphorylation of p62 at serine 349. Free Radic Biol Med 115:471–483
Sun J, Sun G, Meng X, Wang H, Luo Y, Qin M, Ma B, Wang M, Cai D, Guo P, Sun X (2013) Isorhamnetin protects against doxorubicin-induced cardiotoxicity in vivo and in vitro. PLoS ONE 8:e64526
Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, Jensen LJ, von Mering C (2017) The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 45:D362–D368
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461
Upadhyay S, Mantha AK, Dhiman M (2020) Glycyrrhiza glabra (licorice) root extract attenuates doxorubicin-induced cardiotoxicity via alleviating oxidative stress and stabilising the cardiac health in H9c2 cardiomyocytes. J Ethnopharmacol 258:112690
Visavadiya NP, Soni B, Dalwadi N (2009) Evaluation of antioxidant and anti-atherogenic properties of Glycyrrhiza glabra root using in vitro models. Int J Food Sci Nutr 60(Suppl 2):135–149
Wang X, Li Y, Han L, Li J, Liu C, Sun C (2021) Role of flavonoids in the treatment of Iron overload. Front Cell Dev Biol 9:685364
Xiao J, Sun GB, Sun B, Wu Y, He L, Wang X, Chen RC, Cao L, Ren XY, Sun XB (2012) Kaempferol protects against doxorubicin-induced cardiotoxicity in vivo and in vitro. Toxicology 292:53–62
Yang WS, Stockwell BR (2008) Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol 15:234–245
Yin Z, Lv Y, Deng L, Li G, Ou R, Chen L, Zhu Y, Zhong Q, Liu Z, Huang J, Wu H, Zhang Q, Fei J, Liu S (2023) Targeting ABCB6 with nitidine chloride inhibits PI3K/AKT signaling pathway to promote ferroptosis in Multiple Myeloma. Free Radic Biol Med 203:86–101
Yu G, Wang LG, Han Y, He QY (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16:284–287
Zhang G, Yuan C, Su X, Zhang J, Gokulnath P, Vulugundam G, Li G, Yang X, An N, Liu C, Sun W, Chen H, Wu M, Sun S, Xing Y (2022) Relevance of Ferroptosis to Cardiotoxicity caused by anthracyclines: mechanisms to target treatments. Front Cardiovasc Med 9:896792
Acknowledgements
The authors acknowledge the Public Technology Service Center at Fujian Medical University for providing technical support.
Funding
This study was supported by the Natural Science Foundation of Fujian Province of China (2019J01610 and 2019J01608), the Joint Fund for the Innovation of Science and Technology at Fujian Province (2021Y9035), and the Young and Middle-aged Talent Training Project of Fujian Provincial Health Commission (2019ZQN97).
Author information
Authors and Affiliations
Contributions
G.C., S.L., and S.X. worked together on bioinformatics analysis, experiment design and conduct, data collection and analysis, results visualization, figures drawing, and manuscript writing/review/editing. J.L. and H.G. worked together on bioinformatics analysis and manuscript writing/review/editing. All authors listed in the manuscript contributed to the paper and approved the final version. The authors declare that all data were generated in-house and that no paper mill was used.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The animal study protocol for using ICR mice was submitted and approved by the Animal Ethics Committee at Fujian Medical University (IACUCFJMU20230171).
Competing interests
The authors declare no competing interests.
Consent for publication
All the authors agreed on the publication of this manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chen, G., Luo, S., Guo, H. et al. Licochalcone A alleviates ferroptosis in doxorubicin-induced cardiotoxicity via the PI3K/AKT/MDM2/p53 pathway. Naunyn-Schmiedeberg's Arch Pharmacol 397, 4247–4262 (2024). https://doi.org/10.1007/s00210-023-02863-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-023-02863-1