
Abstract
Ferroptosis refers to a novel way of cell death, inconsistent with the conventional concept of apoptosis and necrosis. It shows a close association with iron metabolism and oxidative damage, as marked by the significant increase of reactive oxygen species, the decreases of mitochondrial volume, and the thickening of membrane density. Recent studies confirmed that ferroptosis is closely associated with the occurrence, development, and therapy of the tumors. As impacted by the high levels of reactive oxygen species and lipid peroxides in lung cancer tissues, it is suggested that ferroptosis is more likely to occur in lung cancer tissues, which may act as a novel approach for non-small cell lung cancer (NSCLC) therapy. In the present study, the research achievements in recent years on the regulating mechanism of ferroptosis and its effect on the occurrence and the therapy of lung cancer are reviewed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The growth, development, and even death of life are determined by the growth and death of cells. Apoptosis and necrosis refer to the two major forms of conventional cell death. Over the past few years, with the further development of cell research, increasing ways of cell death have been identified (e.g., autophagy, apoptosis, and necroptosis). They are critical to the physiological or pathological process of cells. In 2012, Dixon et al. (Dixon et al. 2012) reported a novel way of cell death, termed as ferroptosis, when studying the mechanism by which erastin kills fibrosarcoma cells containing RAS mutations in the oncogenes. Ferroptosis is obviously different from cell necrosis, apoptosis, and autophagy in morphology, biochemistry, genetics, and functions (Table 1); it is characterized by a significant increase in intracellular lipid reactive oxygen species, a decrease in the volume of mitochondria, as well as an elevation of membrane density. Because ferroptosis has shown a close association with the treatment of many malignant tumors and neurodegenerative diseases, it has become a research hot pot in recent years. As revealed from recent articles, ferroptosis complexly impacts the occurrence of lung cancer, which presents novel ideas to treat NSCLC.
Features of ferroptosis
Morphological hallmarks
Even though the original study indicated that ferroptosis is morphologically, biochemically, and genetically different from apoptosis, necrosis, and autophagy (Dixon et al. 2012), most researchers consider that cells in which ferroptosis occurred usually show necrosis-like morphological changes (Vanden Berghe et al. 2014). For example, it includes a loss of plasma membrane integrity, cytoplasmic swelling, swelling of the cytoplasmic organelles, and moderate chromatin condensation (Dixon et al. 2012; Dolma et al. 2003; Yagoda et al. 2007; Friedmann Angeli et al. 2014). Under the electronic microscope, cells that undergone ferroptosis exhibit abnormal mitochondria, such as condensation or swelling, increased membrane density, decreased crista, and broken outer member (Dixon et al. 2012; Yagoda et al. 2007; Friedmann Angeli et al. 2014). Although significant changes occur in mitochondrial morphology, the role of these organelles in ferroptosis remains debatable.
Biochemical hallmarks
Ferroptosis is associated with two main biomedical traits, namely iron accumulation and lipid peroxidation.
The accumulation of iron
The classical ferroptosis activators erastin or RSL3 inhibit the antioxidation system by increasing the accumulation of iron in the cells (Dixon et al. 2012). Iron metabolism is one of the important pathways leading to ferroptosis, which will be discussed in more detail later.
Lipid peroxidation
Lipid peroxidation mainly affects unsaturated fatty acids in the cell membrane. During ferroptosis, there is an increasing amount of products of lipid peroxidation, such as the initial lipid hydroperoxides (L-OOH) and subsequent reactive aldehydes.
Genetical hallmarks
Acyl-CoA synthetase long-chain family member 4 (ACSL4) is an enzyme related to fatty acid metabolism. The upregulation of ACSL4 increases the content of polyunsaturated fatty acids (PUFAs) in phospholipids. Therefore, the content of polyunsaturated fatty acids (PUFAs) in phospholipids is increased, which makes ACSL4 a biomarker particular to ferroptosis (Kagan et al. 2017; Yuan et al. 2016; Doll et al. 2017).
The mechanism of regulating ferroptosis in cells
Four main mechanisms can be followed to regulate ferroptosis in cells. To be specific, the first is correlated with oxidative stress, the second shows an association with iron metabolism, the third is correlated with lipid metabolism, and the fourth represents the recently discovered mechanism related to the protein ferroptosis suppressing protein 1 (FSP1)/apoptosis inducing factor mitochondria 2 (AIFM2) inhibiting ferroptosis, which traps lipid peroxides in a GPX4-independent manner. Apart from the fourth mechanism of ferroptosis, the first three processes mentioned are interrelated.
The mechanism regarding oxidative stress
Oxidative stress is a classical pathway, and many other regulatory pathways are closely related to it.
Cystine/glutamate antiporter (cystine/glutamate transporter, System Xc-) consists of two subunits (i.e., SLC7A11 and SLC3A2L) on the cell membrane of the organism (Lin et al. 2016). It is an important antioxidant system in cells, which can excrete glutamate while ingesting cystine. Cystine undergoes reduction in cells to cysteine and the further conversion into glutathione (GSH) (i.e., a vital intracellular free radical scavenger). GSH is capable of reducing reactive oxygen species (ROS) and active nitrogen as impacted by peroxidase system (GPXs), as well as inhibiting the synthesis of GSH, thereby causing oxidative damage and even death of cells, which is termed as the process of oxidative stress (Chen et al. 2015). A process of oxidative stress exists in ferroptosis. GSH is synthesized by glutathione-cysteine ligase; glutathione peroxidase 4 (GPX4) uses GSH as an essential cofactor to catalyze the degradation of hydrogen peroxide and the cytotoxic lipid hydroperoxides (L-OOH) to water and the corresponding alcohols(L-OH) respectively, as well as inhibiting the generation of reactive oxygen species (ROS). According to Yang et al. (Yang et al. 2014), ferroptosis regulated by GPX4 acts as a common mechanism of numerous independent small molecular scaffolds. Knockdown of mouse membrane lipid repairase-GPX4 or directly exploiting GPX4 inhibitors RSL3 can facilitate lipid peroxidation and intracellular ROS accumulation, as well as induce cell death. Jiang et al. (Jiang et al. 2015) found that tumor suppressor P53 can downregulate the expression of SLC7A11, as an attempt to inhibit cells from ingesting cystine by suppressing SystemXc- and thus promoting ferroptosis. Besides, a number of articles (Saint-Germain et al. 2017) suggested that the suppressor of cytokine signaling 1 (SOCS1) is capable of regulating TP53 target genes and inhibiting sensitive cells from ferroptosis by altering the content of GSH based on SLC7A11 downregulation. Poursaitidis et al. (Poursaitidis et al. 2017) reported that in human mammary epithelial cells (HME), by inhibiting the ingestion of cystine, the mutants of cells expressing activated epidermal growth factor receptor (EGFR) generated considerable ROS, thereby resulting in ferroptosis of the cells. NADPH refers to the coenzyme of GSH reductase, which significantly impacts the maintenance of the level of GSH in the body. Shimada et al. (Shimada et al. 2016a) studied the effects of ferroptosis inducers (i.e., erastin and RSL3) in 12 different cell lines. They found that the levels of NAD+ and NADP+ decreased significantly, while the content of ROS was elevated. It was therefore inferred that NADPH may act as a biological index to monitor the sensitivity of ferroptosis inducers.
As revealed from the mentioned articles, the occurrence of ferroptosis in cells is determined by the regulation of oxidative stress of cells. Under the mentioned regulation, ferroptosis of cells can be induced.
Mechanisms related to iron metabolism
Iron, an indispensable trace element in the body, largely exists as ferrous and ferric iron ions in the body. The occurrence of ferroptosis shows a close association with the accumulation of iron and ROS. On a high level, the metabolism of iron in cells comprises three processes, ingestion, transport, and storage. The ferrous iron ions released by the small intestine or red blood cell degradation are oxidized by ceruloplasmin as ferric iron ions. After that, the oxidized iron ions bind to transferrin (TF) and enter the cells via the transferrin receptor 1 (TFR1). As impacted by six-transmembrane epithelial antigen of prostate 3 (STEAP3), Fe3+ is reduced to Fe2+ and then stored in unstable iron pool (LIP) and ferritin; intracellular ferritin is composed of ferritin heavy chain (FTH) and ferritin light chain (FTL), as mediated by the divalent metal transporter 1 (DMT1) or the zinc-ferric regulatory protein family 8/14 (ZIP8/14) involved. Part of the residual divalent iron ions are oxidized to trivalent iron ions as impacted by the membrane iron transport output protein (Fpn), while these ions infiltrate and participate in the recycling of iron in the body (Kazan and Urfali-Mamatoglu 2017; Anderson 2017; Kang 2017). As suggested by Gao et al. (Gao and Monian 2015), heat shock protein B1 (HSPB1) can downregulate the concentration of intracellular iron ions by inhibiting TFR1, and the overexpressed heat shock protein B1 can inhibit ferroptosis. Other articles indicated that (Gammella et al. 2015) inhibition of iron response element-binding protein 2 (IREB2) can significantly upregulate the expression of FTH1 and FTL, lower intracellular ferrous ion concentration and ROS production, and then inhibit erastin-induced ferroptosis. After binding to FTH, nuclear receptor coactivator 4 protein (NCOA4) accumulates ferritin in autophagosome and induces autophagy, thereby elevating the concentration of iron ions in cells and causing oxidative damage in Fenton reaction, which leads to ferroptosis in cells (Kang 2017). Furthermore, research (Chang et al. 2018; Hassannia et al. 2018; Manz et al. 2016) found that heme oxygenase-1 (HO-1) could improve the ability of erastin to induce ferroptosis by iron overloading and subsequently excessive ROS generation and lipid peroxidation. However, some authors also suggest the anti-ferroptosis role of HO-1; HO-1 also plays a negative regulatory role in erastin- and sorafenib-induced hepatocellular carcinoma. The downregulation of the expression of HO-1 could inhibit cell growth with erastin and sorafenib (Sun et al. 2016). The overexpression of the iron–sulfur cluster assembly enzyme (ISCU) alleviates ferroptosis induced by dihydroartemisinin (DHAN) in leukemia cells (Du et al. 2019). As suggested from the mentioned articles, the balance of iron metabolism and the regulation of ferritin may be a regulatory point of ferroptosis, inconsistent with the regulating mechanism of oxidative stress.
Mechanisms related to lipids
The imbalance of lipid metabolism is closely associated with ferroptosis. As indicated by existing articles (Yang et al. 2016), lipid peroxidation of long-chain polyunsaturated fatty acid is capable of inducing ferroptosis in cells, and the degree of ferroptosis is correlated with the content of polyunsaturated fatty acids in cells. In addition, a positive correlation is identified between them. Polyunsaturated fatty acids are first acylated into membrane phospholipids and then oxidized, which can act as an executive signal of ferroptosis. According to Kagan et al. (2017), the vital phospholipids driving ferroptosis in cells are phosphatidylethanolamine (PE), containing either arachidonic acid or adrenalic acid (i.e., an extension product of arachidonic acid). By helping biosynthesize and remodel polyunsaturated fatty acids, long-chain acyl-CoA synthetase-4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are involved in the biosynthesis and remodeling of polyunsaturated fatty acids. Exhibiting a high sensitivity to ferroptosis is considered as a vital step to induce ferroptosis in cells. Through the reduction of ACSL4 or LPCAT3, the substrate of intracellular lipid peroxidation can be reduced, and the occurrence of ferroptosis can be subsequently suppressed. Wenzel et al. (2017) reported that phosphatidylethanolamine-binding protein 1 (PEBP1), a protein kinase cascade stent protein inhibitor, is capable of being combined with two 15-LOXs isozymes and limiting the production of lipid peroxides by altering the ability of its substrate to synthesize PE, thereby avoiding the occurrence of ferroptosis. A recent study by Zou et al. revealed the function of polyunsaturated ether phospholipids (PUFA-ePLs) in ferroptosis (Zou et al. 2020). However, the research by Cui et al in 2021 showed an emerging evidence that long-chain saturated fatty acid (SFA) is related to ferroptosis, and the research revealed a new FAR1-ether lipids-TMEM189 axis-dependent ferroptosis pathway (Cui et al. 2021). According to the comparison of the two studies, most of findings by Zou et al. are highly consistent with those of Cui et al. There was only one obvious difference that TMEM189 was found with no effect on ferroptosis, while Cui et al. found a protective role of TMEM189.
Mechanisms related to FSP1/AIFM2
Two latest articles initially reported the effect of apoptosis inducing factor mitochondria 2 (AIFM2), for example, ferroptosis suppressing protein 1 (FSP1), on ferroptosis, which revealed a novel and effective way to regulate ferroptosis (Bersuker et al. 2019; Doll et al. 2019). Doll et al. (2019) found that FSP1 can protect cells from ferroptosis caused by GPX4 deletion. Cell lines with FSP1 gene knocked out exhibited significantly more sensitivity to ferroptosis inducers, while overexpressed FSP1 gene can reduce the sensitivity of cells to ferroptosis inducers. In addition, the expression of FSP1 was not affected by P53 activation. As revealed from in-depth articles, the myristic acylation of FSP1 is capable of mediating the recruitment of the protein on the plasma membrane, acting as an oxidoreductase on the plasma membrane, reducing coenzyme Q10, as well as inhibiting ferroptosis by blocking lipid peroxidation. Ferroptosis inducers (FINs) restrict the synthesis of coenzyme Q10 via SQS-MVA pathway, facilitate the accumulation of lipid peroxidation, and result in ferroptosis in cells. Statins (e.g., simvastatin) suppress the synthesis of coenzyme Q10 and induce ferroptosis by inhibiting the rate-limiting enzyme HMG-CoA reductase of the MVA pathway. Doll et al. found that FSP1 can catalyze NAD (P) H to promote CoQ10 synthesis and then act in parallel with GPX4 via the FSP1-CoQ10-NAD (P) H pathway to inhibit ferroptosis. This result could account for the effect of NAD(P)H in the MVA pathway through the loss of ubiquinone convergence on FSP1. On that basis, the sensitivity to ferroptosis could be predicted (Doll et al. 2019; Shimada et al. 2016b). Moreover, the resistance to ferroptosis in considerable tumor cell lines displays a positive correlation with the expression of FSP1, and such a correlation is not determined by the level of intracellular glutathione, the activity of GPX4, the expression of ACSL4, or the content of oxidizable fatty acids. FSP1 acts as an NADH-dependent coenzyme Q oxidoreductase inhibitor in vitro, which is capable of inhibiting ferroptosis by reducing coenzyme Q.
Other regulatory pathways of ferroptosis
Some other metabolic pathways are capable of regulating the occurrence of ferroptosis in cells as well.
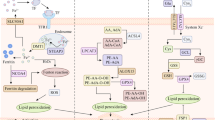
Under oxidative stresses, the sulfur transfer pathway is capable of converting methionine to cystine and subsequently synthesizing GSH to help glutathione peroxidase restrict the production of ROS, avoid oxidative damage, and suppress ferroptosis from cells (McBean 2012). Voltage-dependent anion channel (VDACs) can transport ions and metabolites. Yagoda et al. (2007) reported that RNA interference-mediated knockdown of VDAC2 and VDAC3 leads to resistance to erastin, and erastin can act on VDACs to alter the permeability of the outer membrane of mitochondria, thereby resulting in mitochondrial dysfunction and the release of oxidizing substances, which eventually causes oxidative death. SAT1, the transcriptional target of P53, has been reported as another vital rate-limiting enzyme in polyamine catabolism. The activation of SAT1 can induce the production of lipid peroxide and thus promote ferroptosis, and it exhibits a close association with the expression level of arachidonic acid lipoxygenase 15 (ALOX-15). The participation of SAT1 in the regulation of ferroptosis is done via the P53-SAT1-ALOX15 pathway (Li et al. 2020). According to Mao et al. (2018), the long non-coding RNA (lncRNAs) P53RRA located in the cytoplasm can induce ferroptosis by activating P53 pathway and impacting the transcription of multiple metabolic genes. P53RRA can enhance the growth inhibition induced by eastin. P53RRA can interact with Ras-GTPase activated protein binding to protein 1 (G3BP1) for the replacement of P53 in the G3BP1 complex into the cytoplasm and the regulation of the expressions of P53 target genes in the cytoplasm, thereby elevating the retention rate of P53 in the nucleus and promoting ferroptosis. Chromatin modification-regulated lncRNA P53RRA inhibits ferroptosis regulatory genes by interacting with G3BP1 in a P53-dependent manner. According to other articles, P62-Keap1-NRF232 (Manz et al. 2016), glutamine metabolic pathway (Gao et al. 2015), and ATG5-ATG7-NCOA433 (Hou et al. 2016) are reported to be capable of regulating ferroptosis by regulating the intracellular iron concentration and the formation of ROS. Moreover, Yang et al. found that TAZ, an effector of Hippo pathway, is capable of affecting cell density and lipid ROS synthesis via TAZ-EMP1-NOX4 pathway, thereby regulating the occurrence of ferroptosis (Yang et al. 2019) (Fig. 1).

Regulatory pathways of ferroptosis: the regulatory pathways of ferroptosis, which can fall to four types below: 1. (Shown in blue arrows and boxes) The pathways related to oxidative stress (e.g., inhibition of Xc- system, sulfur transfer pathway, MVA pathway, glutamine pathway, and P53-related regulatory pathway). 2. (Shown in yellow arrows and boxes) The regulatory mechanisms of iron metabolism (e.g., NCOA4 and IREB2 pathways regulating ferritin metabolism) and the regulatory pathways of P62-Keap1-NRF2 and HSPB1 can influence iron. 3. (Shown in orange arrows and boxes) The pathways related to lipid metabolism (e.g., P53-SAT1-ALOX15, ACSL4, LPCAT3, and FAR1-ether lipids-TMEM189 axis) regulate ferroptosis by controlling the production of lipid peroxides. 4. (Shown in pink arrows and boxes) FSP1-CoQ10-NAD (P) H pathway, acting as an independent parallel system, cooperates with GPX4. In addition, erastin regulates ferroptosis via the VDACs pathway, while TAZ, the effector of the Hippo pathway, regulates ferroptosis via the TAZ-EMP1-NOX4 pathway.
Relationship between ferroptosis and lung cancer
Although ferroptosis in lung cancer has not been extensively studied, the high levels of reactive oxygen species and lipid peroxides in lung cancer tissues suggest that ferroptosis is very likely to occur in lung cancer tissues. One previous research demonstrated the erianin-induced ferroptotic cell death in lung cancer cells through affecting Ca2+/CaM signaling (Chen et al. 2020).
The association between gene loci and ferroptosis
Carcinogenic KRAS refers to one of the most common mutated oncogenes in lung cancer. RAS mutations occur in nearly 30% of lung cancer cases and show a significant correlation with poor prognosis (Cox et al. 2014; Román et al. 2018). When RAS mutation promotes cell transformation and proliferation by synthesizing ROS, it can also cause oxidative stress to kill cells. It is unclear whether EGFR mutations in epidermal growth factor cause ROS and oxidative stress in lung cancer. In glioma cells, however, EGFR mutations can synthesize ROS, and EGFR can act as an upstream regulatory molecule of RAS, which is probably achieved by activating RAS to synthesize ROS. Considerable ROS production will promote ferroptosis in cells. P53 gene is capable of suppressing the growth of tumor cells by cell cycle arrest and apoptosis induction. Furthermore, according to the recent articles (Jiang et al. 2015), P53 gene is capable of suppressing the growth of tumor cells by inducing ferroptosis, which demonstrates that ferroptosis critically regulates the growth of tumor cells. Furthermore, Wang et al. reported that (Wang et al. 2019a) LSH regulates the expression of ELAVL1 via P53 signal pathway; however, ELAVL1 can be integrated with LINC00336 to upregulate the expression of LINC00336 via a stable post-transcriptional level, and then, it can competitively inhibit endogenous RNA and the occurrence of ferroptosis in lung cancer.
Signal transducer and activator of transcription 3 (STAT3) critically regulates mitochondrial functions and the cell metabolism. STAT3 is primarily activated in several cancers (e.g., lung cancer), and it regulates physiological regulation systems (e.g., cell growth, differentiation, senescence, and apoptosis) (Lee and Jeong 2019). Most NSCLC have persistent STAT3 activation and regulate vital cancer markers (e.g., cell proliferation, cell differentiation, and evasion of anti-tumor immunity) (Mohrherr et al. 2020). The constitutive activation of STAT3 and P38 shows a close association with the increase of proliferation and metastasis of NSCLC cells. Inhibition of STAT3 and P38 can reduce the metastatic potential of NSCLC (Nyiramana et al. 2020). Several articles (Feng et al. 2020) indicated that the transcriptional activity of STAT3 can regulate some genes associated with mitochondrial respiration, ROS production, and cell death, while considerable ROS production will induce cell ferroptosis, which is of high importance for inhibiting lung cancer.
The relationship between lung environment and ferroptosis
In comparison with other tissues, lung tissue has long been exposed to high oxygen content. In such special scenario, lung cancer cell lines achieved the improved antioxidant capacity of cells by upregulating the expression of SystemXc- (Ji et al. 2018). Moreover, cysteine desulfurase (NFS-1) exhibits high expression in lung cancer tissues, and existing articles proved that NFS-1 can effectively alleviate hyperoxia-induced ferroptosis in lung cancer cells (Alvarez et al. 2017), demonstrating that lung cancer cells are likely to have a positive choice for NFS-1 to avoid ferroptosis in lung hyperoxia environment.
The relationship between iron metabolism and lung cancer
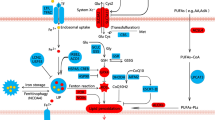
It was reported that the imbalance of iron metabolism has a close relationship to the occurrence and development of lung cancer. The expression of interleukin-6 (IL-6) is upregulated in lung cancer patients, which is related to the occurrence of lung cancer and poor prognosis of patients. By upregulating ferritin, the outflow of iron in cells can be reduced, and the accumulation of iron in cells can be induced. The accumulation of iron will induce ferroptosis. It was reported that the expressions of TFR1 and ferritin were elevated in 88% and 62% of NSCLC patients, respectively. Elevated levels of serum ferritin were identified in patients with NSCLC and small cell lung cancer (SCLC) (Kuang 2019). In NSCLC, the activation of EGFR showed a positive correlation with membrane TFR1 expression and iron level (Wang et al. 2016), which demonstrated that ferroptosis may occur in NSCLC via iron metabolism pathways (Fig. 2).

Relationships between ferroptosis and NSCLC: the possible regulatory pathways of ferroptosis in NSCLC, including links with P53, iron metabolism, Ca2+/CaM signaling, and STAT3.
Therapeutic strategies targeting ferroptosis in NSCLC
The emerging evidence shows that ferroptosis influences an increasing number of oncogenic pathways. For example, P53 regulation promotes tumor cell ferroptosis but also decreases tumor metastases to blood, lung, and liver. Viswanathan et al. affirmed the effect of ferroptosis to drug-resistant phenomenon and cancer immunotherapeutic efficacy (Viswanathan et al. 2017; Wang et al. 2019b; Zheng et al. 2017). Since lung cancer is the most common malignant tumor worldwide, and due to the high levels of reactive oxygen species and lipid peroxides in lung cancer tissues, it may be a prospective strategy for NSCLC through targeting ferroptosis (Table 3).
Ferroptosis induced by small molecules
We consistently consider that small molecules, such as many common ferroptosis inducers (Table 2), are the most likely method for inducing ferroptosis in the clinic. Erastin, a specific ferroptosis-inducing compound, is capable of upregulating the expression of heat shock factor 1 (HSF1)-dependent HSPB1 in cancer cells. The knockout of HSF1 and HSPB1 improves the ability of erastin to induce ferroptosis, while heat shock pretreatment and overexpressed HSPB1 inhibit erastin-induced ferroptosis. Inhibition of HSF1-HSPB1 pathway and phosphorylation of HSPB1 can improve the anticancer activity of erastin in human/mouse transplanted tumor model. Knockout of the mentioned genes in cancer cells may facilitate ferroptosis and improve anticancer activity (Shen et al. 2018). Many erastin derivatives such as piperazine erastin and imidazole ketone erastin (IKE) were designed to solve the disadvantages of erastin (e.g., poor water-solubility and unstable metabolism in vivo). For example, a research in the SUDHL6 xenograft mouse model showed that IKE was successfully used to slow tumor growth of diffuse large B cell lymphoma (DLBCL) (Zhang et al. 2019a), which suggested the potential of erastin and erastin derivatives to treat cancer and its possibility to guide the NSCLC treatment. Eaton et al. (2020) found that nitroisoxazole-containing compound ML210 can kill a therapy-resistant cancer cell as GPX4 inhibitors; this revealed a possibility of treating NSCLC with small molecule.
Another ferroptosis inducer is erianin, and it is reported to exert anti-tumor effects on various cancers. Erianin can induce the ferroptosis of lung cancer cells, promote cycle arrest of lung cancer cells in G2/M phase, and inhibit lung cancer cells from being migrated. After erianin acts on lung cancer cells, Ca2+/CaM signal pathway is activated, thereby inducing ferroptosis in lung cancer cells and exerting an anti-tumor role. Thus, erianin is likely to be a novel ferroptosis inducer and act as a potential compound to treat NSCLC (Chen et al. 2020).
Tyrosine kinase inhibitor (TKI) and specific antibodies are common small molecule medicine in clinic, which can inhibit the proliferation of malignant tumors by blocking the signal transduction of EGFR, HER-2, etc. It has been reported that erlotinib can increase the oxidative stress of head and neck squamous cell carcinoma by acting on EGFR (Orcutt et al. 2011), demonstrating that TKI can induce ferroptosis in cells via oxidative stress. Sorafenib induces ferroptosis in tumor cells by suppressing SystemXc- (Lachaier et al. 2014). It is therefore indicated that TKI drugs have the potential in inducing ferroptosis in lung cancer cells, but further research and verification are required.
Potential gene therapy
FSP1 can act as a biomarker of ferroptosis resistance in numerous cancers, and it is an effective ferroptosis inhibitor, which works jointly with the typical glutathione-dependent GPX4 pathway. As proved by animal experiments, it exerts the mentioned effect in lung cancer as well. The expression of FSP1 is critical to assess the efficacy of ferroptosis-inducing drugs in lung cancer, which demonstrates the potential of FSP1 inhibitors to address ferroptosis resistance in lung cancer; besides, it is expected to use ferroptosis-inducing drugs to treat NSCLC in the future (Doll et al. 2019).
The new role of long non-coding RNA (lncRNAs) in cancer has aroused huge attention, whereas many of its specific roles remain unclear. Long non-coding RNA P53RRA plays a role in lung cancer, and it is an epigenetic regulator promoting ferroptosis (Mao et al. 2018). LncRNAs participates in the formation and progress of NSCLC by mediating ferroptosis. After NSCLC underwent RNA sequencing, it was reported that SLC7A11 could be downregulated by XAV939, the target gene of lncRNAs, and could inhibit the development of NSCLC via ferroptosis pathway (Yu et al. 2019). It is therefore suggested that lncRNAs acts as a regulator of ferroptosis and can be an effective target for NSCLC therapy.
TMEM189, a lastly recognized gene that induces vinyl-ether double bond into alkyl-ether liposomes to produce plasmonogens, can eliminate ferroptosis induced by the FAR1-alkyl-ether lipids axis. The functional consequences required for TMEM189 to produce plasma in a variety of pathological conditions or diseases are unknown. In contrast to the fatality caused by GPX4 deficiency, both loss of TMEM189 mice and wild-type mice survived normally (Werner et al. 2020). Because TMEM189 is enriched in human cancers, and a high TMEM189 expression causes a poor clinical benefit, TMEM189 is suggested to be a very prospective therapeutic target than GPX4 for activating ferroptosis in NSCLC (Cui et al. 2021). Although there is no concrete example of ferroptosis in gene-targeted NSCLC at present, with further research, gene-targeted therapy of NSCLC has certain potential.
Chemotherapy combined with ferroptosis inducers
Targeted therapy is not sensitive to lung cancer cells carrying wild-type EGFR, and platinum-based chemotherapy such as cisplatin (DDP) remains the standard treatment for non-targeted NSCLC. Chemotherapy resistance acts as a major challenge to treat advanced lung cancer. In the previous study, ferroptosis may enhance the anticancer effect of cisplatin in cancer cells (Roh et al. 2017), suggesting that ferroptosis inducers can be adopted to enhance the role of conventional anticancer drugs in lung cancer treatment. Subsequent research found that cisplatin can induce ferroptosis in A549 and HCT116 cells (Guo et al. 2018). As revealed from existing articles, ginkgetin derived from ginkgo biloba leaves promotes ferroptosis in NSCLC under the cooperation with cisplatin, which may be a promising way to enhance the anticancer function of platinum chemotherapeutic drugs (e.g., cisplatin) and is expected to reduce the resistance to chemotherapeutic drugs (Lou et al. 2021). Hangauer et al (Hangauer et al. 2017) reported on chemotherapy persistent cells (including lung cancer cells) that are no longer sensitive to traditional chemotherapy that induces apoptosis but still sensitive to substances that inhibit GPX4 and thus induce ferroptosis. In chemotherapy-resistant cancer cells, Nrf2 targeting genes have the downregulated expressions, while intracellular NF2 and Hippo pathways inhibited can induce ferroptosis Moreover, the levels of glutathione and NADPH in peripheral cells were lowered, and GPX4 inhibitors can cause the death of drug-resistant cancer cells (Jiang et al. 2020). However, co-treatment with multiple drugs could lead to increased toxicity and side effects and thus may compromise the safety and treatment efficacy of patients. Hangauer et al (2017) found that pre- or post-treatment with GPX4 inhibitors, rather than co-treatment, may be enough to deplete the continuous pool of cells that survive targeted therapy or chemotherapy. This staggered therapy strategy may be clinically beneficial in maximizing efficacy while minimizing the potential increase in toxicity associated with simultaneous multi-drug treatment.
Nanoparticle inducing ferroptosis
Ma et al. proposed a sequential drug administration strategy to enhance the sensitivity to cisplatin by exploiting intracellular iron ions released by iron oxide nanoparticles (IO NPs). They consider that cisplatin (IV) prodrug is capable of inducing tumor site-specific ROS production by releasing cisplatin and IO NPs to improve the anticancer activity. However, cisplatin (IV) prodrug can minimize the systemic toxicity, and nanoparticles enter cells through the endocytosis. This helps reverse the resistance of cells to cisplatin (Pa et al. 2017). FePt/MoS2 nanocomposites refer to a novel ferroptosis drug, which causes ferroptosis by inducing Fenton reaction to synthesize ROS. Moreover, as supported by cytosine guanine oligodeoxynucleotides and combined with the systematic checkpoint of cytotoxic T-lymphocyte associated protein 4 (CTLA4) antibody to block immunotherapy, primary tumors are effectively eliminated and destructed, and their metastases are inhibited. In addition, the mentioned strategy exhibits a long-term immune memory function, and it can inhibit tumor recurrence, as verified by animal experiments (Zhang et al. 2019b). It may act as a novel strategy to treat NSCLC. Zhou et al. reported an activatable system to generate singlet oxygen (1O2), thereby inducing ROS and 1O2 to be formed by the reaction of (LAHP) with Fe2+. Such a system has been employed in specific tumor therapies in acidic pH environment (Zhou et al. 2017). SRF@FeIIITA (SFT), a novel type of nanoparticles, critically impacts the inhibition of tumor progression. Methylene blue (MB) was loaded into SRF by depositing tannic acid (TA) and Fe3+, on SFT nanocrystals, and photodynamic therapy (PDT) was effectively integrated with ferroptosis (Liu et al. 2018), which may be employed for treating NSCLC cells as well.
Radiotherapy
Radiotherapy is referred to as the cornerstone of numerous cancer treatments. It is used to treat end-stage lung cancer, where radiation resistance was one of the main causes of radiotherapy failure. Ionizing radiation (IR) can induce the expressions of ROS and ACSL4, which causes lipid peroxidation and ferroptosis to increase. FINs, an inducer of ferroptosis, can enhance the sensitivity of radiation-resistant cancer cells and xenotransplanted tumors to IR after inactivating SLC7A11 or GPX4. Animal experiments indicated a similar radiosensitization effect in lung cancer patients with Keap1 mutant xenograft (PDX) exerted by salicylazosulfapyridine treatment (Lei et al. 2020), demonstrating that FINs combined with IR can improve the effect of radiotherapy, which may serve as a feasible treatment for NSCLC cells resistant to end-stage radiotherapy (Table 3). Another means to increase the radiosensitivity in NSCLC is done via the use of ascorbic acid, which induces cell death via the iron-dependent mechanism (Schoenfeld et al. 2017).
Development and prospect
Ferroptosis, a novel form of cell death, was initially detected in 2012. Ever since its discovery, the mechanisms and signal pathways related to ferroptosis have been extensively studied. Though preliminary insights into the mechanism and targeting effect of ferroptosis have been gained, more specific studies on how to regulate ferroptosis should be conducted before it can be applied to clinical treatment. With an elucidated understanding of the mechanism of ferroptosis, a growing number of researchers are beginning to focus on the emerging field of ferroptosis inducers, which appears very promising in the design and materials used for lung cancer treatment.
Moreover, the treatment for KRAS, a type of mutation that commonly occurs in the case of NSCLC, has not been very successful. Since ferroptosis was initially proposed in the study on small molecular drugs that cause selective death of carcinogenic RAS, it is possible to induce ferroptosis as a potential treatment for KRAS.
This study discussed the recent progress in the mechanism of ferroptosis and how it could affect NSCLC. With a more in-depth understanding of ferroptosis, it may be a novel treatment for NSCLC.
Abbreviations
- NSCLC :
-
non-small cell lung cancer
- ROS :
-
reactive oxygen species
- L-OOH :
-
lipid hydroperoxides
- ACSL4 :
-
acyl-CoA synthetase long-chain family member 4
- PUFAs :
-
polyunsaturated fatty acids
- FSP1 :
-
ferroptosis suppressing protein 1
- AIFM2 :
-
apoptosis inducing factor mitochondria 2
- GSH :
-
glutathione
- GPX 4 :
-
glutathione peroxidase 4
- SOCS1 :
-
suppressor of cytokine signaling 1
- EGFR :
-
epidermal growth factor receptor
- TF :
-
transferrin
- TFR1 :
-
transferrin receptor 1
- FTH :
-
ferritin heavy chain
- STEAP3 :
-
six-transmembrane epithelial antigen of prostate 3
- FTL :
-
ferritin light chain
- DMT1 :
-
divalent metal transporter 1
- HSPB1 :
-
heat shock protein B1
- IREB2 :
-
iron response element-binding protein 2
- NCOA4 :
-
nuclear receptor coactivator 4 protein
- HO-1 :
-
heme oxygenase-1
- ISCU :
-
iron–sulfur cluster assembly enzyme
- DHAN :
-
dihydroartemisinin
- LPCAT3 :
-
lysophosphatidylcholine acyltransferase 3
- PEBP1 :
-
phosphatidylethanolamine binding protein 1
- SFA :
-
saturated fatty acid
- VDACs :
-
voltage-dependent anion channel
- lncRNAs :
-
long non-coding RNA
- G3BP1 :
-
Ras-GTPase activated protein binding to protein 1
- STAT3 :
-
signal transducer and activator of transcription 3
- NFS-1 :
-
cysteine desulfurase
- SCLC :
-
small cell lung cancer
- IO NPs :
-
iron oxide nanoparticles
- CTLA4 :
-
cytotoxic T-lymphocyte-associated protein 4
- IKE :
-
imidazole ketone erastin
- DLBCL :
-
diffuse large B cell lymphoma
- TKI :
-
tyrosine kinase inhibitor
- IR :
-
ionizing radiation
References
Alvarez SW, Sviderskiy VO, Terzi EM et al (2017) NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 551(7682):639–643
Anderson GJ (2017) Frazer DM Current understanding of iron homeostasis. Am J Clin Nutr 106(Suppl 6):1559S–1566S
Bersuker K, Hendricks JM, Li Z et al (2019) The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575(7784):688–692
Chang L-C, Chiang S-K, Chen S-E et al (2018) Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett 416124-137
Chen L, Li X, Liu L et al (2015) Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-γ-lyase function. Oncol Rep 33(3):1465–1474
Chen P, Wu Q, Feng J et al (2020) Erianin, a novel dibenzyl compound in Dendrobium extract, inhibits lung cancer cell growth and migration via calcium/calmodulin-dependent ferroptosis. Signal Transduct Target Ther 5(1):51
Cox AD, Fesik SW, Kimmelman AC et al (2014) Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov 13(11):828–851
Cui W, Liu D, Gu W et al (2021) Peroxisome-driven ether-linked phospholipids biosynthesis is essential for ferroptosis. Cell Death Differ 28(8):2536–2551
Dixon SJ, Lemberg KM, Lamprecht MR et al (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 149(5):1060–1072
Dixon SJ, Patel DN, Welsch M et al (2014) Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3e02523
Dixon SJ, Winter GE, Musavi LS et al (2015) Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol 10(7):1604–1609
Doll S, Proneth B, Tyurina YY et al (2017) ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13(1):91–98
Doll S, Freitas FP, Shah R et al (2019) FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575(7784):693–698
Dolma S, Lessnick SL, Hahn WC et al (2003) Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 3(3):285–296
Du J, Wang T, Li Y et al (2019) DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic Biol Med 131356-369
Eaton JK, Furst L, Ruberto RA et al (2020) Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat Chem Biol 16(5):497–506
Feng J, Jiang W, Liu Y et al (2020) Blocking STAT3 by pyrvinium pamoate causes metabolic lethality in KRAS-mutant lung cancer. Biochem Pharmacol 177113960
Friedmann Angeli JP, Schneider M, Proneth B et al (2014) Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 16(12):1180–1191
Gammella E, Recalcati S, Rybinska I et al (2015) Iron-induced damage in cardiomyopathy: oxidative-dependent and independent mechanisms. Oxidative Med Cell Longev 2015230182
Gao M, Monian P (2015) Jiang X Metabolism and iron signaling in ferroptotic cell death. Oncotarget 6(34):35145–35146
Gao M, Monian P, Quadri N et al (2015) Glutaminolysis and transferrin regulate ferroptosis. Mol Cell 59(2):298–308
Gaschler MM, Andia AA, Liu H et al (2018) FINO initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol 14(5):507–515
Guo J, Xu B, Han Q et al (2018) Ferroptosis: a novel anti-tumor action for cisplatin. Cancer Res Treat 50(2):445–460
Hangauer MJ, Viswanathan VS, Ryan MJ et al (2017) Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551(7679):247–250
Hassannia B, Wiernicki B, Ingold I et al (2018) Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J Clin Invest 128(8):3341–3355
Hou W, Xie Y, Song X et al (2016) Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12(8):1425–1428
Ji X, Qian J, Rahman SMJ et al (2018) xCT (SLC7A11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene. 37(36):5007–5019
Jiang L, Kon N, Li T et al (2015) Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 520(7545):57–62
Jiang M, Qiao M, Zhao C et al (2020) Targeting ferroptosis for cancer therapy: exploring novel strategies from its mechanisms and role in cancers. Transl Lung Cancer Res 9(4):1569–1584
Kagan VE, Mao G, Qu F et al (2017) Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13(1):81–90
Kang R (2017) Tang D Autophagy and ferroptosis - what’s the connection? Curr Pathobiol Rep 5(2):153–159
Kazan HH, Urfali-Mamatoglu C (2017) Gunduz U Iron metabolism and drug resistance in cancer. Biometals. 30(5):629–641
Kuang Y (2019) Wang Q Iron and lung cancer. Cancer Lett 46456-61
Lachaier E, Louandre C, Godin C et al (2014) Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res 34(11):6417–6422
Lee H, Jeong AJ (2019) Ye S-K Highlighted STAT3 as a potential drug target for cancer therapy. BMB Rep 52(7):415–423
Lei G, Zhang Y, Koppula P et al (2020) The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res 30(2):146–162
Li J, Cao F, Yin H-L et al (2020) Ferroptosis: past, present and future. Cell Death Dis 11(2):88
Lin C-H, Lin P-P, Lin C-Y et al (2016) Decreased mRNA expression for the two subunits of system xc(-), SLC3A2 and SLC7A11, in WBC in patients with schizophrenia: evidence in support of the hypo-glutamatergic hypothesis of schizophrenia. J Psychiatr Res 7258-63
Liu T, Liu W, Zhang M et al (2018) Ferrous-supply-regeneration nanoengineering for cancer-cell-specific ferroptosis in combination with imaging-guided photodynamic therapy. ACS Nano 12(12):12181–12192
Lou J-S, Zhao L-P, Huang Z-H et al (2021) Ginkgetin derived from Ginkgo biloba leaves enhances the therapeutic effect of cisplatin via ferroptosis-mediated disruption of the Nrf2/HO-1 axis in EGFR wild-type non-small-cell lung cancer. Phytomedicine 80153370
Louandre C, Ezzoukhry Z, Godin C et al (2013) Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int J Cancer 133(7):1732–1742
Louandre C, Marcq I, Bouhlal H et al (2015) The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett 356(2 Pt B):971–977
Manz DH, Blanchette NL, Paul BT et al (2016) Iron and cancer: recent insights. Ann N Y Acad Sci 1368(1):149–161
Mao C, Wang X, Liu Y et al (2018) A G3BP1-interacting lncRNA promotes ferroptosis and apoptosis in cancer via nuclear sequestration of p53. Cancer Res 78(13):3484–3496
McBean GJ The transsulfuration pathway: a source of cysteine for glutathione in astrocytes. Amino Acids42(1),199-205 (2012).
Mohrherr J, Uras IZ, Moll HP et al (2020) STAT3: versatile functions in non-small cell lung cancer. Cancers (Basel) 12(5)
Nyiramana MM, Cho SB, Kim E-J et al (2020) Sea hare hydrolysate-induced reduction of human non-small cell lung cancer cell growth through regulation of macrophage polarization and non-apoptotic regulated cell death pathways. Cancers (Basel) 12(3)
Orcutt KP, Parsons AD, Sibenaller ZA et al (2011) Erlotinib-mediated inhibition of EGFR signaling induces metabolic oxidative stress through NOX4. Cancer Res 71(11):3932–3940
Pa M, Xiao H, Yu C et al (2017) Enhanced cisplatin chemotherapy by iron oxide nanocarrier-mediated generation of highly toxic reactive oxygen species. Nano Lett 17(2):928–937
Poursaitidis I, Wang X, Crighton T et al (2017) Oncogene-selective sensitivity to synchronous cell death following modulation of the amino acid nutrient cystine. Cell Rep 18(11):2547–2556
Roh J-L, Kim EH, Jang H et al (2017) Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol 11254-262
Román M, Baraibar I, López I et al (2018) KRAS oncogene in non-small cell lung cancer: clinical perspectives on the treatment of an old target. Mol Cancer 17(1):33
Saint-Germain E, Mignacca L, Vernier M et al (2017) SOCS1 regulates senescence and ferroptosis by modulating the expression of p53 target genes. Aging (Albany NY) 9(10):2137–2162
Schoenfeld JD, Sibenaller ZA, Mapuskar KA et al (2017) O and HO-mediated disruption of Fe metabolism causes the differential susceptibility of NSCLC and GBM cancer cells to pharmacological ascorbate. Cancer Cell 31(4)
Shen Z, Song J, Yung BC et al (2018) Emerging strategies of cancer therapy based on ferroptosis. Adv Mater 30(12):e1704007
Shimada K, Hayano M, Pagano NC et al (2016a) Cell-Line selectivity improves the predictive power of pharmacogenomic analyses and helps identify NADPH as biomarker for ferroptosis sensitivity. Cell Chem Biol 23(2):225–235
Shimada K, Skouta R, Kaplan A et al (2016b) Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol 12(7):497–503
Sun X, Ou Z, Chen R et al (2016) Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63(1):173–184
Vanden Berghe T, Linkermann A, Jouan-Lanhouet S et al (2014) Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 15(2):135–147
Viswanathan VS, Ryan MJ, Dhruv HD et al (2017) Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547(7664):453–457
Wang B, Zhang J, Song F et al (2016) EGFR regulates iron homeostasis to promote cancer growth through redistribution of transferrin receptor 1. Cancer Lett 381(2):331–340
Wang M, Mao C, Ouyang L et al (2019a) Long noncoding RNA LINC00336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell Death Differ 26(11):2329–2343
Wang W, Green M, Choi JE et al (2019b) CD8 T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569(7755):270–274
Wenzel SE, Tyurina YY, Zhao J et al (2017) PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell 171(3)
Werner ER, Keller MA, Sailer S et al (2020) The gene encodes plasmanylethanolamine desaturase which introduces the characteristic vinyl ether double bond into plasmalogens. Proc Natl Acad Sci U S A 117(14):7792–7798
Yagoda N, von Rechenberg M, Zaganjor E et al (2007) RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 447(7146):864–868
Yang WS, SriRamaratnam R, Welsch ME et al (2014) Regulation of ferroptotic cancer cell death by GPX4. Cell 156(1-2):317–331
Yang WS, Kim KJ, Gaschler MM et al (2016) Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 113(34):E4966–E4975
Yang W-H, Ding C-KC, Sun T et al (2019) The hippo pathway effector TAZ regulates ferroptosis in renal cell carcinoma. Cell Rep 28(10)
Yu H, Han Z, Xu Z et al (2019) RNA sequencing uncovers the key long non-coding RNAs and potential molecular mechanism contributing to XAV939-mediated inhibition of non-small cell lung cancer. Oncol Lett 17(6):4994–5004
Yuan H, Li X, Zhang X et al (2016) Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun 478(3):1338–1343
Zhang Y, Tan H, Daniels JD et al (2019a) Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell. Chem Biol 26(5)
Zhang D, Cui P, Dai Z et al (2019b) Tumor microenvironment responsive FePt/MoS nanocomposites with chemotherapy and photothermal therapy for enhancing cancer immunotherapy. Nanoscale 11(42):19912–19922
Zheng D-W, Lei Q, Zhu J-Y et al (2017) Switching apoptosis to ferroptosis: metal-organic network for high-efficiency anticancer therapy. Nano Lett 17(1):284–291
Zhou Z, Song J, Tian R et al (2017) Activatable singlet oxygen generation from lipid hydroperoxide nanoparticles for cancer therapy. Angew Chem Int Ed Eng 56(23):6492–6496
Zou Y, Henry WS, Ricq EL et al (2020) Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 585(7826):603–608
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
LM Z and YF Z conceived and designed the research. YF Z, RX G, and J L found some paper, YF Z wrote the paper, and LM Z revised the manuscript. All authors approved of the manuscript. The authors declare that all data were generated in-house and that no paper mill was used.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, Y., Guo, R., Li, J. et al. Research progress on the occurrence and therapeutic mechanism of ferroptosis in NSCLC. Naunyn-Schmiedeberg's Arch Pharmacol 395, 1–12 (2022). https://doi.org/10.1007/s00210-021-02178-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-021-02178-z