
Abstract
Learning and memory deficits appear in chronic diabetes and valproic acid has been proved to be beneficial in neurodegenerative diseases. Hence, the current study investigated the effectiveness of chronic valproate treatment for diabetes-induced memory impairment and increased levels of hippocampal apoptotic caspases. This study was conducted in adult male C57B15/J mice. Diabetes, which was induced by alloxan (150 mg/kg; i.p.), was confirmed when fasting blood sugar (FBS) was > 200 mg/dl. Sodium valproate (100 mg/kg; i.p.) was administrated to the diabetic and non-diabetic groups, every 72 h for 2 months. Next, all groups were evaluated for memory performance using the radial maze and shuttle box. After FBS measurement, animals were killed and the hippocampus was extracted and prepared for ELISA to assess caspase levels. Diabetic animals had significantly high FBS and memory impairment 2 months after the alloxan injection. Hippocampal levels of caspases 3, 6, and 8 were significantly higher in the diabetic group than in the control group. However, valproate treatment of diabetic animals significantly improved memory performance in both the radial maze and shuttle box and reduced the elevated levels of hippocampal apoptotic caspases, in comparison with diabetic animals. Chronic administration of valproate seems to have beneficial effects on diabetic neuropathy.
Similar content being viewed by others
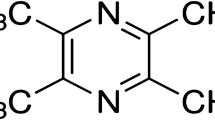

Avoid common mistakes on your manuscript.
Introduction
Uncontrolled hyperglycemia and diabetes induce vascular destruction and damage to the peripheral and central nervous system (Said 2007; Said et al. 2003). Increased formation of reactive oxygen species (ROS) as consequence of chronic hyperglycemia is associated with protein oxidation, DNA damage, and membrane lipid peroxidation, therefore resulting in increased neuronal cell death and damage to the brain areas such as the hippocampus (Nardin et al. 2016), which is essential for learning and memory. Research studies have reported learning and memory deficits and cognitive impairment following diabetes (Deng et al. 2013; Jia et al. 2014).
It has been established that oxidative stress and ROS production change mitochondrial functions and stimulate pathways of apoptotic caspases (Russell et al. 2002). Mitochondrial pathways of apoptosis begin with the release of cytochrome C from mitochondria, which results in an apoptosomal or caspase-activating complex (Talanian et al. 1997). Apoptotic caspases consist of two groups: (1) initiator caspases including caspases 2, 8, 9, and 10; (2) executioner caspases including caspases 3, 6, and 7 (Elmore 2007). Involvement of caspases in diabetic neuropathy has been described previously. It was observed in neuropathic models of diabetes that oxidative damage to the neurons leads to increased levels of caspases 3, 8, and 9 in association with other apoptotic factors (Erbas et al. 2016; Han et al. 2014; Schmeichel et al. 2003).
Valproic acid is a short-chain fatty acid mainly used as an antiepileptic drug. Valproate works as a direct inhibitor of histone deacetylases (HDAC), causing histone hyperacetylation (Phiel et al. 2001). Usually, hyperacetylation is coupled with transcriptional activity, while hypoacetylation is coupled with repression. In neurodegenerative diseases, the acetylation homeostasis changes toward deacetylation (Saha and Pahan 2006). As an HDAC inhibitor, valproate is reported to be neuroprotective and neuroregenerative. Neuroprotective effects of valproate have been reported in neurodegenerative and neuropathic conditions in both the peripheral and central nervous system (Silva et al. 2018; Zhang et al. 2012a). Valproate treatment reduces caspase 3 activation in pathological conditions (Luo et al. 2014; Zhang et al. 2012a). Valproate attenuates memory deficits in an animal model of Alzheimer’s disease (Xuan et al. 2015). In addition, beneficial effects of valproate treatment in diabetes-induced pathology have been demonstrated (Khan et al. 2015; Rabadiya et al. 2017).
In light of the learning and memory impairment produced by chronic diabetes, the important role of apoptotic caspases in the diabetic neuropathy, and the role of valproic acid in repairing neuronal damages and attenuating caspase activity (Zhang et al. 2012a), we evaluated the efficacy of chronic valproate treatment in improving spatial and avoidance memory impairment and aberrant hippocampal levels of apoptotic caspases, in an alloxan-induced model of diabetes.
Materials and methods
Animals
The current study was conducted in male C57B15/J mice (n = 24; age = 8–9 weeks, at the start of the study). Mice were housed under a 12-h light/dark cycle in a controlled temperature (22 ± 2 °C) environment. Food and water were available ad libitum. All research and animal care procedures were performed according to the Guide for the Care and Use of Laboratory Animals (8th edition; National Academies Press; 2011) and approved by the Review Board and Ethics Committee of Arak University of Medical Sciences (agreement number of the ethics committee for this study is “IR.ARAKMU.REC.1395.200”). All efforts were made to minimize the number of animals used and their suffering.
Chemicals
Alloxan (Sigma-Aldrich, USA) was dissolved in saline (0.9%) and injected (150 mg/kg; i.p.). An injectable form of sodium valproate (CONVULEX, Gerot Pharma., Austria) was used (100 mg/kg; i.p.). Fasting blood sugar (FBS) levels were measured using a glucose meter (Accu-Chek, Roche, Germany). Caspase 3, 6, 8, and 9 ELISA kits (all from Abnova, Taiwan) were used for biochemical assessments.
Alloxan diabetes model and valproate treatment
Animals were randomly assigned to four experimental groups (n = 6):
-
(1)
Diabetes group (Diabt): Diabetes was induced by administration of alloxan (150 mg/kg; i.p.) into mice that were fasted for 12 h (Akindele et al. 2015; Sezik et al. 2005). FBS was assessed at 72 h, and animals with FBS > 200 mg/dl were considered as diabetic. If FBS was < 200 mg/dl, the animal received another injection of alloxan (150 mg/kg; i.p.), and FBS was measured 72 h later.
-
(2)
Diabetes+Valproate group (Diabt+Valp): After diabetes was induced and confirmed, diabetic animals were treated with low dose of sodium valproate (100 mg/kg; i.p.), which was indicated neuroprotective (Tai et al. 2014), every 72 h for 2 months.
-
(3)
Valproate group (Valp): In this group, animals received saline (0.9%, i.p.) instead of alloxan and were then treated with sodium valproate (100 mg/kg; i.p.), every 72 h for 2 months.
-
(4)
Control group (Cnt): Animals received saline (0.9%, i.p.) instead of alloxan and were then treated with saline (0.9%, i.p.), every 72 h for 2 months.
Previous reports had been shown (Deng et al. 2013) chronic diabetes and hyperglycemia, when continued more than 1 month, will lead to neuropathologic consequences. Therefore, 2 months of diabetic situation and valproate treatment were designed to provide enough time for appearance of the central neuropathic conditions due to hyperglycemia.
Also, after these 2 months of treatment, FBS of all experimental groups were measured. Then, all animals from the experimental groups were used for the behavioral test of spatial and avoidance memory according to the schematic diagram of Fig. 1. Finally, all rats were sacrificed and the hippocampus was extracted for ELISA assay.

Schematic diagram of the experimental procedure
Spatial learning and radial arm maze
To investigate the effectiveness of valproate treatment on diabetes-induced memory impairment, spatial memory was evaluated using the radial arm maze (RAM). The RAM used in the present study consisted of eight arms (50 × 15 × 15 cm) radiating from a central platform, constructed using black Plexiglas and numbered 1 to 8. Each arm had a removable door for entrance and a well at the end for hiding a food bite. Various intra- and extra-maze visual cues were placed at the same position during the whole study. Experiments were performed daily between 8 a.m. and 11 a.m. A day before the training session, each mouse was placed in the RAM (for 5 min, no arms were baited) to habituate with the environment. During the training sessions, one mouse was placed on the octagonal central platform of the maze and allowed for 30 s to become familiar with the place. Next, all the doors were opened and the test animal was allowed to freely explore the maze for 5 min. Training was performed daily, for 5 continuous days. For the training session, access to food was limited for 2 h before each trial. The maze was cleaned with 70% ethanol after each trial. Only four out of the eight arms were baited during the training sessions and was arranged in the same configuration throughout the entire experiment and all mice were trained on this configuration. The trial ended when the mouse visited all four baited arms or after 5 min, and the time was recorded for future analysis. In addition, two types of errors were calculated:
-
1)
Working memory errors (WME): re-entering a baited arm during the same trial
-
2)
Reference memory errors (RME): entering a non-baited arm during the same trial
Avoidance memory and shuttle box
Passive avoidance test of memory was performed using the shuttle box apparatus. The shuttle box was a two-compartment, dark/lit apparatus with a steel rod grid floor, connected via a guillotine door. After the injections, passive avoidance test of memory was performed over 3 days. For adaptation to the shuttle box, each mouse was placed in the lit chamber and allowed to pass freely between the chambers for 60 s, while the door between the chambers was open. On day 1 (training), the animal was placed in the lit chamber, and after 15 s, the sliding door was opened; the time taken by the animal to enter the dark chamber was recorded (step-through latency). When the animal entered the dark chamber, the door was closed and an electrical shock (1 mA, 2 s) was delivered to the grid floor. Next, the animal was taken out of the chamber and transferred to its home cage. On days 2 and 3 (retrieval), the procedure followed on day 1 was repeated, except that no electrical stimulation was given and the time interval to enter the dark chamber was measured (retention latency). The cut-off time for entering the dark chamber was set at 60 s for all the days.
ELISA assessment of apoptotic caspases
At the end of the behavioral experiments, all animals were deeply anesthetized using diethyl ether and decapitated. The brains were removed and placed in chilled phosphate-buffered saline (pH = 7.4; 100 mM), and the right hippocampus was quickly dissected. All the samples were stored at – 80 °C. Hippocampal tissue was homogenized in phosphate buffer (pH = 7.4) containing the anti-protease cocktail (Sigma-Aldrich, USA) and centrifuged (14,000 rpm, 40 min, 1–4 °C) and the resultant supernatant was isolated. Caspase 3, 6, 8, and 9 ELISA kits (all from Abnova, Taiwan) were used according to the manufacturer’s instructions for biochemical measurements. Standard curves were prepared using the standard solutions provided in the kits. Based on the optical density of the samples and the standard curve, hippocampal levels of caspases were calculated.
Statistical analysis
Data are presented as mean ± SEM. Statistical analyses were performed using GraphPad Prism software (GraphPad Software, CA, USA). Normal distribution of data was evaluated using the Kolmogorov-Smirnov normality test. One-way or two-way ANOVA followed by Bonferroni post hoc test was used to establish comparisons between the experimental groups. Statistical significance was defined as p < 0.05.
Results
Kolmogorov-Smirnov normality test revealed normal distributions of data for FBS, spatial memory, avoidance memory, and caspase values in all the experimental groups (p > 0.05). Therefore, future analyses were continued with parametric tests.
Valproate attenuated alloxan-induced hyperglycemia
Two-way ANOVA analysis followed with Bonferroni post hoc test revealed significant elevations in the FBS level when assessed 72 h after the alloxan injection [F(3, 20) = 22.3; p < 0.001]. As demonstrated in Fig. 2, alloxan injected animals in both the Diabt and Diabt+Valp groups had a significant increase in their FBS levels 72 h after the alloxan injection, when compared to the Cnt group (p < 0.05; 315.8 ± 21.2 for Diabt group; 265.6 ± 28.3 for Diabt+Valp group; 135.3 ± 15.1 for Cnt). FBS levels in the Valp group did not show any significant difference with those in the Cnt group.

Chronic valproate administration attenuates hyperglycemia induced by alloxan. FBS is assessed 72 h and 60 days after the alloxan injection and analyzed by repeated measure 2-way ANOVA, followed by Bonferroni post hoc test. Both the Diabt and Diabt+Valp groups exhibit significant increases in the FBS level, 72 h after the alloxan injection when compared with the Cnt group. Chronic valproate administration in the Diabt+Valp group shows no significant effect on the FBS level, when compared with pre- and post-valproate treatment. Data are presented as mean ± SEM; n = 6 for all groups. *p < 0.05 when compared with the control group
Assessment of FBS 2 months after the diabetes induction revealed hyperglycemic conditions in the Diabt group. FBS levels were significantly increased in the Diabt group (376 ± 84) in comparison with the Cnt group (122 ± 7.2) (Fig. 2; p < 0.05). In addition, chronic treatment with valproate in the Diabt+Valp group did not significantly decrease the mean value of FBS when compared to 72 h after alloxan injection (p > 0.05 when comparing pre- and post-valproate treatment; Fig. 2), although decrease the mean value of FBS below the 200 mg/dl.
In addition, to follow and confirm diabetic situation at 17 days after the first FBS assessment, we repeat this measurement again. The results showed hyperglycemic profile in both the Diabt group and the Diabt+Valp group (data did not present).
Valproate ameliorated alloxan-induced memory impairment
Spatial memory
Mean values of latency, to finish all the four baited arms of the RAM, were analyzed during 5 days between four experimental groups by two-way repeated measures ANOVA followed by Bonferroni post hoc test (Fig. 3). As demonstrated in Fig. 3A, the diabetic condition had a significant effect on latency to finish all the baited arms in the Diabt group [F(4, 80) = 81.2; p < 0.001; n = 6 for all groups]. Bonferroni post-test revealed significant differences at days 1–3 between the Diabt group and the Cnt group (p < 0.05 for day 1; p < 0.001 for days 2 and 3). However, mean values of latency to finish the baited arms in the Diabt+Valp group significantly decreased when compared with the Diabt group (p < 0.05 for days 2, 3, and 5). The Diabt+Valp group showed no significant difference with the Cnt group.

Chronic valproate ameliorates hyperglycemia-induced spatial memory impairment. Repeated measures 2-way ANOVA and Bonferroni post hoc test revealed a significant increase in time to finish baited arms (a) in the Diabt group compared with the Cnt group (p < 0.001). Interestingly, in the Diabt+Valp group, this time significantly reduced when compared with the Diabt group (p < 0.05). One-way ANOVA and Bonferroni post hoc test revealed significant increase in reference memory errors (b) of the Diabt group compared with the Cnt group (p < 0.05). In the Diabt+Valp group, reference memory errors significantly reduced when compared with the Diabt group (p < 0.05). No significant differences were detected in working memory errors between experimental groups (c). Data are presented as mean ± SEM; n = 6 for all groups; *p < 0.05 compared with the control group; ***p < 0.001 compared with the control group; #p < 0.05 compared with the Diabt group
In addition, to analyze working memory errors (WME) or reference memory errors (RME), mean values of total WME or total RME during 5 days of experiment were calculated for each group, then analyzed between the experimental groups by one-way ANOVA and Bonferroni post hoc test. As demonstrated in Fig. 3b, the diabetic condition resulted in a significant increase in the numbers of reference memory errors in the Diabt group when compared with the Cnt group (p < 0.05; n = 6), whereas valproate treatment in the Diabt+Valp group significantly decreased reference memory errors in comparison with the Diabt group (p < 0.05; n = 6). However, working memory errors showed no significant differences between the experimental groups (Fig. 3c).
Avoidance memory
Latency to enter the dark box (where they received the electric shock), 24 and 48 h after training, was analyzed using two-way repeated measures ANOVA [F(2, 40) = 51.9; p < 0.001] and followed by Bonferroni post hoc test. The Cnt group (Fig. 4) displayed significantly increased latency to enter the dark box, 24 and 48 h after training (p < 0.05), indicating successful learning to avoid the unpleasant stimulus. In addition, a significant effect of the diabetic condition on learning avoidance was detected in the Diabt group, while valproate treatment in diabetic animals significantly ameliorated memory impairment and there was a significant difference between the Diabt group and the Diabt+Valp group (p < 0.05).

Chronic treatment with valproate improved hyperglycemia-induced avoidance memory defect. Latency to enter the dark chamber, 24 and 48 h after training, was analyzed by repeated measure 2-way ANOVA followed by Bonferroni post hoc test. The Diabt group shows a decreased latency to enter the dark box, 24 and 48 h after training, when compared with the Cnt group. However, chronic valproate administration in the Diabt+Valp group significantly improved this latency. Data are presented as mean ± SEM; n = 6 for all groups; *p < 0.05 compared control vs Diabt group; #p < 0.05 compared Diabt+Valp group vs Diabt group
Valproate reduced hippocampal levels of apoptosis caspases in diabetic animals
As demonstrated in Fig. 5, hippocampal levels of caspases 3, 6, and 8 were significantly elevated in the Diabt group in comparison with the Cnt group. Diabetic animals, treated for 2 months with sodium valproate (100 mg/kg), in the Diabt+Valp group showed no significant differences with those in the Cnt group. These results suggest that valproate administration modulated the increased levels of hippocampal caspases.

Evaluation of hippocampal levels of apoptotic caspases. One-way ANOVA and Bonferroni post hoc test revealed significant increase in the hippocampal levels of caspase 3 (a) (p < 0.01), caspase 6 (b) (p < 0.05), and caspase 8 (c) (p < 0.05) in the Diabt group compared with the Cnt group (p < 0.05). Interestingly, in the Diabt+Valp group, these apoptotic caspases significantly decreased as revealed by significant difference with Diabt group (p < 0.05); however, there was no significant difference when compared with the Cnt group. No significant differences were detected in caspase 9 between the experimental groups (c). Data are presented as mean ± SEM; n = 6 for all groups; *p < 0.05 and **p < 0.01 compared with the control group; #p < 0.05 and ##p < 0.01 compared with the Diabt group
The hippocampal levels of caspase 3 were significantly increased in the Diabt group (26.2 ± 1.2, n = 6) compared with the Cnt group (19.2 ± 1.1, n = 6) (p < 0.01), while the Diabt+Valp group hippocampal levels of caspase 3 were significantly reduced (21.24 ± 0.70, n = 6) and show no significant difference with the Cnt group (p < 0.05) (Fig. 5a). Hippocampal levels of caspase 6 (Fig. 5b) in the Diabt group (17.7 ± 0.8, n = 6) were compared with those of the Cnt group (15.4 ± 0.9, n = 6) and show a significant increase (p < 0.05). On the other hand, in the Diabt+Valp group, a significant decrease in the hippocampal levels of caspase 6 was observed (14.3 ± 0.51, n = 6) (p < 0.05). Hippocampal levels of caspase 8 were significantly increased in the Diabt group (30.9 ± 1.0, n = 6) compared with the Cnt group (26.2 ± 1.5, n = 6; p < 0.05), while in the Diabt+Valp group, hippocampal levels of caspase 8 were significantly decreased (24.3 ± 0.8, n = 6; p < 0.01) (Fig. 5c). Hippocampal levels of caspase 9 did not show any significant difference between the experimental groups (p > 0.05) (Fig. 5d).
Discussion
The present study demonstrated that chronic diabetes with uncontrolled hyperglycemia produced memory impairment and increased the hippocampal levels of apoptotic caspases 3, 6, and 8. Chronic administration of sodium valproate (100 mg/kg) in these diabetic animals significantly ameliorated memory deficits and reversed the elevated levels of hippocampal caspases, indicating the effectiveness of valproate as a therapeutic candidate for diabetic neuropathy.
The current study revealed that FBS in diabetic animals increased at 72 h after the alloxan injection and remained elevated at 2 months after. Following this uncontrolled chronic hyperglycemia, spatial memory and avoidance memory were assessed and significant memory impairment was observed in both the tests. Spatial memory deficit following chronic diabetes has been reported in many previous studies (Deng et al. 2013; Tuzcu and Baydas 2006; Wang and Jia 2014). However, most of those studies used streptozocin (STZ) to induce diabetes while in this study hyperglycemia was induced using an alloxan injection and then changes in spatial and avoidance memory were investigated. Our data verified damages to both spatial memory and avoidance memory in diabetic animals when assessed 2 months after the chronic hyperglycemia. Neural cell damage associated with the hyperglycemia has been reported in both in vitro and in vivo studies (Oyenihi et al. 2015; Vincent et al. 2011). Therefore, we decided to evaluate apoptotic caspases in the hippocampal tissue following in vivo chronic hyperglycemia. In accordance with the behavioral memory evaluation, increased levels of apoptotic caspases were observed in the hippocampus and provide evidence for neuropathy in the hippocampus which could explain the memory impairments.
Enhanced levels of apoptotic caspases and their involvement in the pathogenesis of diabetic neuropathy have been demonstrated previously (Deng et al. 2013; Han et al. 2014); our data corroborate these previous studies. Increased levels of caspase 3 in the neuronal tissue (Erbas et al. 2016; Kowluru et al. 2004) and in the hippocampus (Wang and Jia 2014) have been reported following chronic hyperglycemia induced by streptozotocin in mice. Since caspases 8 and 9 are activated in the external and internal pathways of cell death, respectively, and lead to conversion of procaspase 3 to caspase 3 through a common pathway (Creagh 2014), and since caspase 3 is involved in the activation of apoptosis signaling (Riedl and Shi 2004), the increased levels of caspases in the hippocampus may indicate increased apoptosis in this region following the chronic diabetic condition. Therefore, the physiological function of hippocampus and its involvement in the memory performance may get disrupted.
Interestingly, our results reveal that chronic administration of sodium valproate (100 mg/kg; i.p.) into the diabetic animals attenuated (not statistically significant) elevated levels of FBS and also reduced levels of initiator caspase 8, and executioner caspases 3 and 6, in the hippocampus. Moreover, chronic valproate treatment ameliorated spatial and avoidance memory deficits in these diabetic animals. Previous studies by Biermann et al. (2011, 2010) have shown neuroprotective and neuroregenerative effects of valproate on the neuropathy of retinal ganglion cells, which was coupled with decreased levels of caspase 3 and increased activity in the cAMP response element-binding (CREB) protein and extracellular signal-regulated kinase (ERK) pathways. CREB has known as a transcription factor mediating expression of critical genes during growth and survival of neurons and decreased following the neurodegeneration (Lonze and Ginty 2002). It has been suggested that the neuroprotective and neuroregenerative effects of valproate might be mediated at least in part by the CREB-induced regulation of gene expression (Rouaux et al. 2007). Valproate has also been shown to stimulate the ERK pathway which involving in neuronal regeneration and survival (Hao et al. 2004). In addition, the study by Zhang et al. (2012b) demonstrated that the neuroprotective effects of valproate in the injured optic nerve model was mediated by inhibiting neuronal apoptosis, via the activation of brain-derived neurotrophic factor (BDNF)-tropomyosin receptor kinase B (TrkB) signaling pathway, the inhibition of caspase 3, and histone deacetylation. Although in the current study we did not examine the involvement of neuroprotective signaling pathways, our behavior data was in accordance with biochemical data and revealed efficiency of chronic valproate treatment to ameliorate memory performance and attenuate elevated levels of apoptotic caspases 3, 6, and 8. Previous reports have shown beneficial effects of valproate administration on different models of memory impairment, possibly mediated through alterations in the brain levels of cytokines (Masuch et al. 2016; Pinheiro et al. 2015).
Hypoglycemic, antilipemic, and antioxidant effects of valproic acid (600 mg/kg; daily for 21 days) have been reported previously in a model of diabetes (Akindele et al. 2015). Additionally, it has been reported that valproic acid treatment (300 mg/kg) attenuates the plasma glucose, via reducing the insulin resistance and modulating insulin signaling (Khan et al. 2016). Our data demonstrated no significant effect of chronic valproate treatment (100 mg/kg) on FBS level and is in disagreement with the previous studies. This conflict might be due to the differences in the dosing regimen of valproate.
In addition, considering the microvascular and macrovascular disruption and blood-brain barrier dysfunction during the diabetic conditions (Alves et al. 2012; Prasad et al. 2014) which may lead to neuropathological conditions for the brain, another interpretation for our behavioral data might be related to the stimulating effects of valproate on angiogenesis. An in vitro study by Jin et al. (2011) has shown that valproate, via histone deacetylase inhibition, increased angiogenesis and endothelial cell sprouting. Chronic treatment with valproate as a histone deacetylase inhibitor promotes functional recovery after brain ischemia through enhancing angiogenesis by upregulation of the proangiogenic factors, vascular endothelial growth factor, and matrix metalloproteinase (Wang et al. 2012). Also, it was reported a single injection of valproate following the STZ-induced hyperglycemic rats ameliorates neuronal degeneration by inhibiting endovascular injury (Suda et al. 2015). In another study, valproic acid attenuated blood-brain barrier disruption, via inhibition of the histone deacetylase (Wang et al. 2011). However, more research to understand the possible effects of valproate on angiogenesis and blood-brain barrier in the diabetes is necessary.
Conclusions
Chronic administration of a moderate dose of valproate in alloxan-induced hyperglycemia attenuates elevated hippocampal levels of apoptotic caspases 3, 6, and 8 and ameliorates memory interruption. Co-administration of valproate and insulin may offer a better therapeutic outcome and should be considered in future investigations.
References
Akindele AJ, Otuguor E, Singh D, Ota D, Benebo AS (2015) Hypoglycemic, antilipidemic and antioxidant effects of valproic acid in alloxan-induced diabetic rats. Eur J Pharmacol 762:174–183
Alves MG, Oliveira PF, Socorro S, Moreira PI (2012) Impact of diabetes in blood-testis and blood-brain barriers: resemblances and differences. Curr Diabetes Rev 8:401–412
Biermann J, Grieshaber P, Goebel U, Martin G, Thanos S, Di Giovanni S, Lagreze WA (2010) Valproic acid-mediated neuroprotection and regeneration in injured retinal ganglion cells. Invest Ophthalmol Vis Sci 51:526–534
Biermann J, Boyle J, Pielen A, Lagreze WA (2011) Histone deacetylase inhibitors sodium butyrate and valproic acid delay spontaneous cell death in purified rat retinal ganglion cells. Mol Vis 17:395–403
Creagh EM (2014) Caspase crosstalk: integration of apoptotic and innate immune signalling pathways. Trends Immunol 35:631–640
Deng W, Lu H, Teng J (2013) Carvacrol attenuates diabetes-associated cognitive deficits in rats. J Mol Neurosci 51:813–819
Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35:495–516
Erbas O, Oltulu F, Yilmaz M, Yavasoglu A, Taskiran D (2016) Neuroprotective effects of chronic administration of levetiracetam in a rat model of diabetic neuropathy. Diabetes Res Clin Pract 114:106–116
Han J, Tan P, Li Z, Wu Y, Li C, Wang Y, Wang B, Zhao S, Liu Y (2014) Fuzi attenuates diabetic neuropathy in rats and protects schwann cells from apoptosis induced by high glucose. PLoS One 9:e86539
Hao Y, Creson T, Zhang L, Li P, du F, Yuan P, Gould TD, Manji HK, Chen G (2004) Mood stabilizer valproate promotes ERK pathway-dependent cortical neuronal growth and neurogenesis. J Neurosci 24:6590–6599
Jia D, Heng LJ, Yang RH, Gao GD (2014) Fish oil improves learning impairments of diabetic rats by blocking PI3K/AKT/nuclear factor-kappaB-mediated inflammatory pathways. Neuroscience 258:228–237
Jin G, Bausch D, Knightly T, Liu Z, Li Y, Liu B, Lu J, Chong W, Velmahos GC, Alam HB (2011) Histone deacetylase inhibitors enhance endothelial cell sprouting angiogenesis in vitro. Surgery 150:429–435
Khan S, Jena G, Tikoo K (2015) Sodium valproate ameliorates diabetes-induced fibrosis and renal damage by the inhibition of histone deacetylases in diabetic rat. Exp Mol Pathol 98:230–239
Khan S, Kumar S, Jena G (2016) Valproic acid reduces insulin-resistance, fat deposition and FOXO1-mediated gluconeogenesis in type-2 diabetic rat. Biochimie 125:42–52
Kowluru RA, Chakrabarti S, Chen S (2004) Re-institution of good metabolic control in diabetic rats and activation of caspase-3 and nuclear transcriptional factor (NF-kappaB) in the retina. Acta Diabetol 41:194–199
Lonze BE, Ginty DD (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35:605–623
Luo HM, Hu S, Bai HY, Wang HB, du MH, Lin ZL, Ma L, Wang H, Lv Y, Sheng ZY (2014) Valproic acid treatment attenuates caspase-3 activation and improves survival after lethal burn injury in a rodent model. J Burn Care Res 35:e93–e98
Masuch A, Shieh CH, van Rooijen N, van Calker D, Biber K (2016) Mechanism of microglia neuroprotection: involvement of P2X7, TNFα, and valproic acid. Glia 64:76–89
Nardin P, Zanotto C, Hansen F, Batassini C, Gasparin MS, Sesterheim P, Goncalves CA (2016) Peripheral levels of AGEs and astrocyte alterations in the hippocampus of STZ-diabetic rats. Neurochem Res 41:2006–2016
Oyenihi AB, Ayeleso AO, Mukwevho E, Masola B (2015) Antioxidant strategies in the management of diabetic neuropathy. Biomed Res Int 2015:515042
Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS (2001) Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem 276:36734–36741
Pinheiro RMC, de Lima MNM, Portal BCD, Busato SB, Falavigna L, Ferreira RDP, Paz AC, de Aguiar BW, Kapczinski F, Schröder N (2015) Long-lasting recognition memory impairment and alterations in brain levels of cytokines and BDNF induced by maternal deprivation: effects of valproic acid and topiramate. J Neural Transm 122:709–719
Prasad S, Sajja RK, Naik P, Cucullo L (2014) Diabetes mellitus and blood-brain barrier dysfunction: an overview. Aust J Pharm 2:125
Rabadiya S, Bhadada S, Dudhrejiya A, Vaishnav D, Patel B (2017) Magnesium valproate ameliorates type 1 diabetes and cardiomyopathy in diabetic rats through estrogen receptors. Biomed Pharmacother 97:919–927
Riedl SJ, Shi Y (2004) Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol 5:897–907
Rouaux C, Panteleeva I, Rene F, Gonzalez de Aguilar JL, Echaniz-Laguna A, Dupuis L, Menger Y, Boutillier AL, Loeffler JP (2007) Sodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model. J Neurosci 27:5535–5545
Russell JW, Golovoy D, Vincent AM, Mahendru P, Olzmann JA, Mentzer A, Feldman EL (2002) High glucose-induced oxidative stress and mitochondrial dysfunction in neurons. FASEB J 16:1738–1748
Saha RN, Pahan K (2006) HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ 13:539–550
Said G (2007) Diabetic neuropathy—a review. Nat Rev Neurol 3:331
Said G, Lacroix C, Lozeron P, Ropert A, Planté V, Adams D (2003) Inflammatory vasculopathy in multifocal diabetic neuropathy. Brain 126:376–385
Schmeichel AM, Schmelzer JD, Low PA (2003) Oxidative injury and apoptosis of dorsal root ganglion neurons in chronic experimental diabetic neuropathy. Diabetes 52:165–171
Sezik E, Aslan M, Yesilada E, Ito S (2005) Hypoglycaemic activity of Gentiana olivieri and isolation of the active constituent through bioassay-directed fractionation techniques. Life Sci 76:1223–1238
Silva MR, Correia AO, Dos Santos GCA, Parente LLT, de Siqueira KP, Lima DGS, Moura JA, da Silva Ribeiro AE, Costa RO, Lucetti DL, Lucetti ECP, Neves KRT, de Barros Viana GS (2018) Neuroprotective effects of valproic acid on brain ischemia are related to its HDAC and GSK3 inhibitions. Pharmacol Biochem Behav 167:17–28
Suda S, Ueda M, Nito C, Nishiyama Y, Okubo S, Abe A, Aoki J, Suzuki K, Sakamoto Y, Kimura K (2015) Valproic acid ameliorates ischemic brain injury in hyperglycemic rats with permanent middle cerebral occlusion. Brain Res 1606:1–8
Tai YT et al (2014) Low dose of valproate improves motor function after traumatic brain injury. Biomed Res Int 2014:980657
Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D, Ghayur T, Brady KD, Wong WW (1997) Substrate specificities of caspase family proteases. J Biol Chem 272:9677–9682
Tuzcu M, Baydas G (2006) Effect of melatonin and vitamin E on diabetes-induced learning and memory impairment in rats. Eur J Pharmacol 537:106–110
Vincent AM, Callaghan BC, Smith AL, Feldman EL (2011) Diabetic neuropathy: cellular mechanisms as therapeutic targets. Nat Rev Neurol 7:573–583
Wang SB, Jia JP (2014) Oxymatrine attenuates diabetes-associated cognitive deficits in rats. Acta Pharmacol Sin 35:331–338
Wang Z, Leng Y, Tsai L-K, Leeds P, Chuang D-M (2011) Valproic acid attenuates blood–brain barrier disruption in a rat model of transient focal cerebral ischemia: the roles of HDAC and MMP-9 inhibition. J Cereb Blood Flow Metab 31:52–57
Wang Z, Tsai LK, Munasinghe J, Leng Y, Fessler EB, Chibane F, Leeds P, Chuang DM (2012) Chronic valproate treatment enhances postischemic angiogenesis and promotes functional recovery in a rat model of ischemic stroke. Stroke 43:2430–2436
Xuan A-G, Pan XB, Wei P, Ji WD, Zhang WJ, Liu JH, Hong LP, Chen WL, Long DH (2015) Valproic acid alleviates memory deficits and attenuates amyloid-β deposition in transgenic mouse model of Alzheimer’s disease. Mol Neurobiol 51:300–312
Zhang Z, Qin X, Zhao X, Tong N, Gong Y, Zhang W, Wu X (2012a) Valproic acid regulates antioxidant enzymes and prevents ischemia/reperfusion injury in the rat retina. Curr Eye Res 37:429–437
Zhang ZZ, Gong YY, Shi YH, Zhang W, Qin XH, Wu XW (2012b) Valproate promotes survival of retinal ganglion cells in a rat model of optic nerve crush. Neuroscience 224:282–293
Funding
This study was supported by Arak University of Medical Sciences (Funding Nos. 2655 and 2688).
Author information
Authors and Affiliations
Contributions
MS was responsible for the study concept and design. PZ, MG, BA, and SH performed the acquisition of animal data. PZ, MG, and MS contributed to the analysis and interpretation of findings. PZ, MG, and MS drafted and revised the manuscript for important intellectual content. All authors critically reviewed and revised content, and approved the final version for publication.
Corresponding author
Ethics declarations
Statement on the welfare of animals
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted. This article does not contain any studies with human participants performed by any of the authors.
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Parvin Zareie and Mahsa Gholami had the same cooperation in this work.
Rights and permissions
About this article

Cite this article
Zareie, P., Gholami, M., Amirpour-najafabadi, B. et al. Sodium valproate ameliorates memory impairment and reduces the elevated levels of apoptotic caspases in the hippocampus of diabetic mice. Naunyn-Schmiedeberg's Arch Pharmacol 391, 1085–1092 (2018). https://doi.org/10.1007/s00210-018-1531-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-018-1531-3