
Abstract
The present study explored the protective effects of cannabinoid receptor agonist WIN55,212-2 (WIN) and fatty acid amide hydrolase inhibitor URB597 (URB) against neuroinflammation in rats with chronic cerebral hypoperfusion (CCH). Activated microglia, astrocytes, and nuclear factor kappa B (NF-κB) p65-positive cells were measured by immunofluorescence. Reactive oxygen species (ROS) was assessed by dihydroethidium staining. The protein levels of cluster of differentiation molecule 11b (OX-42), glial fibrillary acidic protein (GFAP), NF-κB p65, inhibitor of kappa B alpha (IκB-a), IκB kinase a/β (IKK a/β), phosphorylated IKK a/β (p-IKK a/β), cyclooxygenase-2 (COX-2), inducible nitric oxide synthase (iNOS), tumor necrosis factor (TNF)-α, and interleukin-1β (IL-1β) were examined by western blotting or enzyme-linked immunosorbent assay. All the protein levels of OX-42, GFAP, TNF-a, IL-1β, COX-2, and iNOS are increased in CCH rats. WIN and URB downregulated the levels of OX-42, GFAP, TNF-α, IL-1β, COX-2 and iNOS and inhibited CCH-induced ROS accumulation in CCH rats, indicating that WIN and URB might exert their neuroprotective effects by inhibiting the neuroinflammatory response. In addition, the NF-κB signaling pathway was activated by CCH in frontal cortex and hippocampus, while the aforementioned changes were reversed by WIN and URB treatment. These findings suggest that WIN and URB treatment ameliorated CCH-induced neuroinflammation through inhibition of the classical pathway of NF-κB activation, resulting in mitigation of chronic ischemic injury.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A substantial amount of evidence indicates that chronic cerebral hypoperfusion (CCH) is a common pathological process associated with many cerebrovascular diseases (Hasumi et al. 2007; Hainsworth and Markus 2008; Hai et al. 2009; Su et al. 2013, 2014). CCH, a chronic state of reduced cerebral blood flow, may cause neural impairment and delayed apoptotic cell death in brain regions that are vulnerable to cerebral hypoperfusion (Smith et al. 1984). Studies on the effects of CCH on the central nervous system showed that microglial and astrocytic responses were enhanced and that both microglia and astrocytes released various neurotoxic factors, such as tumor necrosis factor (TNF)-α, interleukin-1β (IL-1β), cyclooxygenase-2 (COX-2), inducible nitric oxide synthase (iNOS), and reactive oxygen species (ROS) (Liu and Hong 2003; Block and Hong 2005; Farkas et al. 2006). Microglial and astrocytic activation along with neuroinflammatory responses were shown to induce inflammatory neuronal cell death (Boje and Arora 1992; Abramov et al. 2004), which could further result in clinical symptoms such as cognitive dysfunction, neurological deficits, neurodegeneration, etc. Collectively, neuroinflammation might be an important mechanism associated with the initiation and progression of neuronal cell damage. Nuclear factor kappa B (NF-κB), a family of DNA-binding proteins, is involved in regulating many aspects of cellular activity including cellular differentiation, proliferation, apoptosis, oxidative response, and immune response. Activation of the NF-κB pathway has been reported to induce transcriptions of many pro-inflammatory mediators such as TNF-α, IL-1β, COX-2, and iNOS (Surh et al. 2001). In the central nervous system, NF-κB modulates neuroinflammatory responses by inducing nuclear translocation (Lu et al. 2010, 2011). Hence, protection against neuroinflammation and blockage of NF-κB-mediated inflammatory signaling are considered potential therapeutic strategies for CCH.
The endocannabinoid system (ECS) is a group of elements including the endogenous ligands (e.g., anandamide (AEA) and 2-arachidonoyl glycerol (2-AG)), endocannabinoid modulatory enzymes regulating ligand biosynthesis and degradation (e.g., fatty acid amide hydrolase (FAAH)) and two cannabinoid receptors (i.e., CB1 and CB2). Both receptors have been implicated in the modulation of neurodevelopment, synaptic transmission, synaptic plasticity, inflammation, and cognitive development (Demuth and Molleman 2006; Tagliaferro et al. 2006; Bosier et al. 2010; Hill et al. 2010). Recent data suggested that pharmacological agents that enhance ECS signaling provide a novel approach to the treatment of neurological disorders after CCH (Lin et al. 2010; Hai et al. 2013; Su et al. 2015, 2016). In our previous studies (Su et al. 2015, 2016), we found that WIN55,212-2 (WIN), a non-selective cannabinoid receptor agonist and URB597 (URB), an FAAH inhibitor which facilitates CB1 signaling through a more subtle mechanism (Hwang et al. 2010), exerted neuroprotective effects on neuronal cells in frontal cortex and hippocampus in response to CCH-induced neuronal apoptosis, cognitive dysfunction, and impaired neuronal plasticity. In addition, Naidu et al. (2010) reported that WIN and URB ameliorated chronic inflammatory pain in peripheral systems. Martín-Moreno et al. (2012) demonstrated that WIN showed beneficial effects concomitant with reduced neuroinflammation and increased Aβ clearance in a mice model of Alzheimer’s diseases. Rezende et al. (2012) presented that URB was involved in the analgesic effects of celecoxib in the central nervous system in a rat model of inflammatory pain. However, to the best of our knowledge, the animal models which were used in the published studies regarding the effects of WIN and URB were totally in acute ischemic phase. Little is known about the effects of WIN and URB on neuroinflammation induced by chronic ischemic insult in the central nervous system. Based on these considerations, this study attempted to explore the aforementioned issue and investigate the potential mechanisms underlying WIN and URB action in the frontal cortex and hippocampus of rats with CCH.
Materials and methods
Animal administration and drug treatment
Male Sprague-Dawley rats (240 g ± 10) were purchased from the experimental animal center of Shanghai Sippr-BK Laboratory Animals Ltd. (Shanghai, China). Prior to experiments, the rats were group-housed in climate-controlled facilities with a 12:12-h light/dark cycle and with free access to food and water. After acclimatization to these conditions for 1 week, rats were randomly divided into five treatment groups (10 rats per group). Based on our pilot studies, low doses of WIN (0.3 mg/kg) and URB (0.06 mg/kg) had little effect while median doses (1.0 and 0.3 mg/kg, respectively) alleviated the neurological impairment induced by CCH. However, high doses (3.0 and 1.5 mg/kg, respectively) induced side effects in 40% of rats. WIN and URB were therefore administered at 1 and 0.3 mg/kg body weight/day using 0.1% dimethyl sulfoxide (DMSO) as vehicle, respectively, in our study, which are doses similar to those reported in other studies (Baek et al. 2009; Hu et al. 2010; Slusar et al. 2013; Katz et al. 2015). Each group received one of the following treatments: (1) Sham-operated group: daily administration of vehicle; (2) Bilateral common carotid artery occlusion (BCCAo) group: daily administration of vehicle; (3) BCCAo + WIN (Sigma, St. Louis, MO, USA) group: daily administration of WIN (1 mg/kg/day, i.p.); (4) BCCAo + URB (Cayman Chemicals, Tallinn, Estonia) group: daily administration of URB (0.3 mg/kg/day, i.p.); (5) BCCAo + WIN + URB group: daily administration of WIN (1 mg/kg/day, i.p.) and URB (0.3 mg/kg/day, i.p.). All the drugs were given after surgery procedure of animal model. After 12 weeks of treatment, rats were sacrificed and brain tissues were immediately collected for experiments or stored at − 70 °C for later use.
All experiments were performed according to international guidelines and the guidelines of Chinese legislation on the use and care of laboratory animals and were approved by Tongji university and Shanghai Jiao Tong university for animal experiments (No.: 81271212).
Surgery procedure
CCH in rats was conducted based on previously described protocols (Ni et al. 1994). Twelve weeks is the longest time for a CCH model of rats with BCCAo (Farkas et al. 2007), which was used in this study. Each rat was anesthetized with pentobarbital-sodium (50 mg/kg, i.p.). Through a midline cervical incision, both common carotid arteries were exposed and ligated with 5–0 silk sutures in the BCCAo group. Sham-operated animals were similarly subjected to the surgery without undergoing carotid arteries ligations.
Brain tissue preparation
As described previously (Su et al. 2015, 2016), four rats in each groups were deeply anesthetized with an overdose of sodium pentobarbital and perfused transcardially with 200 ml 0.1-M phosphate-buffered saline (PBS, pH 7.4) at 4 °C followed by 200 ml 4% buffered paraformaldehyde phosphate. The brains were removed and postfixed for 24 h in 4% paraformaldehyde and then divided into two hemibrains along the midline. The hemibrains were serially dehydrated, embedded in paraffin, and then cut into 5-μm-thick coronal sections for immunofluorescence. The other groups were also deeply anesthetized with an overdose of sodium pentobarbital and sacrificed. Hippocampi and frontal cortices were dissected and homogenized in ice-cold buffer (20 mM Tris–HCl, pH 7.5, 1 mM EGTA, 1 mM EDTA, 25 μg/ml aprotinin, 25 μg/ml leupeptin, 1 mM Na4P2O7, 500 μM phenylmethylsulfonyl fluoride, 4 mM para-nitrophenylphosphate, 1 mM sodium orthovanadate, and 20 μl of protease inhibitor mix per gram of tissue). The homogenates were then centrifuged at 10,000 rpm for 5 min at 4 °C. Supernatants were collected and stored at − 70 °C for western blot studies. NF-κB p65 expression in cytoplasm and nucleus was extracted from brain tissues using a nuclear/cytoplasmic isolation kit (Beyotime Biotech Inc., Nangjing, China). Protein levels of the supernatants were determined using the bicinchoninic acid (BCA) protein determination assay (Pierce Biotechnology, Inc., Rockford, IL, USA).
Immunofluorescence
Briefly, after dewaxing, paraffin-embedded sections were washed three times with PBS for 5 min. Then, the sections were immersed in EDTA-Tris solution (pH 9.0) for 30 min at 98 °C for antigen retrieval and rinsed three times with PBS for 5 min. Subsequently, the slides were incubated with 10% non-immune goat serum for 30 min at room temperature to block non-specific staining before overnight incubation with primary antibodies: rabbit anti-cluster of differentiation molecule 11b (OX-42) (1:80, Santa Cruz, Santa Cruz, CA, USA), rabbit anti-GFAP (1:200, Santa Cruz) and rabbit anti-NF-κB p65 (1:550, Cell Signaling Technology, Beverly, MA, USA), in humidified chambers at 4 °C. After washing these sections in PBS, tetramethylrhodamine isothiocyanate (TRITC)-conjugated anti-rabbit secondary antibodies (1:200, Santa Cruz) were then applied for 1 h at 37 °C. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (KeyGen Biotech, Nanjing, China). All stained specimens were observed under a fluorescent microscope (Olympus IX71, Olympus Optical Co. Ltd., Tokyo, Japan). For the quantitative analysis, the average score of six randomly selected areas (three slides for each brain) was calculated using National Institutes of Health (NIH) Image Pro Plus 6.0 software.
Dihydroethidium staining
ROS was assessed by dihydroethidium (DHE) staining as described previously (Poulet et al. 2005). Briefly, the sections were incubated in 10 mmol/l DHE (Sigma) at room temperature for 30 min and protected from light. All stained slices were observed under a fluorescent microscope (Olympus IX71, Olympus Optical Co. Ltd). For the quantitative analysis, the average score of six randomly selected areas (three slides for each brain) was calculated using NIH Image Pro Plus 6.0 software. The ROS level (DHE-positive cells/mm2) in the sham group was normalized to 1.
Enzyme-linked immunosorbent assay
Quantitative measurements of TNF-α and IL-1β were performed by an enzyme-linked immunosorbent assay kit (R&D Systems, Minneapolis, MN, USA), according to the manufacturer’s instructions. Briefly, equal amounts of proteins were loaded into all wells, followed by measurements of optical density (OD) in a plate reader at a wavelength of 450 nm, with analyses for concentrations based on a standard curve. For convenience, all results were expressed as picogram per milligram protein.
Western blot analysis
Equal amounts of samples (20 μg proteins) were separated by 10% sodium dodecyl sulfate (SDS)-polyacrylamide gels and transferred onto nitrocellulose membranes. The membrane was blocked with 5% non-fat milk and 0.1% Tween-20 in Tris-buffered saline (TBS-T) for 1 h, incubated overnight with appropriate primary antibodies against OX-42 (1:600, Santa Cruz); glial fibrillary acidic protein (GFAP) (1:1000, Santa Cruz); NF-κB p65 (1:2000, Cell Signaling Technology); inhibitor of kappa B alpha (IκB-a) (1:2000, Cell Signaling Technology); IκB kinase a/β (IKK a/β) (1:500, Bioworld, MN, USA); phosphorylated IKK a/β (p-IKK a/β) (1:500, Bioworld), COX-2 (1:1500, Cell Signaling Technology); iNOS (1:1000, Chemicon International Inc., Temecula, CA, USA); β-actin (1:5000, Abcam, Cambridge, UK); and Histone-H3 (1:800, Santa Cruz), followed by detection with an enhanced chemiluminescent substrate solution (Millipore, Watford, UK) after incubation with horseradish peroxidase-conjugated goat anti-rabbit or mouse IgG secondary antibody for 1 h at room temperature. The proteins were quantified by OD ratio using β-actin as a control. In case of nuclear NF-κB p65, histone-H3 was employed as the loading control. The extent of phosphorylation of IKK a/β was evaluated with respect to IKK a/β abundances (p-IKK a/β/IKK a/β).
Statistical analysis
The data are reported as mean ± standard error of the mean (SEM). Statistical analyses were double-blinded and were performed by one-way ANOVA, followed by Dunnett’s test as the post hoc analysis. P values of less than 0.05 were used as the criterion for significance in all types of data analyses.
Results
WIN and URB treatment decreases the number of activated microglia and activated astrocytes in frontal cortex and hippocampus of CCH-induced rats
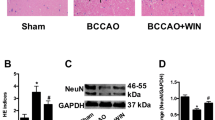
Previous studies suggested that microglial and astrocytic activation are being considered as a pathological hallmark in neurodegenerative disorders and chronic ischemic injury (Kwak et al. 2012). In the present study, activated microglia (OX-42-stained cells) and astrocytes (glial fibrillary acidic protein (GFAP)-stained cells) were rarely detected in frontal cortex and hippocampal CA1 region in sham control rats, while CCH significantly increased the number of OX-42-positive microglial cells and GFAP-positive astrocytes as compared with the control group (Figs. 1 and 2). Nevertheless, treatment with WIN, URB, or WIN + URB abrogated the CCH-induced increase in the number of OX-42-positive microglia and GFAP-positive astrocytes in the frontal cortex and hippocampus relative to the BCCAo group.

Effects of WIN55,212-2 and URB597 on the protein expressions of GFAP in rats hippocampus and frontal cortex after chronic cerebral hypoperfusion. a Representative immunofluorescence for GFAP in hippocampal CA1 region (magnification = × 200, scale bars = 50 μm). b Representative immunofluorescence for GFAP in frontal cortex (magnification = × 200, scale bars = 50 μm). c Representative immunoblot and relative optical density analysis for GFAP in hippocampus (F[4, 20] = 45.122). d Representative immunoblot and relative optical density analysis for GFAP in frontal cortex (F[4, 20] = 99.360). Data are expressed as mean ± SEM (n = 5). * P < 0.05 versus sham group. ** P < 0.05 versus BCCAo group. BCCAo bilateral common carotid artery’s occlusion; GFAP glial fibrillary acidic protein

Effects of WIN55,212-2 and URB597 on the protein expressions of OX-42 in rats hippocampus and frontal cortex after chronic cerebral hypoperfusion. a Representative immunofluorescence for OX-42 in hippocampal CA1 region (magnification = × 200, scale bars = 50 μm). b Representative immunofluorescence for OX-42 in frontal cortex (magnification = × 200, scale bars = 50 μm). c Representative immunoblot and relative optical density analysis for OX-42 in hippocampus (F[4, 45] = 67.869). d Representative immunoblot and relative optical density analysis for OX-42 in frontal cortex (F[4, 20] = 494.77). Data are expressed as mean ± SEM (n = 5). * P < 0.05 versus sham group. ** P < 0.05 versus BCCAo group. BCCAo bilateral common carotid artery’s occlusion, OX-42 cluster of differentiation molecule 11b
WIN and URB treatment reduces ROS accumulation in rat frontal cortex and hippocampus
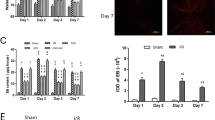
Dihydroethidium (DHE) is oxidized by intracellular ROS to produce fluorescent ethidium that is subsequently intercalated in DNA, further amplifying its fluorescence, indicating that an increase in DHE oxidation and a subsequent increase in fluorescence are highly suggestive of ROS generation (Gross et al. 2003). As shown in Fig. 3, compared with the sham group, the levels of ROS in the CCH group were significantly increased. Notably, WIN as well as URB inhibited CCH-induced ROS accumulation markedly.

Effects of WIN55,212-2 and URB597 on the level of ROS in rats hippocampus and frontal cortex after chronic cerebral hypoperfusion. a Representative DHE fluorescence staining for ROS in hippocampal CA1 region (magnification = × 200, scale bars = 50 μm). b Representative DHE fluorescence staining for ROS in frontal cortex (magnification = × 200, scale bars = 50 μm). c Relative level of ROS (DHE-positive cells/mm2) in hippocampal CA1 region (F[4, 20] = 380.736). d Relative level of ROS (DHE-positive cells/mm2) in frontal cortex (F[4, 20] = 715.586). The ROS level in sham group were normalized to 1. Data are expressed as mean ± SEM (n = 5). * P < 0.05 versus sham group. ** P < 0.05 versus BCCAo group. BCCAo bilateral common carotid artery’s occlusion; DHE dihydroethidium
WIN and URB treatment suppresses the expression of inflammatory markers in frontal cortex and hippocampus of CCH-induced rats
We further examined the expression of inflammatory markers, including TNF-α, IL-1β, COX-2, and iNOS in frontal cortex and hippocampus of each group (Fig. 4). CCH induced a significant increase of TNF-α, IL-1β, COX-2, and iNOS levels in rat frontal cortex and hippocampus as compared with the sham group. Administration of WIN and URB significantly attenuated these increases in the frontal cortex and hippocampus of the BCCAo group. However, protein levels in the frontal cortex and hippocampus of BCCAo + WIN, BCCAo + URB, and BCCAo + WIN + URB groups were not restored to a normal level.

Effects of WIN55,212-2 and URB597 on the expression of inflammatory markers in rats hippocampus and frontal cortex after chronic cerebral hypoperfusion. a, b The protein levels of TNF-a in hippocampus and frontal cortex, respectively. c, d The protein levels of IL-1β in hippocampus and frontal cortex, respectively. e, f Representative western blot and relative optical density analysis of COX-2 and β-actin in hippocampus and frontal cortex respectively. g, h Representative western blot and relative optical density analysis of iNOS and β-actin in hippocampus and frontal cortex, respectively. Data are expressed as mean ± SEM (n = 5). *P < 0.05 versus sham group. **P < 0.05 versus BCCAo group. a F(4, 20) = 343.672; b F(4, 20) = 89.979; c F(4, 20) = 332.599; d F(4, 20) = 159.676; e F(4, 20) = 13.583; f F(4, 20) = 63.252; g F(4, 20) = 191.409; h F(4, 20) = 472.357; BCCAo bilateral common carotid artery’s occlusion
.WIN and URB treatment blocks NF-κB activation through inhibiting NF-κB p65 nuclear translocation in frontal cortex and hippocampus of CCH-induced rats
NF-κB signaling is involved in the regulation of several pathological conditions associated with inflammation, including ischemic stroke and neurodegeneration (Pizzi et al. 2009; Khan et al. 2012). Recent studies showed that CCH activated NF-κB-mediated inflammatory pathways, resulting in neuronal damage (Fu et al. 2014). In the present study, we evaluated the activation of NF-κB p65 and its nuclear translocation using immunofluorescence and western blot (Figs. 5 and 6). In agreement with the aforementioned results, CCH significantly increased NF-κB activation by increasing the phosphorylation of IKK a/β, inducing the degradation of IκB-a and stimulating nuclear translocation of NF-κB p65 in rat frontal cortex and hippocampus. However, WIN and URB treatment significantly reversed these changes.

Effects of WIN55,212-2 and URB597 on the NF-κB signaling alterations in rats hippocampus and frontal cortex after chronic cerebral hypoperfusion. a, e Representative western blot and relative optical density analysis of p-IKK a/β, IKK a/β, and β-actin in hippocampus and frontal cortex, respectively. b, f Representative western blot and relative optical density analysis of IκB-a and β-actin in hippocampus and frontal cortex, respectively. c, g Representative western blot and relative optical density analysis of cytoplasmic NF-κB p65 and β-actin in hippocampus and frontal cortex respectively. d, h Representative western blot and relative optical density analysis of nuclear NF-κB p65 and Histone-H3 in hippocampus and frontal cortex respectively. Data are expressed as mean ± SEM (n = 5). *P < 0.05 versus sham group. **P < 0.05 versus BCCAo group. a F(4, 20) = 32.660; b F(4, 20) = 60.274; c F(4, 20) = 41.523; d F(4, 20) = 234.127; e F(4, 20) = 105.729; f F(4, 20) = 15.335; g F(4, 20) = 19.517; h F(4, 20) = 95.100; BCCAo bilateral common carotid artery’s occlusion

Immunofluorescence staining of the nuclear translocation of NF-κB p65 in each group. a Representative immunofluorescence for NF-κB p65 in hippocampal CA1 region (magnification = × 200, scale bars = 50 μm). b Representative immunofluorescence for NF-κB p65 in frontal cortex (magnification = × 200, scale bars = 50 μm). In sham group, the immunoreactive staining occurred less in the cytoplasm and nucleus, while in the BCCAo group, strong immunoreactive staining occurred in the cytoplasm and nucleus, especially in the nucleus. However, in the BCCAo + WIN55,212-2, BCCAo + URB597, and BCCAo + WIN55,212-2 + URB597 groups, immunoreactive staining was predominantly detected in the cytoplasm rather than in the nucleus. BCCAo bilateral common carotid artery’s occlusion
Discussion
A robust inflammatory response is one of the most important delayed mechanisms after the onset of brain ischemia (Wang et al. 2007). Chronic neuroinflammation is tightly associated with neuronal loss, neuronal cell death, cognitive dysfunction, and neurodegeneration (Bond and Rex 2014; Hovens et al. 2015; Lozano et al. 2015; Macchi et al. 2015). Therapeutic strategies targeting the delayed inflammatory reaction may inhibit the progression of the tissue damage and provide an extended therapeutic window for neuroprotection. According to our previous studies (Su et al. 2015, 2016), WIN and URB could exert neuroprotective effects against CCH-induced neuronal apoptosis, cognitive dysfunction, and impaired neuronal plasticity in the frontal cortex and hippocampus. However, limited studies have examined the effects of WIN and URB on neuroinflammation induced by CCH in the central nervous system. Therefore, the major focus of the present study was to determine the role of WIN and URB in regulating CCH-induced neuroinflammation in vivo.
It has been reported that cerebral ischemic injury may trigger the activation of microglia and astrocytes (Kato et al. 2003), which can release pro-inflammatory cytokines including TNF-α, IL-1β, COX-2, and iNOS (Pickering and O’Connor 2007; Kaushal and Schlichter 2008; Kim et al. 2009; Jin et al. 2010). Subsequently, the accumulation of these pro-inflammatory factors could further exacerbate ischemic damages (Batti and O’Connor 2010). Taken together, neuroinflammation is a key contributor in the ischemic cascade after cerebral ischemia that leads to neuronal damage and death. Consistent with these previous reports, we observed that CCH could efficiently induce microglial and astrocytic activation. Furthermore, CCH increased the expression of TNF-α, IL-1β, COX-2, and iNOS. However, WIN and URB treatment decreased the number of activated microglia and astrocytes in frontal cortex and hippocampal CA1 region. Both WIN and URB significantly downregulated the levels of the aforementioned inflammatory factors, indicating that WIN and URB might exert their neuroprotective effects via inhibition of the neuroinflammatory response.
Oxidative stress occurs when the intracellular antioxidant defense system is overwhelmed by the generation of ROS, leading to the increase of toxic molecules (Lee et al. 2014). ROS is one of the crucial signaling molecules in the oxidative stress response. Cerebral ischemic injury may evoke many intracellular events including oxidative damage and the accumulation of ROS (Geng et al. 2015), which cause the destruction of cell proteins, lipids, nucleic acids, and DNA (Chan 2001). Post-ischemic ROS generation may eventually lead to neuronal death (Chan 2001; Ye et al. 2014). Moreover, ROS may contribute to inflammation through the involvement of specific signaling pathways (Wang et al. 2004). In agreement with these studies, we found that CCH significantly increased the level of ROS in frontal cortex and hippocampal CA1 region. Nevertheless, WIN as well as URB inhibited CCH-induced ROS accumulation markedly, suggesting that WIN and URB may have antioxidant properties, which activated its neuroprotective mechanisms against the adverse effects of ischemia.
NF-κB p65 is a major subunit that is sequestered in the cytoplasm and kept inactive through its association with IκB-a. The stimulation of pro-inflammatory cytokines induces a rapid degradation of IκB-a, causing the release and nuclear translocation of NF-κB for gene regulation. Activated NF-κB may be one of the crucial regulators of inflammatory damage by acting as an essential transcription factor for further promotion of target genes such as TNF-α, IL-1β, COX-2, and iNOS (Hayden and Ghosh 2004; Lawrence and Fong 2010), thereby triggering the vicious cycle and exacerbating inflammatory injury (Denes et al. 2011). Furthermore, ROS overproduction, which exacerbates the oxidative stress and mitochondrial damage, can serve as a second messenger to diverse active downstream signaling molecules including the nuclear translocation of NF-κB (Flohé et al. 1997). In addition, the activation of NF-κB signaling is associated with apoptosis and cognitive deficits (Kuhad et al. 2009; Tusi et al. 2010), whereas inhibition of the NF-κB pathway may not only decrease inflammatory responses but also prevent apoptosis and cognitive dysfunction (Lawrence et al. 2001; Kuhad et al. 2009). In our previous studies, we also confirmed that neuronal apoptosis and impaired learning and memory induced by CCH was accompanied by DNA fragmentation, a decrease of Bcl-2 to Bax ratio and activation of caspase-3 (Su et al. 2015, 2016). Hence, we further investigated whether CCH could activate NF-κB signaling and whether this activation was modulated by WIN and URB treatment. In the current study, we showed that the NF-κB signaling pathway was activated by CCH in frontal cortex and hippocampus, as evidenced by marked upregulation of p-IKK a/β, degradation of IκB-a, and nuclear translocation of NF-κB p65, while the aforementioned changes were reversed by WIN and URB treatment. These results demonstrated that WIN and URB might exert neuroprotective effects in part by suppressing the classical pathway of NF-κB activation.
As described previously (Su et al. 2015, 2016), FAAH inhibition (which blocks AEA inactivation) is a possible therapeutic strategy for indirectly enhancing cannabinergic signaling by targeting regions of endocannabinoid synthesis and release (Hwang et al. 2010). FAAH inhibitors may thus offer greater protection without the undesirable side effects observed with non-specific cannabinoid receptor agonists (Janero and Makriyannis 2009). Thus, it may not be surprising that we found superiority of URB to WIN in terms of neuroprotection. Although we observed anti-inflammatory function of WIN and URB in the present study, other studies on the contributions of WIN and URB to neuroinflammation remain scarce. Hence, future investigations are necessary to gain more information to verify our viewpoints. In particular, the mechanisms behind the WIN- and URB-mediated inhibition of neuroinflammatory responses require further clarification.
In conclusion, our in vivo data showed a key role for the NF-κB signaling pathway in mediating CCH-induced neuroinflammation. WIN and URB treatment reduced CCH-induced activation of microglia and astrocytes, decreased ROS generation, and reduced the expression of inflammatory markers through inhibition of the NF-κB pathway, resulting in mitigation of chronic ischemic injury. Combined with our previous studies (Su et al. 2015, 2016), we proposed a signaling schematic diagram for neuroprotective effects of WIN and URB against CCH-induced neuronal apoptosis, cognitive dysfunction, and inflammatory responses (Fig. 7). Collectively, these findings suggest therapeutic effects for WIN and URB in chronic cerebral ischemic insults.

Schematic diagram for neuroprotective effects of WIN and URB against CCH-induced neuronal apoptosis, cognitive dysfunction, and inflammatory responses. WIN and URB blocked JNK phosphorylation as well as the decrease in Bcl-2/Bax ratio and caspase-3 activation induced by CCH. WIN and URB reversed the decreased levels of phosphorylated AKT (p-AKT), p-CREB, p-GSK-3β, p-FOXO3A, p-BAD, and BDNF and synapse loss induced by CCH. WIN and URB suppressed the nuclear translocation of NF-κB p65, downregulated the generation of ROS, decreased the accumulation of pro-inflammatory cytokines and inhibited the activation of microglia and astrocytes induced by CCH
References
Abramov AY, Canevari L, Duchen MR (2004) Beta–amyloid peptides induce mitochondrial dysfunction and oxidative stress inastrocytes and death of neurons through activation of NADPH oxidase. J Neurosci 24:565–575
Baek J, Zheng Y, Darlington CL, Smith PF (2009) The CB1 receptor agonist, WIN 55,212-2, dose-dependently disrupts object recognition memory in adult rats. Neurosci Lett 464:71–73
Batti L, O’Connor JJ (2010) Tumor necrosis factor-alpha impairs the recovery of synaptic transmission from hypoxia in rat hippocampal slices. J Neuroimmunol 218:21–27
Block ML, Hong JS (2005) Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 76:77–98
Boje KM, Arora PK (1992) Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronalcell death. Brain Res 587:250–256
Bond WS, Rex TS (2014) Evidence that erythropoietin modulates neuroinflammation through differential action on neurons, astrocytes, and microglia. Front Immunol 5:523
Bosier B, Muccioli GG, Hermans E, Lambert DM (2010) Functionally selective cannabinoid receptor signalling: therapeutic implications and opportunities. Biochem Pharmacol 80:1–12
Chan PH (2001) Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab 21:2–14
Demuth DG, Molleman A (2006) Cannabinoid signalling. Life Sci 78:549–563
Denes A, Pinteaux E, Rothwell NJ, Allan SM (2011) Interleukin-1 and stroke: biomarker, harbinger of damage, and therapeutic target. Cerebrovasc Dis 32:517–527
Farkas E, Institóris A, Domoki F, Mihály A, Bari F (2006) The effect of pre- and posttreatment with diazoxide on the early phase of chronic cerebral hypoperfusion in the rat. Brain Res 1087:168–174
Farkas E, Luiten PG, Bari F (2007) Permanent, bilateral common carotid artery occlusion in the rat: a model for chronic cerebral hypoperfusion-related neurodegenerative diseases. Brain Res Rev 54:162–180
Flohé L, Brigelius-Flohé R, Salious C, Traber MG, Packer L (1997) Redox regulation of NF-kappaB activation. Free Radic Biol Med 22:1115–1126
Fu X, Zhang J, Guo L, Xu Y, Sun L, Wang S, Feng Y, Gou L, Zhang L, Liu Y (2014) Protective role of luteolin against cognitive dysfunction induced by chronic cerebral hypoperfusion in rats. Pharmacol Biochem Behav 126:122–130
Geng X, Elmadhoun O, Peng C, Ji X, Hafeez A, Liu Z, Du H, Rafols JA, Ding Y (2015) Ethanol and normobaric oxygen: novel approach in modulating pyruvate dehydrogenase complex after severe transient and permanent ischemic stroke. Stroke 46:492–499
Gross ER, LaDisa JF Jr, Weihrauch D, Olson LE, Kress TT, Hettrick DA, Pagel PS, Warltier DC, Kersten JR (2003) Reactive oxygen species modulate coronary wall shear stress and endothelial function during hyperglycemia. Am J Physiol Heart Circ Physiol 284:H1552–H1559
Hai J, Wan JF, Lin Q, Wang F, Zhang L, Li H, Zhang L, Chen YY, Lu Y (2009) Cognitive dysfunction induced by chronic cerebral hypoperfusion in a rat model associated with arteriovenous malformations. Brain Res 8(1301):80–88
Hai J, Lin Q, Wu YF, Huang XS, Zhang GY, Wang F (2013) Effects of N-stearoyl-L-tyrosine on the hippocampal ubiquitin-proteasome system in rats with chronic cerebral hypoperfusion. Neurol Res 35:734–743
Hainsworth AH, Markus HS (2008) Do in vivo experimental models reflect human cerebral small vessel disease? A systematic review. J Cereb Blood Flow Metab 28:1877–1891
Hasumi T, Fukushima T, Haisa T, Yonemitsu T, Waragai M (2007) Focal dural arteriovenous fistula (DAVF) presenting with progressive cognitive impairment including amnesia and alexia. Intern Med 46:1317–1320
Hayden MS, Ghosh S (2004) Signaling to NF-kappaB. Genes Dev 18:2195–2224
Hill MN, Patel S, Campolongo P, Tasker JG, Wotjak CT, Bains JS (2010) Functional interactions between stress and the endocannabinoid system: from synaptic signaling to behavioral output. J Neurosci. 30:4980–4986
Hovens IB, van Leeuwen BL, Nyakas C, Heineman E, van der Zee EA, Schoemaker RG (2015) Postoperative cognitive dysfunction and microglial activation in associated brain regions in old rats. Neurobiol Learn Mem 118:74–79
Hu B, Wang Q, Chen Y, Du J, Zhu X, Lu Y, Xiong L, Chen S (2010) Neuroprotective effect of WIN 55,212-2 pretreatment against focal cerebral ischemia through activation of extracellular signal-regulated kinases in rats. Eur J Pharmacol 645:102–107
Hwang J, Adamson C, Butler D, Janero DR, Makriyannis A, Bahr BA (2010) Enhancement of endocannabinoid signaling by fatty acid amide hydrolase inhibition: a neuroprotective therapeutic modality. Life Sci 86:615–623
Janero DR, Makriyannis A (2009) Cannabinoid receptor antagonists: pharmacological opportunities, clinical experience, and translational prognosis. Expert Opin Emerg Drugs 14:43–65
Jin R, Yang G, Li G (2010) Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol 87:779–789
Kato H, Takahashi A, Itoyama Y (2003) Cell cycle protein expression in proliferating microglia and astrocytes following transient global cerebral ischemia in the rat. Brain Res Bull 60:215–221
Katz PS, Sulzer JK, Impastato RA, Teng SX, Rogers EK, Molina P (2015) Endocannabinoid degradation inhibition improves neurobehavioral function, blood brain barrier integrity, and neuroinflammation following mild traumatic brain injury. J Neurotrauma 32:297–306
Kaushal V, Schlichter LC (2008) Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J Neurosci. 28:2221–2230
Khan MM, Gandhi C, Chauhan N, Stevens JW, Motto DG, Lentz SR, Chauhan AK (2012) Alternatively-spliced extra domain a of fibronectin promotes acute inflammation and brain injury after cerebral ischemia in mice. Stroke 43:1376–1382
Kim DH, Kim S, Jung WY, Park SJ, Park DH, Kim JM, Cheong JH, Ryu JH (2009) The neuroprotective effects of the seeds of Cassia obtusifolia on transient cerebral global ischemia in mice. Food Chem Toxicol 47:1473–1479
Kuhad A, Bishnoi M, Tiwari V, Chopra K (2009) Suppression of NF-kappa beta signaling pathway by tocotrienol can prevent diabetes associated cognitive deficits. Pharmacol Biochem Behav 92:251–259
Kwak PA, Lim SC, Han SR, Shon YM, Kim YI (2012) Supra-additive neuroprotection by renexin, a mixed compound of ginkgo biloba extract and cilostazol, against apoptotic white matter changes in rat after chronic cerebral hypoperfusion. J Clin Neurol 8:284–292
Lawrence T, Fong C (2010) The resolution of inflammation: anti-inflammatory roles for NFkappaB. Int J Biochem Cell Biol 42:519–523
Lawrence T, Gilroy DW, Colville-Nash PR, Willoughby DA (2001) Possible new role for NF-kappaB in the resolution of inflammation. Nat Med 7:1291–1297
Lee JE, Park JH, Jang SJ, Koh HC (2014) Rosiglitazone inhibits chlorpyrifos-induced apoptosis via modulation of the oxidative stress and inflammatory response in SH-SY5Y cells. Toxicol Appl Pharmacol. 278:159–171
Lin Q, Hai J, Yao LY, Lu Y (2010) Neuroprotective effects of NSTyr on cognitive function and neuronal plasticity in rats of chronic cerebral hypoperfusion. Brain Res 1325:183–190
Liu B, Hong JS (2003) Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther 304:1–7
Lozano D, Gonzales-Portillo GS, Acosta S, de la Pena I, Tajiri N, Kaneko Y, Borlongan CV (2015) Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr Dis Treat 11:97–106
Lu J, Wu DM, Zheng YL, Hu B, Zhang ZF, Ye Q, Liu CM, Shan Q, Wang YJ (2010) Ursolic acid attenuates D-galactose-induced inflammatory response in mouse prefrontal cortex through inhibiting AGEs/RAGE/NF-κB pathway activation. Cereb Cortex 20:2540–2548
Lu J, Wu DM, Zheng YL, Hu B, Cheng W, Zhang ZF, Shan Q (2011) Ursolic acid improves high fat diet-induced cognitive impairments by blocking endoplasmic reticulum stress and IκB kinase β/nuclear factor-κB-mediated inflammatory pathways in mice. Brain Behav Immun 25:1658–1667
Macchi B, Paola DR, Marino-Merlo F, Felice MR, Cuzzocrea S, Mastino A (2015) Inflammatory and cell death pathways in brain and peripheral blood in Parkinson’s disease. CNS Neurol Disord Drug Targets 14:313–324
Martín-Moreno AM, Brera B, Spuch C, Carro E, García-García L, Delgado M, Pozo MA, Innamorato NG, Cuadrado A, de Ceballos ML (2012) Prolonged oral cannabinoid administration prevents neuroinflammation, lowers β-amyloid levels and improves cognitive performance in Tg APP 2576 mice. J Neuroinflammation 9:8
Naidu PS, Kinsey SG, Guo TL, Cravatt BF, Lichtman AH (2010) Regulation of inflammatory pain by inhibition of fatty acid amide hydrolase. J Pharmacol Exp Ther 334:182–190
Ni J, Ohta H, Matsumoto K, Watanabe H (1994) Progressive cognitive impairment following chronic cerebral hypoperfusion induced by permanent occlusion of bilateral carotid arteries in rats. Brain Res 653:231–236
Pickering M, O’Connor JJ (2007) Pro-inflammatory cytokines and their effects in the dentate gyrus. Prog Brain Res 163:339–354
Pizzi M, Sarnico I, Lanzillotta A, Battistin L, Spano P (2009) Post-ischemic brain damage: NF-kappaB dimer heterogeneity as a molecular determinant of neuron vulnerability. FEBS J 276:27–35
Poulet R, Gentile MT, Vecchione C, Distaso M, Aretini A, Fratta L, RussoG EC, Maffei A, De Simoni MG, Lembo G (2005) Acute hypertension induces oxidative stress in brain tissues. J Cereb Blood Flow Metab 26:253–262
Rezende RM, Paiva-Lima P, Dos Reis WG, Camêlo VM, Faraco A, Bakhle YS, Francischi JN (2012) Endogenous opioid and cannabinoid mechanisms are involved in the analgesic effects of celecoxib in the central nervous system. Pharmacology 89:127–136
Slusar JE, Cairns EA, Szczesniak AM, Bradshaw HB, Di Polo A, Kelly ME (2013) The fatty acid amide hydrolase inhibitor, URB597, promotes retinal ganglion cell neuroprotection in a rat model of optic nerve axotomy. Neuropharmacology 72:116–125
Smith ML, Auer RN, Siesjö BK (1984) The density and distribution of ischemic brain injury in the rat following 2-10 min of forebrain ischemia. Acta Neuropathol 64:319–332
Su SH, Hai J, Zhang L, Yu F, Wu YF (2013) Assessment of cognitive function in adult patients with hemorrhagic moyamoya disease who received no surgical revascularization. Eur J Neurol 20:1081–1087
Su SH, Xu W, Hai J, Yu F, Wu YF, Liu YG, Zhang L (2014) Cognitive function, depression, anxiety and quality of life in Chinese patients with untreated unruptured intracranial aneurysms. J Clin Neurosci 21:1734–1739
Su SH, Wu YF, Lin Q, Yu F, Hai J (2015) Cannabinoid receptor agonist WIN55,212-2 and fatty acid amide hydrolase inhibitor URB597 suppress chronic cerebral hypoperfusion-induced neuronal apoptosis by inhibiting c-Jun N-terminal kinase signaling. Neuroscience 301:563–575
Su SH, Wang YQ, Wu YF, Lin Q, Hai J (2016) Cannabinoid receptor agonist WIN55,212-2 and fatty acid amide hydrolase inhibitor URB597 may protect against cognitive impairment in rats of chronic cerebral hypoperfusion via PI3K/AKT signaling. Behav Brain Res 313:334–344
Surh YJ, Chun KS, Cha HH, Han SS, Keum YS, Park KK, Lee SS (2001) Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: down-regulation of COX-2 and iNOS through suppression of NF-kappa B activation. Mutat Res 480-481:243–268
Tagliaferro P, Javier Ramos A, Onaivi ES, Evrard SG, Lujilde J, Brusco A (2006) Neuronal cytoskeleton and synaptic densities are altered after a chronic treatment with the cannabinoid receptor agonist WIN 55,212-2. Brain Res 1085:163–176
Tusi SK, Ansari N, Amini M, Amirabad AD, Shafiee A, Khodagholi F (2010) Attenuation of NF-kappa B and activation of Nrf2 signaling by 1,2,4-triazine derivatives, protects neuron-like PC12 cells against apoptosis. Apoptosis 15:738–751
Wang T, Qin L, Liu B, Liu T, Wilson B, Eling TE, Langenbach R, Taniira S, Hong JS (2004) Roles of reactive oxygen species in LPS induced production of prostaglandin E2 in microglia. J Neurochem 88:939–947
Wang Q, Tang XN, Yenari MA (2007) The inflammatory response in stroke. J Neuroimmunol 184:53–68
Ye M, Yang W, Ainscough JF, Hu XP, Li X, Sedo A, Zhang XH, Zhang X, Chen Z, Li XM, Beech DJ, Sivaprasadarao A, Luo JH, Jiang LH (2014) TRPM2 channel deficiency prevents delayed cytosolic Zn2+ accumulation and CA1 pyramidal neuronal death after transient global ischemia. Cell Death Dis 5:e1541
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant no.: 81271212 to J. Hai and no.: 81601146 to S.H. Su)
Ethics declarations
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Su, SH., Wu, YF., Lin, Q. et al. Cannabinoid receptor agonist WIN55,212-2 and fatty acid amide hydrolase inhibitor URB597 ameliorate neuroinflammatory responses in chronic cerebral hypoperfusion model by blocking NF-κB pathways. Naunyn-Schmiedeberg's Arch Pharmacol 390, 1189–1200 (2017). https://doi.org/10.1007/s00210-017-1417-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-017-1417-9