
Abstract
Coronary heart disease remains a leading cause of death in the world. The demand on targeting therapy to reduce myocardial ischemia/reperfusion (I/R) injury is still urgent. The pathogenesis of I/R-induced myocardial injury is complicated. Reactive oxygen species (ROS) generation and inflammatory response activation participate in the development of I/R injury. Cell death occurs and finally leads to myocardial infarction. A newly phenolic aporphine alkaloid derivative, TM-1-1DP, was synthesized in our team. We aimed to investigate the effect of novel compound on myocardial I/R injury. Rats were subjected to 1-h coronary artery occlusion and followed by 2-h reperfusion. Adult rat cardimoycyte was isolated for the cell study, and H2O2 was added into culture medium to induce ROS stress. As compared to the sham group, TM-1-1DP-treated rats had better cardiac performance in association with less infarct size and cardiac injury markers after myocardial I/R. The protective effect is associated with the inhibition of inflammatory response, cell death-related pathway (caspase-3 and TNF-α), and the activation of AKT-eNOS pathway. The finding was further coincided with the cell study. TM-1-1DP treatment significantly alleviated ROS production and improved cell viability in cardiomyocyte after H2O2 exposure. The action of TM-1-1DP is via a nitric oxide (NO)-dependent manner, since NOS inhibitor, L-NAME, abolished the protective effect. We provide a new insight into this therapeutic potential for phenolic aporphine alkaloid in myocardial I/R.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Coronary heart disease remains a leading cause of death in the world. Ischemia in coronary heart disease is defined as lack of the blood flow, which leads to insufficient oxygen and nutrient supply to the heart tissue (Korantzopoulos and Goudevenos 2007). The re-establishing of blood flow to ischemic zone called reperfusion, which triggers cell death in cardiomyocyte (Yellon and Hausenloy 2007). This circumstance is known as ischemia reperfusion (I/R) injury, which is often encountered in clinical intervention, such as thrombolysis, angioplasty, and heart transplantation (Zahn et al. 1997; Yellon and Hausenloy 2007). There is considerable evidence indicating that reactive oxygen species (ROS) generation as an initial cause of the injury (Ramaraj 2007; Yellon and Hausenloy 2007). ROS induce functional and structural damages of lipids, nucleic acids, and proteins in cardiomyocyte (Dhalla et al. 2000). Reperfusion itself can lead to an acceleration of ROS production, which inhibit mitochondrial electron transport chain and result in cell damage and cardiac dysfunction (Galiuto et al. 2000). The trigger of inflammatory response also involved in the development of I/R injury. Neutrophils that infiltrate to infarct myocardium mediate tissue damage by releasing ROS and matrix-degrading enzymes (Carbone et al. 2013). Cell apoptosis and death occur and finally lead to myocardial infarction (Zalesskii et al. 2002).
Targeting antiapoptotic mechanisms offer a potential approach to attenuate I/R-induced cell death. There are two major signaling involved in apoptotic cell death. One is intrinsic pathway via mitochondria, and the other is extrinsic pathway via Fas ligand or TNF-α (Peter 2011). In the intrinsic pathway, the proapoptotic bcl-2 family enhances the permeability of the mitochondrial outer membrane, releases of cytochrome c to cytoplasm, and finally activates caspase-3 to induce apoptosis (Marsden et al. 2002; Peter 2011). On the other hand, the extrinsic pathway is activated when death ligands, such as Fas or TNF-α, bind to the receptors and result in the recruitment and activation of caspase-8 to induce cell death (Ashkenazi and Dixit 1998; Scott et al. 2009). AKT regulates a variety of responses in mammalian cells, including inhibition of apoptosis and activation of survival pathway (Fujio et al. 2000). Activating the prosurvival kinase cascades during reperfusion has been demonstrated to protect against I/R-induced injury.
Thaliporphine, a phenolic aporphine alkaloid isolated from plants of several families (Kiryakov et al. 1981), possess anti-inflammatory, antioxidant response and exerts protective effect on cardiovascular system, including endotoxemia and I/R-induced cardiac dysfunction (Chiao et al. 2005; Chen et al. 2010; Lee et al. 2012). Our previous study proved that thaliporphine protected heart against I/R injury in a Langendorff perfused heart model, via a nitric oxide (NO)-dependent manner (Huang et al. 2001). In vivo study also demonstrated that thaliporphine decreased infarct size via activating opioid receptor and opening mitochondrial KATP channel during I/R injury (Chang et al. 2005). Our newly synthetic derivative of thaliporphine is TM-1-1DP synthesized by SS Lee’s Lab (Fig. 1). TM-1-1DP possesses greater water solubility than thaliporphine and is rapidly converted into TM-1-1. The distribution of TM-1-1 in the brain after intravenous injection of TM-1-1DP is less than 1 % of that in the peripheral blood (unpublished data). The effect of TM-1-1DP on cardiovascular function in animal is unknown; therefore, we aimed to investigate the response of TM-1-1DP in both animal and cell model. Accordingly, we assessed the effect of TM-1-1DP on myocardial I/R injury in rat and on hydrogen peroxide (H2O2)-induced oxidative stress in isolated adult rat cardiomyocytes.

Structure of thaliporphine and TM-1-1DP. The chemical name of thaliporphine is (R)-2,9,10-trimethoxy-6-methyl-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-1-ol. The chemical name of TM-1-1DP is 1,10-dimethoxy-6-(2-(2-methoxyphenoxy)ethyl)-5,6,6a,7-tetrahydro-4H-dibenzol[de,g]quinoline-2,9-diy1 bis(dihydrogen phosphate)
Materials and methods
Experimental animals
The research was conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85–23, revised 1996). Eight-week-old male Sprague–Dawley rats (BioLASCO Taiwan, Co., Ltd, Taipei, Taiwan) were used. Animals were maintained under a 12-h light/dark cycle at a controlled temperature (21 ± 2 °C) with free access to food and tap water.
Experimental model of myocardial I/R injury in vivo
Rats were anesthetized and were subjected to 1-h coronary artery occlusion, and followed by 2-h reperfusion as described previously (Ku et al. 2011). Sham groups received the same surgery process without ligation. TM-1-1DP was intravenously injected 10 min before reperfusion started in coronary artery occluded animals, which is similar and available in the clinical condition. Three different doses of TM-1-1DP were chosen to estimate the potency of the drug: low dose (0.005 mg/kg), medium dose (0.015 mg/kg), and high dose (0.05 mg/kg). After 2 h of reperfusion, the rats were sacrificed, and the hearts were harvested. The blood plasma samples were collected immediately and stored at −80 °C until used.
Determination of cardiac function in vivo
Cardiac function was estimated in vivo by a pressure–volume catheter (1.9 F; Scisense Instruments, Ontario, Canada) as described previously (Ku et al. 2010; Ku et al. 2011; Ku and Su 2014). The catheter was inserted into the carotid artery and then advanced into the left ventricle for the measurement of pressure and volume. Several parameters were chosen for analysis: left ventricular end-systolic (LVESP), left ventricular end-diastolic pressure (LVEDP), and stroke work (SW). SW is used to assess ventricular function by calculating the area within the PV loop. PV Loops were then determined under condition of preload change, elicited by inferior vena cava occlusion. The end-systolic pressure–volume relation (ESPVR) and end-diastolic pressure–volume relation (EDPVR) were, respectively, examined by the slope of end-systolic and end-diastolic pressure–volume points, which are load-independent indexes of myocardial contractility and ventricular compliance.
Determination of the myocardial risk size and the infarct size
At the end of reperfusion, the heart was excised and perfused on a Langendorff system with 95 % O2, 5 % CO2 Tyrode’s solution (137 mM NaCl, 11.9 mM NaHCO3, 11 mM glucose, 2 mM CaCl2, 1.1 mM MgCl2, 0.33 mM NaH2PO4, and 5.4 mM KCl). After the blood was washed out, the coronary artery was ligated again and perfused with 1 % evan blue to determine the risk area. The nonischemic myocardium stained blue, and the ischemic myocardium remained red. The risk size was calculated as a percentage of the red area to total ventricular area. Each heart was then sliced horizontally, and the slices were incubated in 1 % triphenyltetrazolium chloride (Sigma, St. Louis, MO, USA) prepared in normal saline for 30 min at 37 °C, and then placed in 10 % formaldehyde for 3 days. The infarct area appeared white, and the infarct size was calculated as a percentage of the white area to the total risk area.
Determination of plasma LDH, CK-MB, troponin concentration
Plasma CK-MB, LDH, and troponin concentration were measured by ELISA kit (Life Science, USA).
Protein extraction from cardiac tissue
Left ventricles were homogenized in RIPA buffer (Tris–HCl 50 mM, NaCl 150 mM, EGTA 1 mM, EDTA 1 mM, NP-40 1 %, sodium deoxycholate 1 %) containing cocktail protease and phosphatase inhibitor (Sigma, St. Louis, MO, USA). The supernatant of tissue homogenate was collected after centrifugation (800 × g, 10 min at 4 °C) and defined as total cardiac protein. Protein concentrations were determined by BCA protein assay kit (Thermo Fisher Scientific Inc., Rockford, IL, USA).
Detection of myeloperoxidase (MPO) activity in the heart
Cardiac tissue homogenate was used to detect MPO activity by using ELISA kit (Santa Cruz Biotechnology, Inc.).
Western blotting
The method was described in previous study (Ku et al. 2013). Cardiac protein expression was measured for TNF-α (Santa Cruz Biotechnology, Inc.), GAPDH (Santa Cruz Biotechnology, Inc.), AKT, p-AKT, (serine-473) (Cell Signaling Technology, Inc.), eNOS, p-eNOS (serine-1177) (Cell Signaling Technology, Inc.), and caspase-3 (Cell Signaling Technology, Inc.).
Adult rat cardiomyocyte isolation
Adult ventricular cardiomyocytes were isolated from Sprague–Dawley rats as described previously (Ku et al. 2013). Briefly, the hearts were excised and perfused with enzyme mixtures containing 0.04 % w/v collagenase (type II, Washington Biochemical Corp.) and 0.02 % w/v hyaluronidase for 20 min. After dissecting to small pieces, the heart was shaken in flasks with enzyme mixture for another 40 min. The collagenase digested ventricle suspensions were then filtered through a mesh to remove remnants of connective tissues. Cell suspensions were placed on top of BSA solution where noncardiomyocytes were removed by a density gradient. Cells were cultured in medium with Dulbecco’s Modified Eagle’s Medium supplemented with antibiotics (100 μg/ml penicillin and 100 μg/ml streptomycin) at 37 °C under a 5 % CO2–95 % air atmosphere. After the initial incubation period, quiescent cardiomyocytes were exposed to H2O2 (300 μM) for 1 h. TM-1-1DP (70 nM) was pretreated 1 h before H2O2 exposure.
Detection of cell viability
Cell viability was determined by MTT assay. Cells were treated with MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide) at 0.5 mg/ml. The purple formazan crystals were dissolved in DMSO. Solutions were then loaded in a 96-well plate and determined on an automated microplate spectrophotometer at 570 nm. Each condition tested was performed in triplicate in each experiment.
Detection of endogenously produced reactive oxygen species (ROS) in cardiomyocytes
ROS generation in cardiomyocytes was detected by labeling with fluorescence dye CM-H2DCFDA (5-(6)-chloromethyl-2′7′-dihydrofluoresceindiacetate). By using fluorescence microscopy, cardiomyocyte ROS level could be monitored at 488-nm excitation and 515-nm emission. Fluorescence intensity was calculated by averaging fluorescence intensity of numerous outlined cells using Imagequant (Molecular Dynamics, Inc., Sunnyvale, CA, USA).
Detection of leukocyte count and platelet bound to neutrophil
EDTA-anticoagulated blood was collected. Leukocyte count was by Coulter counter STKS (Coulter Electronics, Krefeld, Germany). Erythrocytes were lysed by lysing buffer. Mouse anti-rat CD42d-FITC was used to label platelets, and mouse anti-rat RP1-PE was used to label neutrophils (all from BD Biosciences Franklin Lakes, NJ). Flow cytometry assay was developed to analyze platelet and neutrophil complex in whole blood. The FSC/SSC gate around neutrophil population is selected, and 10,000 cells within the gated region were analyzed. Platelet bound to neutrophil was assessed by counting CD42d-FITC-positive events in the RP1-PE double-positive gate. In all experiments, suitable isotype controls were used to adjust for nonspecific antibody binding.
Statistical analysis
All values were presented as means ± SE. The results were analyzed using ANOVA followed by Bonferroni’s post hoc tests. P < 0.05 was considered as significant difference.
Results
TM-1-1DP alleviated injury after rat subjected to myocardial I/R
The risk size was similar among groups (Fig. 2a, b), indicating that the position of coronary artery occlusion was not significantly different. Rats subjected to coronary artery occlusion for 1 h and followed by reperfusion for 2 h resulted in myocardial infarction (Fig. 2a, c). Low dose of TM-1-1DP (0.005 mg/kg) treatment did not affect infarct size. However, high dose of TM-1-1DP (0.05 mg/kg) was shown to exert greater reduction on infarct size than medium dose (0.015 mg/kg), being 32.9 and 57.8 % of I/R group, respectively.

Effect of TM-1-1DP on myocardial I/R-induced injury. Cardiac injury was shown after rats subjected to 1-h coronary artery occlusion and followed by 2-h reperfusion. a Representative cardiac slices after I/R were shown. b Risk size and c infarct size were reported. The nonischemic myocardium stained blue, and the remnants indicated ischemic myocardium. The infarct myocardium stained white, and the noninfarct myocardium stained red. Ratio of red plus white zone to total zone was defined as risk size. Ratio of white zone to red plus white zone was defined as infarct size. d Plasma CK-MB, e LDH, and f troponin concentration were measured. (n = 6) *P < 0.05 vs sham, #P < 0.05 vs I/R
CK-MB and LDH are markers of myocardial injury. I/R injury resulted in the release of CK-MB and LDH in blood, which were 3.7- and 2.9-fold higher than sham, respectively (Fig. 2d, e). High dose of TM-1-1DP significantly ameliorated CK-MB and LDH content. Since the protective effect of TM-1-1DP was only significant at high dose, the dosage used by the following study was chosen in high dose (0.05 mg/kg) through the experiment.
Troponin is a specific marker of myocardial injury. I/R injury resulted in a prominent elevation of troponin in blood, which were significantly alleviated by TM-1-1DP (0.05 mg/kg) (Fig. 2f).
TM-1-1DP improved cardiac function after rat subjected to myocardial I/R
The cardiac functions were measured by PV loops after rat subjected to myocardial I/R (Fig. 3), and the data were all quantified (Table 1). There was no significant difference in PV loops between sham and sham + TM-1-1DP in rat. I/R caused a reduction of LVESP (63.5 % of sham) and an elevation of LVEDP (2.1-fold of sham), indicating that cardiac dysfunction occurred after I/R. Stroke work markedly decreased by 51.8 % after I/R, compared to the sham group. Furthermore, load-independent cardiac function indexes attenuated after I/R, both ESPVR and EDPVR values markedly decreased, being 70.2 and 10.1 % of sham group, respectively.

Effect of TM-1-1DP on myocardial I/R-induced cardiac dysfunction. Cardiac function was measured by PV loop after rats subjected to 1-h coronary artery occlusion and followed by 2-h reperfusion. The representative PV loops were shown among groups
TM-1-1DP-treated rats had better cardiac performance after I/R. LVESP attenuated less in TM-1-1DP-treated group, being 77.1 % of sham, along with LVEDP elevated less (1.4-fold of sham). Load-independent values were also preserved by TM-1-1DP treatment; both ESPVR and EDPVR values remained by 93.4 and 35.6 % of sham, respectively, implying that TM-1-1DP treatment markedly improved cardiac systolic and diastolic function after rat subjected to I/R.
TM-1-1DP improved AKT-eNOS pathway after rat subjected to myocardial I/R
The levels of cardiac AKT and eNOS phosphorylation in rats subjected to I/R were decreased, being 31.7 and 40.5 % of sham group, respectively (Fig. 4a–c). The phosphorylation of AKT and eNOS was both markedly recovered by TM-1-1DP treatment. The reduction of eNOS phosphorylation in I/R rats was correlated with the NO content. Plasma NO concentration was decreased by 57.8 % after rat subjected to I/R (Fig. 4d), compared to the sham group. TM-1-1DP-treated rats augmented plasma NO concentration after I/R, which was coincided with the preservation of eNOS phorphorylation.

Effect of TM-1-1DP on myocardial I/R-induced changes of AKT-eNOS pathway. Proteins were detected after rats subjected to I/R. a Original Western blots of AKT and eNOS phosphorylation were shown. Ratios of b p-AKT to AKT and c p-eNOS to eNOS were calculated. d Plasma NO concentration was also measured after I/R. (n = 5) *P < 0.05 vs sham, #P < 0.05 vs I/R
TM-1-1DP alleviated myocardial I/R-induced death-related protein
I/R resulted in a 4.6- and 4.9-fold elevation of cardiac caspase-3 and TNF-α expression (Fig. 5), respectively, compared to the sham group. Treatment with TM-1-1DP alleviated both I/R-induced caspase-3 and TNF-α expression.

Effect of TM-1-1DP on myocardial I/R-induced changes of caspase-3 and TNF-α protein expression. Proteins were detected after rats subjected to I/R. a Original Western blots of casepase-3 and TNF-α were shown. Ratios of b caspase-3 to GAPDH and c TNF-α to GAPDH were calculated. (n = 5) *P < 0.05 vs sham, #P < 0.05 vs I/R
TM-1-1DP alleviated inflammatory response in myocardial I/R
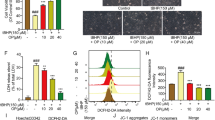
MPO activity, a marker of neutrophil accumulation, was increased in I/R heart, which was attenuated by TM-1-1DP (Fig. 6a). Blood samples were collected after I/R. Leukocyte count markedly increased after I/R, which was diminished by TM-1-1DP treatment (Fig. 6b). Platelet bound to neutrophil was assessed by counting the percentage of CD42d and RP1 double-positive cell (Fig. 6c). IR induced the formation of platelet and neutrophil complex, which were attenuated by TM-1-1DP treatment.

Effect of TM-1-1DP on myocardial I/R-induced changes of inflammatory response. a Myeloperoxidase (MPO) activity was detected in I/R-injured cardiac tissue. b Blood was collected after rats subjected to I/R. Leukocyte count in blood was measured. c Platelet was labeled by CD42d-FITC, and neutrophil was labeled by RP1-PE. Platelet bound to neutrophil was assessed by counting CD42d-FITC-positive events in the RP1-PE double-positive gate. The original flow cytrometry plots were shown, and the percentage of platelet and neutrophil complex was also calculated. (n = 4) *P < 0.05 vs sham, #P < 0.05 vs I/R
TM-1-1DP decreased ROS stress and improved cell vitality in isolated adult rat cardiomyocyte after H2O2 exposure
H2O2 exposure resulted in an elevation of ROS production in adult rat cardiomyocyte (Fig. 7a, b), along with an augmentation of caspase-3 activity (Fig. 7c). ROS and caspase-3 activtity induced by H2O2 were both ameliorated by TM-1-1DP treatment. Cell viability decreased after H2O2 exposure in adult rat cardiomyocyte (Fig. 7d). Treatment with TM-1-1DP was capable of improving cell viability, while L-NAME, a NOS inhibitor, abrogated the protective effect.

Effect of TM-1-1DP on H2O2-induced ROS stress in isolated adult rat cardiomyocyte. Isolated adult rat cardiomyocyte underwent H2O2 treatment for 1 h. a Original microscopy photos were reported for ROS production in cardiomyocyte and b results of densitometry. c Caspase-3 activity and d cell viability were measured after H2O2-induced stress. L-NAME is a nitric oxide synthesis inhibitor. Bar = 100 μm (n = 4) *P < 0.05 vs control, #P < 0.05 vs H2O2, @P < 0.05 vs H2O2 + TM-1-1DP
Discussion
In our present study, we demonstrated that TM-1-1DP, a newly phenolic aporphine alkaloid derivative, exerted cardioprotective effect. TM-1-1DP-treated rats had better cardiac performance after I/R injury, including the alleviation of infarct size, cardiac injury marker, and the improvement of cardiac function. The protective effect is associated with the activation of AKT-eNOS pathway, the alleviation of inflammatory response, and the inhibition of cell death-related pathway. The finding was further confirmed with the cell study, in which TM-1-1DP treatment significantly improved cell viability during H2O2-induced oxidative stress in a NO-dependent mechanism. TM-1-1DP possess higher potency on treatment of myocardial I/R than its original compound, thaliporphine. In the previous study, thaliporphine were found to reduce infarct size at the dose of 0.05–0.5 mg/kg after myocardial I/R [12]. This report demonstrated that TM-1-1DP decreased infarct size at the dose of 0.015–0.05 mg/kg. TM-1-1DP is a newly potential drug for the treatment of myocardial infarction.
ROS are the major initiators of myocardial damage during I/R (Hori and Nishida 2009). The deleterious radicals are hydroxyl radical, superoxide, and H2O2, which are derived from a variety of sources such as activated neutrophils, xanthine oxidase system, and electron transport chain of mitochondria (Nayler 1992). ROS production is eliminated by endogenous antioxidant enzymes, such as superoxide dismutase, catalase, glutathione peroxidase, and thioredoxin reductase systems (Dhalla et al. 2000). Overexpression of superoxide dismutase protected against myocardial I/R injury in mice (Chen et al. 1998). In this regard, targeting ROS with various antioxidant agents or upregulation of endogenous free radical scavengers has been shown to reduce injury during oxidative stress and preserve cardiac function from I/R injury (Dhalla et al. 2000; Hori and Nishida 2009). ROS stimulate the generation of inflammatory cytokines, such as TNF-α and interleukin, whereas inflammatory cytokines stimulate ROS formation and lead to a consequence of vicious cycle (Zhang and Chen 2008). Antioxidant and anti-inflammatory treatments are promising approach for myocardial infarction (Hori and Nishida 2009). In our study, TM-1-1DP was shown to alleviate ROS concentration in cardiomyocyte after H2O2 exposure. The antioxidant effect of TM-1-1DP was correlated with the anti-inflammatory response in the heart after I/R.
An elevation of leukocyte count after myocardial infarction is associated with poor outcome in clinic (Madjid et al. 2004). The inflammatory response of I/R injury mediated largely by neutrophil, including ROS generation, release of proteases, arachidonic metabolites, and other proinflammatory mediators (Carbone et al. 2013). The upregulation of cellular adhesion molecules promotes the recruitment and infiltration of neutrophils to injury sites (Golias et al. 2007). Infiltration of neutrophil into ischemic cardiac tissue aggravates I/R injury and leads to infarct expansion, while neutrophil depletion markedly reduced infarct size in animal model (Litt et al. 1989). Our investigation observed that the elevation of leukocyte count was parallelized with the increased level of MPO activity in the heart after I/R, indicating the role of inflammatory response and neutrophil infiltration. Activated platelets participate in the process of neutrophil recruitment and aggravate myocardial tissue damage (Xu et al. 2006). The formation of platelet–neutrophil complex (PNC) promotes neutrophil activation, along with the release of cytokines, and the extension of tissue injury (Zarbock et al. 2007; Kohler et al. 2011). Since the alleviation of PNC formation is protective (Zarbock et al. 2007), TM-1-1DP that attenuated the percentage of PNC after I/R may contribute to its cardioprotective effect. The role of vasodilator-stimulated phosphoprotein (VASP) in regulation of PNC formation has recently been proposed (Kohler et al. 2011). VASP can be phosphorylated in a cAMP or cGMP-dependent manner (Walter and Gambaryan 2009). The phosphorylation results in conformational changes of the cellular surface (Ferron et al. 2007) and influences the presence of the CD11b receptor on neutrophils and of GP IIb/IIIa on platelets (Anderson et al. 2003; Kohler et al. 2011). VASP phosphorylation resulted in reduced formation of PNC and dampened the extent of myocardial I/R injury (Kohler et al. 2011).
The activation of AKT can stimulate the survival pathway to protect myocardium from apoptosis during I/R injury (Mullonkal and Toledo-Pereyra 2007; Ravingerova et al. 2007). Inhibition of AKT signaling by PI3K/AKT inhibitor or genetic deletion ablated the protective effect (Mullonkal and Toledo-Pereyra 2007). Conversely, overexpression of AKT limited myocardial infarct size (Hua et al. 2007). AKT phosphorylation activates several downstream targets including eNOS. AKT phosphorylates eNOS at serine-1177 residue (Dimmeler et al. 1999) and mediates rapid eNOS activation in response to stress of in vitro and in vivo studies (Dimmeler et al. 1999; Oudit et al. 2004). Activation of eNOS plays an important role on mediating the cardioprotective effect against I/R injury (Korantzopoulos and Goudevenos 2007; Kukreja and Xi 2007). Furthermore, it has been demonstrated that pharmacological agents enhancing the activity of eNOS during myocardial I/R is beneficial to the heart for the preservation of cardiac function (Laufs et al. 2002; Li et al. 2012). In our study, AKT and eNOS phorphorylation improved by TM-1-1DP during myocardial I/R injury, along with elevation of NO concentration. Furthermore, the protective effect of TM-1-1DP was abolished by L-NAME, a NOS inhibitor, in isolated cardiomyocytes exposed to H2O2. TM-1-1DP not only activate survival pathway but also inhibit apoptotic pathway. In our study, TNF-α and caspase-3 expressions were ameliorated both by TM-1-1DP treatment in myocardial I/R injury, which were coincided with the alleviation of caspase-3 activity by TM-1-1DP in cardiomyocyte after H2O2 exposure. The improvement of AKT-eNOS signaling contributed to the protective action of TM-1-1DP.
In conclusion, the present study identified the cardioprotective mechanisms of TM-1-1DP. TM-1-1DP improved cardiac function and reduced infarct size via antioxidant, anti-inflammatory effects, and activating AKT-eNOS signaling. We provide a new insight into this therapeutic potential for phenolic aporphine alkaloid in myocardial I/R.
References
Anderson SI, Behrendt B, Machesky LM, Insall RH, Nash GB (2003) Linked regulation of motility and integrin function in activated migrating neutrophils revealed by interference in remodelling of the cytoskeleton. Cell Motil Cytoskeleton 54:135–146
Ashkenazi A, Dixit VM (1998) Death receptors: signaling and modulation. Science 281:1305–1308
Carbone F, Nencioni A, Mach F, Vuilleumier N, Montecucco F (2013) Pathophysiological role of neutrophils in acute myocardial infarction. Thromb Haemost 110:501–514
Chang WL, Lee SS, Su MJ (2005) Attenuation of post-ischemia reperfusion injury by thaliporphine and morphine in rat hearts. J Biomed Sci 12:611–619
Chen Z, Siu B, Ho YS, Vincent R, Chua CC, Hamdy RC, Chua BH (1998) Overexpression of MnSOD protects against myocardial ischemia/reperfusion injury in transgenic mice. J Mol Cell Cardiol 30:2281–2289
Chen WP, Tzeng HJ, Ku HC, Ho YJ, Lee SS, Su MJ (2010) Thaliporphine ameliorates cardiac depression in endotoxemic rats through attenuating TLR4 signaling in the downstream of TAK-1 phosphorylation and NF-kappaB signaling. Naunyn Schmiedebergs Arch Pharmacol 382:441–453
Chiao CW, Lee SS, Wu CC, Su MJ (2005) Thaliporphine increases survival rate and attenuates multiple organ injury in LPS-induced endotoxaemia. Naunyn Schmiedebergs Arch Pharmacol 371:34–43
Dhalla NS, Elmoselhi AB, Hata T, Makino N (2000) Status of myocardial antioxidants in ischemia-reperfusion injury. Cardiovasc Res 47:446–456
Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM (1999) Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399:601–605
Ferron F, Rebowski G, Lee SH, Dominguez R (2007) Structural basis for the recruitment of profilin-actin complexes during filament elongation by Ena/VASP. EMBO J 26:4597–4606
Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K (2000) Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation 101:660–667
Galiuto L, DeMaria AN, Iliceto S (2000) Microvascular damage during myocardial ischemia-reperfusion: pathophysiology, clinical implications and potential therapeutic approach evaluated by myocardial contrast echocardiography. Ital Heart J 1:108–116
Golias C, Tsoutsi E, Matziridis A, Makridis P, Batistatou A, Charalabopoulos K (2007) Review. Leukocyte and endothelial cell adhesion molecules in inflammation focusing on inflammatory heart disease. In Vivo 21:757–769
Hori M, Nishida K (2009) Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc Res 81:457–464
Hua F, Ha T, Ma J, Li Y, Kelley J, Gao X, Browder IW, Kao RL, Williams DL, Li C (2007) Protection against myocardial ischemia/reperfusion injury in TLR4-deficient mice is mediated through a phosphoinositide 3-kinase-dependent mechanism. J Immunol 178:7317–7324
Huang LM, Lee SS, Chen JK, Huang SS, Su MJ (2001) Thaliporphine protects ischemia and ischemia-reperfused rat hearts via an NO-dependent mechanism. Drug Dev Res 52:446–453
Kiryakov HG, Iskrenova E, Kuzmanov B, Evstatieva L (1981) Alkaloids from Corydalis bulbosa. Planta Med 43:51–55
Kohler D, Straub A, Weissmuller T, Faigle M, Bender S, Lehmann R, Wendel HP, Kurz J, Walter U, Zacharowski K, Rosenberger P (2011) Phosphorylation of vasodilator-stimulated phosphoprotein prevents platelet-neutrophil complex formation and dampens myocardial ischemia-reperfusion injury. Circulation 123:2579–2590
Korantzopoulos PG, Goudevenos JA (2007) Myocardial reperfusion injury. N Engl J Med 357:2409, author reply 2409-2410
Ku HC, Su MJ (2014) DPP4 deficiency preserved cardiac function in abdominal aortic banding rats. PLoS One 9:e85634
Ku HC, Chen WP, Su MJ (2010) GLP-1 signaling preserves cardiac function in endotoxemic Fischer 344 and DPP4-deficient rats. Naunyn Schmiedebergs Arch Pharmacol 382:463–474
Ku HC, Chen WP, Su MJ (2011) DPP4 deficiency preserves cardiac function via GLP-1 signaling in rats subjected to myocardial ischemia/reperfusion. Naunyn Schmiedebergs Arch Pharmacol 384:197–207
Ku HC, Chen WP, Su MJ (2013) DPP4 deficiency exerts protective effect against H2O2 induced oxidative stress in isolated cardiomyocytes. PLoS One 8:e54518
Kukreja RC, Xi L (2007) eNOS phosphorylation: a pivotal molecular switch in vasodilation and cardioprotection? J Mol Cell Cardiol 42:280–282
Laufs U, Gertz K, Dirnagl U, Bohm M, Nickenig G, Endres M (2002) Rosuvastatin, a new HMG-CoA reductase inhibitor, upregulates endothelial nitric oxide synthase and protects from ischemic stroke in mice. Brain Res 942:23–30
Lee AS, Chen WP, Kuo YL, Ho YJ, Lee SS, Su MJ (2012) Thaliporphine preserves cardiac function of endotoxemic rabbits by both directly and indirectly attenuating NFkappaB signaling pathway. PLoS One 7:e39174
Li XD, Yang YJ, Geng YJ, Zhao JL, Zhang HT, Cheng YT, Wu YL (2012) Phosphorylation of endothelial NOS contributes to simvastatin protection against myocardial no-reflow and infarction in reperfused swine hearts: partially via the PKA signaling pathway. Acta Pharmacol Sin 33:879–887
Litt MR, Jeremy RW, Weisman HF, Winkelstein JA, Becker LC (1989) Neutrophil depletion limited to reperfusion reduces myocardial infarct size after 90 minutes of ischemia. Evidence for neutrophil-mediated reperfusion injury. Circulation 80:1816–1827
Madjid M, Awan I, Willerson JT, Casscells SW (2004) Leukocyte count and coronary heart disease: implications for risk assessment. J Am Coll Cardiol 44:1945–1956
Marsden VS, O'Connor L, O'Reilly LA, Silke J, Metcalf D, Ekert PG, Huang DC, Cecconi F, Kuida K, Tomaselli KJ, Roy S, Nicholson DW, Vaux DL, Bouillet P, Adams JM, Strasser A (2002) Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature 419:634–637
Mullonkal CJ, Toledo-Pereyra LH (2007) Akt in ischemia and reperfusion. J Invest Surg 20:195–203
Nayler WG (1992) The role of oxygen radicals during reperfusion. J Cardiovasc Pharmacol 20(Suppl 5):S14–S17
Oudit GY, Sun H, Kerfant BG, Crackower MA, Penninger JM, Backx PH (2004) The role of phosphoinositide-3 kinase and PTEN in cardiovascular physiology and disease. J Mol Cell Cardiol 37:449–471
Peter ME (2011) Programmed cell death: apoptosis meets necrosis. Nature 471:310–312
Ramaraj R (2007) Myocardial reperfusion injury. N Engl J Med 357:2408, author reply 2409-2410
Ravingerova T, Matejikova J, Neckar J, Andelova E, Kolar F (2007) Differential role of PI3K/Akt pathway in the infarct size limitation and antiarrhythmic protection in the rat heart. Mol Cell Biochem 297:111–120
Scott FL, Stec B, Pop C, Dobaczewska MK, Lee JJ, Monosov E, Robinson H, Salvesen GS, Schwarzenbacher R, Riedl SJ (2009) The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature 457:1019–1022
Walter U, Gambaryan S (2009) cGMP and cGMP-dependent protein kinase in platelets and blood cells. Handb Exp Pharmacol 2009:533–548
Xu Y, Huo Y, Toufektsian MC, Ramos SI, Ma Y, Tejani AD, French BA, Yang Z (2006) Activated platelets contribute importantly to myocardial reperfusion injury. Am J Physiol Heart Circ Physiol 290:H692–H699
Yellon DM, Hausenloy DJ (2007) Myocardial reperfusion injury. N Engl J Med 357:1121–1135
Zahn R, Koch A, Rustige J, Schiele R, Wirtzfeld A, Neuhaus KL, Kuhn H, Gulker H, Senges J (1997) Primary angioplasty versus thrombolysis in the treatment of acute myocardial infarction. ALKK Study Group. Am J Cardiol 79:264–269
Zalesskii VN, Gavrilenko TI, Fil'chenkov AA (2002) [Apoptosis in myocardial ischemia and reperfusion]. Lik Sprava: 8–15
Zarbock A, Polanowska-Grabowska RK, Ley K (2007) Platelet-neutrophil-interactions: linking hemostasis and inflammation. Blood Rev 21:99–111
Zhang M, Chen L (2008) Status of cytokines in ischemia reperfusion induced heart injury. Cardiovasc Hematol Disord Drug Targets 8:161–172
Acknowledgments
The authors’ work was supported by the grant from National Science Council of Taiwan, ROC (MOST-100-2325-B-002-066, MOST-101-2325-B-002-058, MOST-102-2325-B-002-086, MOST 103-2325-B-002-020), and National Taiwan University (98A801).
Conflicts of interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
Shih-Yi Lee is the co-first author.
Rights and permissions
About this article

Cite this article
Ku, HC., Lee, SY., Chen, CH. et al. TM-1-1DP exerts protective effect against myocardial ischemia reperfusion injury via AKT-eNOS pathway. Naunyn-Schmiedeberg's Arch Pharmacol 388, 539–548 (2015). https://doi.org/10.1007/s00210-015-1098-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-015-1098-1