
Abstract
The misuse of novichok agents in assassination attempts has been reported in the international media since 2018. These relatively new class of neurotoxic agents is claimed to be more toxic than the agents of the G and V series and so far, there is no report yet in literature about potential antidotes against them. To shed some light into this issue, we report here the design and synthesis of NTMGMP, a surrogate of A-242 and also the first surrogate of a novichok agent useful for experimental evaluation of antidotes. Furthermore, the efficiency of the current commercial oximes to reactivate NTMGMP-inhibited acetylcholinesterase (AChE) was evaluated. The Ellman test was used to confirm the complete inhibition of AChE, and to compare the subsequent rates of reactivation in vitro as well as to evaluate aging. In parallel, molecular docking, molecular dynamics and MM-PBSA studies were performed on a computational model of the human AChE (HssAChE)/NTMGMP complex to assess the reactivation performances of the commercial oximes in silico. Experimental and theoretical studies matched the exact hierarchy of efficiency and pointed to trimedoxime as the most promising commercial oxime for reactivation of AChE inhibited by A-242.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
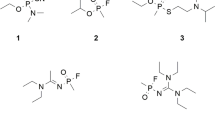
The nerve agents of the A-series are organophosphorus (OP) compounds regarded as strong acetylcholinesterase (AChE) inhibitors, which were recently added to Schedule 1 of the Annex on Chemicals of the Chemical Weapons Convention—CWC (https://www.opcw.org/chemical-weapons-convention/annexes/annex-chemicals/schedule-1). These substances were allegedly created between the 1970s and 1990s, during the Cold War, in an attempt to develop a new powerful class of neurotoxic chemical agents (Franca et al. 2019; Kloske and Witkiewicz 2019; Mirzayanov 2009; Nepovimova and Kuca 2018, 2019). The most likely structures of the compounds developed in this context are the phosphoramidates listed in Fig. 1 (Mirzayanov 2009).

Structures of the A-series agents and the clinically available pyridinium oximes: A-230 (1), A-232 (2), A-234 (3), A-242 (4), A-262 (5), 2-PAM (6), trimedoxime (7), obidoxime (8) and HI-6 (9)
Recent events involving A-series agents have highlighted the urgency of a better understanding of their toxic profile to develop more efficient medical countermeasures, particularly antidotes capable of reactivating the inhibited AChE (Lewis 2018; Steindl et al. 2021). Due to the potential threat posed by these toxic chemicals, different research groups around the world have been studying the subject, aiming to overcome the lack of knowledge about their properties, deleterious effects and plausible countermeasures (Bolt and Hengstler 2022; Imrit et al. 2020; Jeong and Choi 2019; Nepovimova and Kuca 2018). Nevertheless, there is still an absence of information regarding the effectiveness of the existing reactivators (Fig. 1) against intoxications with the A-series agents (Gorecki et al. 2016; Kucera et al. 2019). Although pyridinium oximes such as pralidoxime (6, 2-PAM), trimedoxime (7, TMB), obidoxime (8, OBD) and HI-6 (9, asoxime) are expected to have reasonable results reactivating the inhibited AChE, their poor ability to permeate into the blood–brain barrier still poses a limitation, and therefore, the search for a universal antidote is an unmet need. Thus, research on novel medical countermeasures and better comprehension of the adducts and interactions of those agents inside the active site and other relevant pockets of AChE are clearly warranted.
Considering the high risks involved with the handling of these substances, demanding complex infrastructure and specialized training, one alternative resource is the use of surrogates as toxicological tools. Surrogates are molecules that, in general, are able to afford similar enzymatic adducts of the actual nerve agents but usually have features that allow safer handling for trained personnel (de A Cavalcante et al. 2019a, b). In addition, molecular modeling studies are a valuable instrument to investigate and elucidate the interactions of chemical agents with biological systems. In this context, the use of surrogates and molecular modeling are powerful combinations to advance antidote research, with reduced risks, allowing a reliable extrapolation of the results to actual hazardous substances (Santos et al. 2022) (Cavalcante et al. 2019b).
In this work, we synthesized a surrogate for A-242 and evaluated the efficacy of the clinically available pyridinium oximes in the reactivation of AChE inhibited by this surrogate using an enzyme model (Electrophorus eel acetylcholinesterase, EeAChE) and our in-house methodology for Ellman’s assay (De A Cavalcante et al. 2018). In addition to in vitro data, we also performed computational simulations using the human AChE (HssAChE) as an enzyme model, comprising docking studies followed by molecular dynamics (MD) simulations and molecular mechanics—Poisson Boltzmann surface area (MM-PBSA) calculations (Homeyer and Gohlke 2012; Kumari et al. 2014). Using the near-attack conformation (NAC) criterion (Allgardsson et al. 2016; da Silva et al. 2019; Santos et al. 2022 (Fig. 2), we compared the in silico and in vitro results, assessing the reliability of the biochemical tool designed and the potential use of current oximes as efficient medical countermeasures.

Illustration of the NAC concept using A-242 (4) as the OP and trimedoxime (7) as the reactivator. According to this approach, to enable the attack of the oxime on OP-inhibited AChE, the distance between the O atom of the oxime and the P atom of the OP-Ser203 adduct (dOP) should be close to the O–P bond van der Waals contact of 3.3 Å (Johnson et al. 2018), while the angle among the O atom of the oxime group, the P of the OP-Ser203 adduct, and the O of Ser203 (θOPO) should be close to 180° (da Silva et al. 2019). Therefore, the optimum angle/distance ratio would be approximately 54.55
Results
Design and synthesis of the A-242 surrogate: 4-nitrophenyl N-(bis(dimethylamino)methylene)-P-methylphosphonamidate (NTMGMP, 10)
NTMGMP (10) was designed by essentially changing the leaving group (fluoride in the actual agent, 4-nitrophenolate in our surrogate), in accordance with the literature (Cavalcante et al. 2019a; Kitagawa et al. 2021; Meek et al. 2012). Therefore, NTMGMP is a molecular hybrid between A-242 (4) and the pesticide paraoxon (11), according to Scheme 1.

Rational design of the NTMGMP (10)
The one-pot synthesis of NTMGMP is depicted in Scheme 2, starting from dimethylphosphonic dichloride (MPDC, 12). Due to the potential toxicity of this surrogate, we opted to not purify the compounds for safety reasons, as they are the major impurities 1,1,3,3-tetramethylguanidine (TMG) and 4-nitrophenol (PNP).

Synthetic route to NTMGMP (10)
After completion of the reaction (by HPLC-MS (ESI +)), the reaction mixture was filtered through a 0.22 µm syringe filter, transferred into a round-bottomed flask, and evaporated carefully to dryness. After treating the residue with hot hexane and chilling at − 25 °C overnight, a yellow oil was obtained. After HPLC-MS-MS (ESI +) analysis (Fig. 3), the presence of the desired compound was confirmed. A proposal for fragmentation is given in Scheme 3.

HPLC-MS-MS of crude NTMGMP

Proposed MS–MS fragmentation of NTMGMP (10)
Analysis of NMR data confirmed the structure of NTMGMP. The 1H-NMR spectrum showed a doublet at δ 1.66 ppm (H3C-P, 2JHP 16.37 Hz). In addition to all other relevant resonances related to the surrogate, the methyl groups related to 1,1,3,3-tetramethylguanidine can be found at δ 3.12 ppm, and two aromatic doublets at 7.38 ppm (2H, 3JHH 9.14 Hz) and 8.25 ppm (2H, 3JHH 9.15 Hz), related to the 4-nitrophenol moiety. The 31P NMR spectrum showed only one signal at 20.96 ppm. Purity was estimated as 50% by 1H-NMR, which was used for toxicological assessments. NMR data are available in Figures S1-S7 of the supplementary material.
Ellman’s test
A modified Ellman’s spectrophotometric assay was employed to estimate the inhibition caused by our crude surrogate and the reactivation using clinical oximes (De A Cavalcante et al. 2018). We prepared seven test solutions to identify which one could afford the highest AChE inhibition at the lowest concentration after 10 min of incubation (1 μmol/L). The results are given in Table 1. This concentration was used for reactivation assays and proved that our toxicological tool could be useful for this kind of assessment.
The in vitro reactivating potential of the clinical oximes 2-PAM (6), trimedoxime (7), obidoxime (8) and HI-6 (9) (Table 2) was also evaluated through our modified Ellman’s spectrophotometric assay (De A Cavalcante et al. 2018). All tested compounds presented reactivation at a maximum of 100 μM (highest concentration tolerated in vivo) (Bajgar 2004; Bajgar et al. 2007; Tattersall 1993).
Since trimedoxime (7) presented the best results of reactivation of the complex AChE/NTMGMP at 100 μmol/L, it was chosen for the aging estimation of this complex. After enzyme inhibition for 10 and 90 min, reactivation assays were performed accordingly, and no aging was observed.
Molecular modeling studies
The model constructed for the HssAChE/NTMGMP complex (see “methodology” section for more details) obtained a Ramachandran plot, with no residue found in disallowed regions, 98.6% in allowed regions and 1.3% in generously allowed regions (data not shown) (Ramachandran et al. 1963). Additionally, the server Verify 3D (Bowie et al. 1991; Lüthy et al. 1992) showed that 98.17% of the residues had an average 3D-1D score higher than or equal to 0.2 (data not shown). These results were satisfactory for validating this model in further studies.
The results of ionization studies of the commercial oximes under physiological pH (7.4) suggest that all observed deprotonations occur at the hydroxyl group. 2-PAM and HI-6 (9) showed only one deprotonated form, while obidoxime (8) and trimedoxime (7) showed more than one ionizable hydrogen. For these last two oximes, however, forms with more than one lost hydrogen were not considered in this study, since the percentages of those coexisting species are negligible. Therefore, only the species shown in Table 1 were used in our modeling studies.
Molecular docking
The redocking procedure performed to validate the docking protocol used (see “methodology”) resulted in a pose with an RMSD of 0.88 Å between HI-6 and its position in the crystallographic structure. Considering the limit of 2.00 Å, which is well established in the literature (Kontoyianni et al. 2004), the docking parameters were considered adequate to simulate the interactions inside the HssAChE/NTMGMP/oxime complexes.
All ionization forms from Table 3 were submitted to the docking studies using the software Molegro Virtual Docker (MVD®), where all angles between the O of the oxime group, the P of the OP-Ser203 adduct, and the O of Ser203 (θOPO) were recorded, together with the distances between the O of the oxime group and the P of the OP (dOP), to calculate the respective values of Rθd. The results of the best pose obtained for each ligand were ranked based on the higher values of Rθd, according to the NAC criterion (see Fig. 2). As seen in Table 4, HI-6 (9), in both protonated and deprotonated forms, was the oxime presenting the Rθd values closest to the ideal value of 54.55, followed by 2-PAM (6), trimedoxime (7) and obidoxime (8). The poses from Table 4 were exported from MVD® as .mol2 files to be used in further steps of MD simulations.
MD simulations and MM-PBSA calculations
The eight HssAChE/NTMGMP/oxime complexes presented quick stabilization during the MD simulations, as shown in the energy and RMSD plots in Figs. S8 and S9 of the supplementary material. The plots of total and mean energy of each complex over time show that they were able to reach stable values of energy with low standard deviations. The corresponding average energy values are presented in the bar chart in Fig. 4.

Average values of energy for each HssAChE/NTMGMP/oxime complex (D = deprotonated)
To evaluate the results through the NAC criterion, dOP and θOPO were registered along the simulation. The results are presented in Figs. 5 and 6, respectively, separating protonated and deprotonated oximes for better visualization and analysis.

Distances over time during MD simulation: a protonated oximes and b deprotonated oximes

Angles over time during MD simulation: a protonated oximes and b deprotonated oximes
According to the MD simulations, HI-6 (9) continued to present the most promising result among the protonated oximes, with higher values of θOPO. Among the deprotonated oximes, obidoxime (8) was slightly better than 2-PAM (6) and HI-6 (9). As seen in the dOP results, trimedoxime (7) was less affected by deprotonation. The average values of dOP and θOPO are presented in Table 5, with the resulting values of Rθd.
Along with the Rθd results, the binding free energies estimated by the MM-PBSA method (Fig. 7) provide useful information to evaluate the affinity between oximes and the inhibited HssAChE. Corroborating what was observed for the dOP results, deprotonated oximes have much less affinity with the active site than their respective protonated forms. Trimedoxime (7) showed the best result, followed by HI-6 (9) and obidoxime (8).

Average values of binding free energy for each HssAChE/NTMGMP/oxime complex (D = deprotonated)
The hydrogen bonds formed during the simulation were also analyzed, and the graphics are shown in Figure S10 in the supplementary material. Table 6 presents the relevant average number of H-bonds per timeframe (values under 0.1 were disregarded).
Discussion
In contrast to the literature (Hosseini et al. 2016), the synthetic route devised for NTMGMP in this work (Scheme 2) used methylphosphonic dichloride (12) as the starting material. This chemical is usually in stock in our lab due to the work on the synthesis of reference compounds related to CWC as part of our activities as an OPCW Designated Laboratory for the analysis of environmental samples. Therefore, the research group at IDQBRN is used to work with such toxic compounds (Cavalcante et al. 2019a; Kitagawa et al. 2021) and possesses the necessary preparedness to safely perform all operations.
Considering the current knowledge on the reactions involving OP nerve agents, it is expected that the phosphoramido moiety of NTMGMP (10) will bind to the hydroxyl group of the serine residue of the AChE active site, in accordance with Scheme 4 below, affording the enzymatic adduct (22) similar to that yielded by authentic A-242, pivotal for the assessment of the reactivation potential of clinical oximes. Therefore, 22 was the HssAChE/NTMGMP complex used in our modeling studies.

Proposed reaction of NTMGMP (10) with AChE
Based on the design of the surrogate, available literature and experience with NEMP, a VX surrogate (25, Fig. 8), four different synthetic approaches were explored (procedures A-D). The first attempt to synthesize NTMGMPs was simultaneously conducted in two different ways. In procedure A, at room temperature, a solution of dimethylphosphonic dichloride (100 mg, 1 eq.) was prepared in dry dichloromethane. After cooling at 0 °C, we added 4-nitrophenol (PNP, 1.2 eq) and triethylamine (1.5 eq.) in dichloromethane (1 mL), and then, after 2 h, 1,1,3,3-tetramethylguanidine (TMG, 1.2 eq.) and triethylamine (1.5 eq.) in dry dichloromethane (1 mL) were added at 0 °C and stirred at this temperature for 4 h and at room temperature for 30 min. In procedure B, it was added dropwise into a solution of dimethylphosphonic dichloride in dichloromethane, the same reactants, but in reverse order, keeping the same reaction times and molar equivalents. At the completion of the reaction (confirmed by LC–MS (ESI +), m/z 315, [M + H]+), an orange color developed in the system. A third attempt (Procedure C), using 1,1,3,3-tetramethylguanidine as the nucleophile and base (2.5 eq.), was unsuccessful, leading to the formation of a bisphosphoramido compound (by LC–MS (ESI +), data not shown).

2D structure of NEMP
The experiment that succeeded (Procedure D) consisted of the preparation of a solution of methylphosphonic dichloride (12) (100 mg, 1.0 eq.) in dry dichloromethane (500 µL) at room temperature. After chilling the mixture at 0 °C, solutions of triethylamine (1.2 eq. in 500 µL of dry dichloromethane) and 1,1,3,3-tetramethylguanidine (1.1 eq. in 500 µL of dry dichloromethane), with the last one added slowly to avoid an increase in temperature and controlled formation of a white cloud. After stirring for 2 h at 0 °C (reaction followed either by GC–MS, checking by consumption of dichloride), a dichloromethane suspension (1 mL) of 4-nitrophenol (1.25 eq.) and sodium hydride (1.3 eq) was carefully added at 0 °C. After 4 h of reaction (followed by GC–MS and LC–MS), the reaction was allowed to reach room temperature and stirred for additional 30 min. After the solids settled, the reaction mixture was filtered using a syringe filter (0.22 micron), transferred to a 4-mL vial and gently evaporated in a rotatory evaporator (bath at 25 °C), yielding a yellow oil.
To remove volatiles, liquid–liquid extraction was performed using two different approaches to clean up the title compound from byproducts. The residue from evaporation was carefully transferred into a separatory funnel and washed with deionized water (2 × 20 mL). The organic layer was collected, dried over anhydrous sodium sulfate and carefully evaporated to dryness, yielding a yellow slurry. Another sample treated similarly, except for washing with 1% v/v HCl aqueous solution, afforded a similar material. HPLC–MS analysis of slurries indicated a small amount of the compound and extensive degradation. Surprisingly, and connected to what has been disclosed on the A-series agents, the compound of interest partitioned into the aqueous phase, confirming the risks associated with such chemicals being detected after HPLC–MS analysis. Recovering the material was considered worthless.
Therefore, to avoid loss of material and further risks, the crude material from evaporation was taken into hot hexane followed by chilling overnight at − 25 °C, yielding the crude A-242 surrogate NTMGMP (10) as a yellow oil (Cavalcante et al. 2019a; Kitagawa et al. 2021; Meek et al. 2012). The expected impurities (detected by different analytical techniques) were TMG and PNP.
The results of Elmann’s tests demonstrated that clinical oximes could be a strategy for the treatment of intoxication by OPs of the A-series (Oh et al. 2006). Trimedoxime (7) showed higher reactivation and was the only one that exhibited reactivation at 1 μM, showing up as a promising AChE reactivator against intoxications with A-242. These results are similar to those reported by Malinak et al. (Malinak et al. 2018) employing tabun as an inhibitor, suggesting a similarity between the A-series agents and other phosphonamidates. Additionally, the ageing results suggest that oximes can be valuable compounds to address A-series poisoning, which is important information for first responders and professionals in related fields.
The docking results presented in Table 4 suggest that HI-6 (9), in its protonated form, is able to adopt a position very close to the NAC, with an Rθd of 53.72. Even its deprotonated form also had an excellent result, with an Rθd of 50.26, pointing to this oxime as the most promising one for HssAChE reactivation, according to the NAC criterion. 2-PAM (6) was also able to adopt good approximation angles but could not get as close to the active site as HI-6 (9) did, despite its reduced dimensions compared to the other oximes.
HI-6 (9) showed the most promising results among the protonated oximes, with lower and more stable values of dOP. Among the deprotonated oximes, however, trimedoxime (7) was able to move closer to the active site. It is also noticeable that deprotonated oximes had much more difficulty approximating the active site. This observation corroborates the results obtained by Gonçalves et al. (Goncalves et al. 2006), indicating that the active site of HssAChE is a continuous region of negative electrostatic potential and, therefore, protonated oximes have more chances to access the site and adopt the NAC.
Table 5, from the MD simulations, reports the decrease in the results of Rθd values when compared to the docking results. It is noticeable that the decrease in the deprotonated oximes was much more pronounced than in the protonated oximes. This observation indicates that the docking algorithm, which is simpler than MD, is less accurate when simulating the changes in affinity between the anionic site and oximes due to deprotonation.
Although the Rθd results, considering both docking and MD simulations, suggest that HI-6 (9) would be the most promising oxime according to the NAC criterion to reactivate the HssAChE inhibited by NTMGMP, it was necessary to consider its high rate of deprotonation under physiological pH (Table 1). The second better Rθd result was obtained by 2-PAM (6), which has a lower rate of deprotonation but higher values of energy in MM-PBSA calculations. To assess the combination of these factors, we defined two products, P1 and P2. P1 Takes into account the results from Table 4, multiplying Rθd results by their respective percentages of ionization. This consideration adjusts the relevance of ligands according to their existence under physiological pH. The second product, P2, multiplies P1 by the modular MM-PBSA results as an approximation to consider the importance of the binding free energy in each HssAChE/NTMGMP/oxime complex. Table 7 below presents these outcomes, together with the successive changes in the ranking of better oxime when Rθd, P1 or P2 are considered.
Just after P1 consideration, protonated HI-6 (9), initially considered the most promising oxime to reactivate the HssAChE inhibited by NTMGMP, dropped to the seventh position in rank. The ionization form of interest, which keeps its positive electrostatic potential to facilitate access to the active site, would be present in very low proportion and therefore demand a prohibitive dose of the reactivator to be effective. 2-PAM (6) ascended to first position in P1, but its lower affinity to the active site, demonstrated by the higher value of binding free energy, dropped its position in the final rank, with P2. Although far from the optimum Rθd, trimedoxime (7) reached the best result when considering all factors, demonstrating good stability under physiological pH and good affinity for the active site.
In addition to the analysis shown above, the plots of RMSD for the eight complexes (Fig. S8) show no deviation exceeding 0.2 nm. This suggests that the oximes accommodate well inside the active site during the simulation. The root-mean-square fluctuations (RMSF) of HssAChE residues over the simulation time were also analyzed and are shown in Fig. S11. All ligands presented a similar interaction profile and low RMSF values in the three residues of the active site (Ser203, Glu334 and His447), suggesting reduced mobility, probably due to interaction with the ligands.
Regarding H-bonds, Glu202 was the residue that presented interactions with the higher number of ligands. Different authors (Allgardsson et al. 2016; Kua et al. 2003) report the importance of this residue to stabilize the quaternary nitrogen in the anionic subsite, supporting the docking of ACh or oximes before binding to Ser203. Considering this, some observations can be made: (a) trimedoxime (7) presented the higher number of H-bonds per timeframe, highlighting once more its performance when compared to the other oximes; (b) no deprotonated oxime presented a higher number of H-bonds per timeframe than its protonated form, confirming their difficulties to stabilize in the NAC; (c) HI-6 was the only oxime with no H-bond with Glu202, which indicates its worse result and corroborates the previous results; (d) the descending list of oximes with more interactions with Glu202 matches exactly the hierarchy of better ligands from Table 4.
The lack of data regarding HssAChE reactivation in the case of intoxications with the A-series agents and the consequent vulnerability against this class of nerve agents demands continuous research and global efforts on this subject. In this article, we designed and synthesized an A-242 surrogate, NTMGMP (the first surrogate of an A series of agents useful for reactivation assays), used it to inhibit HssAChE in vitro and evaluated the efficacy of 2-PAM (6), trimedoxime (7), obidoxime (8), and HI-6 (9) to reactivate the enzyme. In parallel, we performed a computational simulation and evaluated the results based on the NAC criterion, RMSD and RMSF criteria, and MM-PBSA calculations. After the successful in vitro steps of synthesis and enzyme inhibition, the results converged with the in silico outcomes, pointing to trimedoxime (7) as the most promising oxime against A-242 poisoning, followed by obidoxime (8) and 2-PAM (6). This conclusion not only indicates that the surrogate was suitable for the reactivation study, but also indicates that the computational method was able to simulate the biological interactions between HssAChE/inhibitor/oxime, proving to be a valuable instrument to precede in vitro procedures. Another relevant finding was the absence of aging in in vitro test conditions, indicating that trimedoxime (7) (and other oximes) is of clinical importance in the first response and medical management of intoxication.
Methodology
Study design
All chemicals used in this work were purchased from commercial suppliers and used as received. EeAChE (C2888, Type V-S, 1000 U/mg protein), pralidoxime iodide, acetylthiocholine iodide (ATCI), DTNB (Ellman’s reagent, 5,5′-dithio-bis-(2-nitrobenzoic acid)), DMSO (dry, biological assays grade), 1,1,3,3-tetramethylguanidine, 4-nitrophenol, dry triethylamine and inorganic compounds (sodium hydride 60% in mineral oil, phosphate salts for buffer solutions, anhydrous sodium sulphate, molecular sieves) were purchased from Sigma Aldrich Brazil (São Paulo, Brazil). Dichloromethane was purchased from Anidrol (Diadema, São Paulo, Brazil) and dried over 4 Å molecular sieves. Methylphosphonic dichloride, trimedoxime dibromide, obidoxime dichloride and asoxime (HI-6) dichloride were synthesized in-house, and analytical data were compatible with the literature. Absolute ethanol and acetonitrile for chromatographic analysis were purchased from Merck Brasil (São Paulo, Brazil). Deuterated chloroform (1% tetramethylsilane as internal standard) was purchased from Cambridge Isotopes Laboratories (Tewksbury, Massachusetts, USA). Purified water was obtained from a Millipore Milli-Q system (18.2 MΩ.cm at 25 °C, Millipore Brazil, São Paulo, Brazil). NMR spectra were obtained from Varian Unity 400 MHz (Santa Clara, CA, USA) and referred to tetramethylsilane for 1H- and 13C-NMR spectra, as 31P-NMR spectra were referred using secondary referencing with a unified chemical shift scale. Gas chromatography coupled to mass spectrometry (GC-MS) data were obtained from an Agilent 6890 GC system equipped with a 5975C mass spectrometer detector (Billerica, MA, USA). LC-MS (liquid chromatography coupled to mass spectrometry) and LC-MS-MS (liquid chromatography tandem mass spectrometry) data were obtained from an Agilent 1210 LC system equipped with a 6410B triple quadrupole (QqQ) mass spectrometer detector (Billerica, MA, USA). A SpectraMax Plus 384 microplate reader (Molecular Devices, San Jose, CA, USA) was used in all in vitro assays. Ninety-six-well microplates were purchased from Kasvi Brasil (São José dos Pinhais, Paraná, Brazil). Gilson single channel pipettes were purchased from Gilson Inc. (Middleton, WI, USA), and Eppendorf 8-channel pipettes were acquired from Eppendorf Brasil (São Paulo, Brazil). Ellman’s tests were performed in triplicate in three different assays by at least three different operators measured at 25 ± 2 °C. Microsoft Excel® 2010 was used for all calculations. All disposable materials and glassware in contact with OP compounds were decontaminated with aqueous solution containing 10% w/v NaOH and 10% w/v NaClO for 48 h at room temperature in a fume hood before correct destination. MKTOP (Ribeiro et al. 2008) and ACPYPE (Da Silva and Vranken 2012) software were used to build the model of HssAChE inhibited by NTMGP. The ionization states of the selected oximes were estimated with the online platform Chemicalize (Swain 2012). Molegro Virtual Docker (MVD) (Thomsen and Christensen 2006) was used for docking simulations, and GROMACS 5.1.4 (Abraham et al. 2015) was used for MD simulations. The MM-PBSA method was applied through the g_mmpbsa (Kumari et al. 2014) tool. Data from MD were analyzed with Visual Molecular Dynamics (VMD) (Humphrey et al. 1996) and XMGRACE (https://plasma-gate.weizmann.ac.il/Grace/) 2005 software. Full spectroscopic data of all synthesized compounds can be found in the Supplementary material.
Synthesis of NTMGMP
Important
The synthetic procedure must be conducted under strict safety requirements due to the toxicity of nerve agents’ surrogates. All personnel must be well trained in the techniques, and unit operations must be carried out in a good fume hood. All glassware and disposable materials must be decontaminated before correct destination.
In a round-bottomed reaction flask (dried in an oven at 120 °C for 20 min and cooled under N2 flow) equipped with N2 flow (balloon), methylphosphonic dichloride (100 mg, 1.0 eq.) and 500 µL of dichloromethane were added and kept at 0 °C with the aid of an ice-water bath. Triethylamine (TEA, 170 µL in 500 µL of dichloromethane, 1.2 eq.) and 1,1,3,3-tetramethylguanidine (TMG, 140 µL in 500 µL of dichloromethane, 1.1 eq.), with the last one added slowly. White fumes evolved during the addition of the TMG. After 2 h (reaction followed either by GC-MS, checked by consumption of dichloride), a dichloromethane suspension (1 mL) of 4-nitrophenol (PNP, 130 mg, 1.25 eq.) and sodium hydride (40 mg, 60% in mineral oil, 1.3 eq., previously treated with hexane for mineral oil removal) was carefully added at 0 °C. After 4 h of reaction (followed by GC-MS and LC-MS), the reaction was allowed to reach room temperature and stirred for additional 30 min. After the solids settled, the reaction mixture was filtered using a syringe filter (0.22 micron), transferred to a 4-mL vial and gently evaporated in a rotatory evaporator (bath at 25 °C), yielding a yellow oil. This crude material was treated with hot hexane and chilled overnight at − 25 °C, affording the crude A-242 surrogate NTMGMP (yellow oil). As it is a potential cholinesterase inhibitor, it must be manipulated with precaution, and it was not purified to avoid increasing toxicity risks to all operators. TMG and PNP are major impurities (detected by GC-MS, LC-MS and NMR techniques).
Ellman’s tests
Ellman’s test was performed with slight modifications from some of our published papers (De A Cavalcante et al. 2018) using 96-well microplates and a maximum volume of 200 µL, followed by UV spectrophotometry at 412 nm. After addition of the enzyme substrate (ATCI), absorbance was read for 1 h (intervals of 5 min). All measurements were done in triplicate, performed by three different operators, with mean values used for final calculations.
Briefly, the negative control (maximum activity of the enzyme, native enzyme) was measured by pipetting 30 µL of phosphate buffer solution (PBS, pH 7.60 ± 0.10), 70 µL of AChE solution (2.14 U/mL in each well, prepared in PBS) and 80 µL of DTNB 0.4 mg/mL (in PBS). Then, 20 µL of 1 mmol/L ATCI (in PBS) was added, followed by UV (A0).
For the positive control (i.e., inhibition assays), we prepared seven solutions of crude A-242 surrogate in absolute ethanol (final concentration of the inhibitor in each well: 1000, 100, 10, 1, 0.1, 0.01, 0.001 µmol/L) and determined the level of EeAChe inhibition after 10 min of incubation (inhibition reaction), selecting the minor concentration that retrieved 85–95% of inhibition for reactivation studies (for safety reasons). It was measured by pipetting 20 µL of phosphate buffer solution, 70 µL of AChE solution, 80 µL of DTNB solution and 10 µL of inhibitor solution (in the three concentrations mentioned above). After 10 min of inhibition, 20 µL of ATCI solution was added, followed by UV (Ai). Equation 2 was used to calculate the enzyme inhibition (%I).
For reactivation studies using the clinically available oximes (prepared in three different concentrations in PBS, final concentrations in each well: 100, 10 and 1 µmol/L, prepared from stock solutions in DMSO), 70 µL of EeAChE solution, 80 µL of DTNB solution and 10 µL of inhibitor solution were pipetted, followed by 10 min of inhibition reaction. After this time, 20 µL of solutions of the standard antidotes were added, and 30 min were waited for the reactivation reaction. Finally, 20 µL of ATCI solution were added, and the absorbance was read to calculate the enzyme reactivation (Ar). Equation 3 was used to calculate the enzyme reactivation (%R).
For estimation of AChE aging caused by the surrogate, the enzyme was reactivated following the reactivation procedure after enzyme inhibition for 10 and 90 min using trimedoxime (7) at 100 μmol/L as a reactivator. The difference in reactivation rates was considered the aging estimation.
It is noteworthy to state that all impurities from the synthetic process, namely, PNP and TMG, were tested in Ellman’s conditions and did not inhibit the enzyme, as found for all solvents involved in the experimental steps (DMSO, ethanol, dichloromethane).
HssAChE/NTMGMP model
A model of HssAChE inhibited by the surrogate was necessary for the computational simulations. The construction was made through mutations on the crystallographic structure of recombinant HssAChE inhibited by VX, available in the RCSB-Protein Data Bank (Berman et al. 2002) with the code 6CQW (Bester et al. 2018). The software MKTOP (Ribeiro et al. 2008) and AnteChamber PYthon Parcer InterfacE (ACPYPE) (Da Silva and Vranken 2012) were used to parameterize the tridimensional structure of the complex. In addition to parameterization, all bond information, angles, proper and improper dihedrals and third neighbors needed to be manually added to the topology file before docking and MD simulations, so the nerve agent could be recognized as part of the protein. Prior to computer calculations, the model was submitted to a validation procedure with the use of a Ramachandran plot (Ramachandran et al. 1963) and Verify 3D server (Bowie et al. 1991; Lüthy et al. 1992).
Estimation of ionization state and construction of oximes
To determine which ionization forms of the selected oximes should be constructed, the online platform Chemicalize (available at https://chemicalize.com/welcome) (Swain 2012) was used, simulating the physiological pH (7.4). The molecular structures were then built and optimized with the software Spartan 08 (Hehre and Johnson 2006), and the semiempirical method Recife Model 1 (RM1) (Goncalves et al. 2010; Rocha et al. 2006) was used to calculate the respective partial atomic charges.
Molecular docking
The docking protocol was validated using a redocking procedure with the HssAChE/NTMGMP model. The HI-6 oxime, present in the original crystallographic structure from PDB, was submitted to docking simulations with the software MVD® using the MolDock algorithm (Thomsen and Christensen 2006). The coordinates obtained were then compared to the experimental data from crystallographic structure, with the use of root-mean square deviation (RMSD), to check if the procedure was able to predict the position of the ligand in the protein.
The ionization forms of the oximes, suggested by the Chemicalize platform, were then imported to MVD® for docking simulations. The ligands were submitted to six runs each, varying between three different population sizes (50, 200 and 350) and two iteration limits (2000 and 3000). Thirty poses with lower Moldock scores in each run were selected for further analysis following the NAC criterion. For that purpose, considering the van der Waals distance for the O – P bond as 3.3 Å (Johnson et al. 2018) and the ideal angle for the substitution reaction as 180º, we defined the optimum ratio angle/distance as approximately 54.55, as shown in Eq. 1.
This definition was then used to rank the 180 poses generated per ligand, selecting the best pose from each oxime to proceed to MD simulations.
MD simulations and MM-PBSA calculations
The chosen poses for MD simulations were prepared with ACPYPE (Da Silva and Vranken 2012) to parameterize the poses and create topology and coordinate files. This step was necessary to allow the forcefield OPLSA/AA (Jorgensen et al. 1996) in GROMACS 5.1.4 software (Abraham et al. 2015) to recognize the ligands. MKTOP software (Ribeiro et al. 2008) was used to identify the unknown atom types in the topology files. Semiempirical quantum (SQM) chemistry software (Walker et al. 2008) was then used to calculate the atomic partial charges. The protein topology and coordinate files were also generated with the use of GROMACS, with the same forcefield, to be merged with ligand files and produce the HssAChE/NTMGMP/oxime systems to be simulated. The systems were then simulated in the center of dodecahedron boxes under periodic boundary conditions, with a volume of 1088.15 nm3. The boxes were filled with approximately 33,000 TIP3P water molecules, and the system charge was neutralized through the substitution of the proper number of solvent molecules by Na+ counterions. As before (da Silva et al. 2019), energy minimization was performed in three steps. The first one was a steepest descent (ST) minimization with position restriction (PR) of protein and ligand, up to a maximum force of 600 kJ mol−1 nm−1, to accommodate the solvent molecules inside the box. The second was also an ST, but with no PR, up to 300 kJ mol−1, aiming at a local minimum in the system potential energy surface. The last minimization was a limited-memory Broyden-Fletcher-Goldfarb-Shanno (L-BFGS) step, up to 41.84 kJ mol−1 nm−1. Then, the temperature was equilibrated to 310 K by an isothermal-isochoric ensemble, and the pressure was equilibrated to 1 bar by an isothermal-isobaric ensemble. Temperature coupling used a velocity-rescale thermostat (Bussi et al. 2007), and pressure coupling used a Parrinello-Rahman barostat (Parrinello and Rahman 1981). MD was then performed in two different stages. The first one was destinated to accommodate water molecules, keeping PR for protein and ligand molecules, in a 500 ps simulation. The second stage had no restriction in a 100 ns simulation, with 2 fs as integration time, leapfrog integrator and a cutoff of 1,0 nm for short-range (Lennard–Jones and Coulomb) interactions. The coordinates of the complexes were stored every 20 ps, and the resulting data were analyzed using XMGRACE (Turner 2005) and Visual Molecular Dynamics (VMD) (Humphrey et al. 1996) software.
To estimate the binding free energy of each HssAChE/NTMGMP/oxime complex after MD simulations, the MM-PBSA method (Hansen and Van Gunsteren 2014) was employed with the use of the g_mmpbsa tool (Kumari et al. 2014). The calculations considered the potential energy in the vacuum, including both bonded and nonbonded interactions, and desolvation of the different species, including both polar and nonpolar solvation energies. The polar solvation energy was estimated by solving the Poisson-Boltzmann equation, and the nonpolar was calculated through the solvent accessible surface area (SASA) method (Kumari et al. 2014).
References
Abraham MJ, Murtola T, Schulz R et al (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2:19–25. https://doi.org/10.1016/j.softx.2015.06.001
Allgardsson A, Berg L, Akfur C et al (2016) Structure of a prereaction complex between the nerve agent sarin, its biological target acetylcholinesterase, and the antidote HI-6. Proc Natl Acad Sci 113(20):5514–5519
Bajgar J (2004) Organophosphates/nerve agent poisoning: mechanism of action, diagnosis, prophylaxis, and treatment. Adv Clin Chem 38(1):151–216
Bajgar J, Fusek J, Kuca K, Bartosova L, Jun D (2007) Treatment of organophosphate intoxication using cholinesterase reactivators: facts and fiction. Mini Rev Med Chem 7(5):461–466
Berman HM, Battistuz T, Bhat TN et al (2002) The protein data bank. Acta Crystallogr D Biol Crystallogr. https://doi.org/10.1107/S0907444902003451
Bester SM, Guelta MA, Cheung J et al (2018) Structural Insights of Stereospecific Inhibition of Human Acetylcholinesterase by VX and Subsequent Reactivation by HI-6. Chem Res Toxicol. https://doi.org/10.1021/acs.chemrestox.8b00294
Bolt HM, Hengstler JG (2022) Recent research on Novichok. Arch Toxicol 96:1137–1140
Bowie JU, Lüthy R, Eisenberg D (1991) A method to identify protein sequences that fold into a known three-dimensional stucture. Science. https://doi.org/10.1126/science.1853201
Bussi G, Donadio D, Parrinello M (2007) Canonical sampling through velocity rescaling. J Chem Phys. https://doi.org/10.1063/1.2408420
Cavalcante SF, Kitagawa DA, Rodrigues RB et al (2019a) One-pot synthesis of NEMP, a VX surrogate, and reactivation of NEMP-inhibited Electrophorus eel acetylcholinesterase by current antidotes. J Brazil Chem Soc 30:1095–1102
Cavalcante SFD, Kitagawa DAS, Rodrigues RB et al (2019b) Synthesis and in vitro evaluation of neutral aryloximes as reactivators of Electrophorus eel acetylcholinesterase inhibited by NEMP, a VX surrogate. Chem-Biol Interact. https://doi.org/10.1016/j.cbi.2019.05.048
da Silva JAV, Nepovimova E, Ramalho TC, Kuca K, Celmar Costa França T (2019) Molecular modeling studies on the interactions of 7-methoxytacrine-4-pyridinealdoxime, 4-PA, 2-PAM, and obidoxime with VX-inhibited human acetylcholinesterase: a near attack conformation approach. J Enzym Inhib Med Ch. https://doi.org/10.1080/14756366.2019.1609953
Da Silva AWS, Vranken WF (2012) ACPYPE-Antechamber python parser interface. BMC Res Notes 5(1):1–8
De A, Cavalcante SF, Kitagawa DAS, Rodrigues RB et al (2018) Straightforward, economical procedures for microscale ellman⇔s test for cholinesterase inhibition and reactivation. Quim Nova. https://doi.org/10.21577/0100-4042.20170278
de Cavalcante ASF, Simas AB, Kuča K (2019) Nerve agents’ surrogates: invaluable tools for development of acetylcholinesterase reactivators. Curr Organ Chem 23(14):1539–1559
Franca TCC, Kitagawa DAS, Cavalcante SFD, da Silva JAV, Nepovimova E, Kuca K (2019) Novichoks: the dangerous fourth generation of chemical weapons. Int J Mol Sci. https://doi.org/10.3390/ijms20051222
Goncalves AD, Franca TCC, Wilter A, Figueroa-Villar JD (2006) Molecular dynamics of the interaction of pralidoxime and deazapralidoxime with acetylcholinesterase inhibited by the neurotoxic agent tabun. J Brazil Chem Soc 17(5):968–975. https://doi.org/10.1590/s0103-50532006000500022
Goncalves AD, Franca TCC, Figueroa-Villar JD, Pascutti PG (2010) Conformational analysis of toxogonine, TMB-4 and HI-6 using PM6 and RM1 methods. J Brazil Chem Soc 21(1):179-U82. https://doi.org/10.1590/s0103-50532010000100025
Gorecki L, Korabecny J, Musilek K et al (2016) SAR study to find optimal cholinesterase reactivator against organophosphorous nerve agents and pesticides. Arch Toxicol 90(12):2831–2859
Hansen N, Van Gunsteren WF (2014) Practical aspects of free-energy calculations: a review. J Chem Theory Comput 10:2632–2647
Hehre WOSKPDBDA, Johnson JOP (2006) Spartan ‘08 Tutorial and User’s Guide. Wavefunction, Irvine
Homeyer N, Gohlke H (2012) Free energy calculations by the molecular mechanics poisson-boltzmann surface area method. Mol Inf. https://doi.org/10.1002/minf.201100135
Hosseini SE, Saeidian H, Amozadeh A, Naseri MT, Babri M (2016) Fragmentation pathways and structural characterization of organophosphorus compounds related to the Chemical Weapons Convention by electron ionization and electrospray ionization tandem mass spectrometry. Rapid Commun Mass Spectrom. https://doi.org/10.1002/rcm.7757
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. Graphics 14:33–38. https://doi.org/10.1016/0263-7855(96)00018-5
Imrit YA, Bhakhoa H, Sergeieva T et al (2020) A theoretical study of the hydrolysis mechanism of A-234; the suspected novichok agent in the Skripal attack. RSC Adv 10(47):27884–27893. https://doi.org/10.1039/D0RA05086E
Jeong K, Choi J (2019) Theoretical study on the toxicity of ‘Novichok’ agent candidates. R Soc Open Sci. https://doi.org/10.1098/rsos.190414
Johnson LA, Robertson AJ, Baxter NJ et al (2018) Van der Waals contact between nucleophile and transferring phosphorus is insufficient to achieve enzyme transition-state architecture. ACS Catal. https://doi.org/10.1021/acscatal.8b01612
Jorgensen WL, Maxwell DS, Tirado-Rives J (1996) Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. https://doi.org/10.1021/ja9621760
Kitagawa DA, Rodrigues RB, Silva TN et al (2021) Design, synthesis, in silico studies and in vitro evaluation of isatin-pyridine oximes hybrids as novel acetylcholinesterase reactivators. J Enzym Inhib Med Ch 36(1):1370–1377
Kloske M, Witkiewicz Z (2019) Novichoks—the A group of organophosphorus chemical warfare agents. Chemosphere 221:672–682. https://doi.org/10.1016/j.chemosphere.2019.01.054
Kontoyianni M, McClellan LM, Sokol GS (2004) Evaluation of docking performance: comparative data on docking algorithms. J Med Chem. https://doi.org/10.1021/jm0302997
Kua J, Zhang Y, Eslami AC, Butler JR, McCammon JA (2003) Studying the roles of W86, E202, and Y337 in binding of acetylcholine to acetylcholinesterase using a combined molecular dynamics and multiple docking approach. Protein Sci 12(12):2675–2684
Kucera T, Gorecki L, Soukup O et al (2019) Oxime K203: a drug candidate for the treatment of tabun intoxication. Arch Toxicol 93(3):673–691
Kumari R, Kumar R, Consortium OSDD, Lynn A (2014) g _ mmpbsa - A GROMACS tool for MM-PBSA and its optimization for high-throughput binding energy calculations. J Chem Inf Model. https://doi.org/10.1021/ci500020m
Lewis S (2018) Salisbury, Novichok and international law on the use of force. RUSI J 163(4):10–19. https://doi.org/10.1080/03071847.2018.1529889
Lüthy R, Bowie JU, Eisenberg D (1992) Assessment of protein models with three-dimensional profiles. Nature. https://doi.org/10.1038/356083a0
Malinak D, Nepovimova E, Jun D, Musilek K, Kuca K (2018) Novel group of AChE reactivators—synthesis, in vitro reactivation and molecular docking study. Molecules 23(9):2291
Meek EC, Chambers HW, Coban A et al (2012) Synthesis and in vitro and in vivo inhibition potencies of highly relevant nerve agent surrogates. Toxicol Sci. https://doi.org/10.1093/toxsci/kfs013
Mirzayanov VS (2009) State secrets: an insider`s chronicle of the Russian Chemical Weapons Program. Outskirts Press Inc, Parker
Nepovimova E, Kuca K (2018) Chemical warfare agent NOVICHOK—mini-review of available data. Food Chem Toxicol 121:343–350. https://doi.org/10.1016/j.fct.2018.09.015
Nepovimova E, Kuca K (2019) The history of poisoning: from ancient times until modern ERA. Arch Toxicol 93(1):11–24
Oh K-A, Yang GY, Jun D, Kuca K, Jung Y-S (2006) Bis-pyridiumaldoxime reactivators connected with CH2O (CH2) nOCH2 linkers between pyridinium rings and their reactivity against VX. Bioorg Med Chem Lett 16(18):4852–4855
Parrinello M, Rahman A (1981) Polymorphic transitions in single crystals: A new molecular dynamics method. J Appl Phys. https://doi.org/10.1063/1.328693
Ramachandran GN, Ramakrishnan C, Sasisekharan V (1963) Stereochemistry of polypeptide chain configurations. J Mol Biol 7:95–99
Ribeiro AAST, Horta BAC, De Alencastro RB (2008) MKTOP: A program for automatic construction of molecular topologies. J Brazil Chem Soc. https://doi.org/10.1590/S0103-50532008000700031
Rocha GB, Freire RO, Simas AM, Stewart JJ (2006) Rm1: a reparameterization of am1 for h, c, n, o, p, s, f, cl, br, and i. J Comput Chem 27(10):1101–1111. https://doi.org/10.1002/jcc.20425
Santos MC, Botelho FD, Gonçalves AS, Kuca K, Nepovimova E, Cavalcante SFA, Lima ALS, França TCC (2022) Theoretical assessment of the performances of commercial oximes on the reactivation of acetylcholinesterase inhibited by the nerve agent A-242 (novichok). Food Chem Toxicol 165:113084. https://doi.org/10.1016/j.fct.2022.113084
Steindl D, Boehmerle W, Körner R et al (2021) Novichok nerve agent poisoning. Lancet 397(10270):249–252
Swain M (2012) Chemicalize. org. vol 52. ACS Publications, p 613–615
Tattersall JE (1993) Ion channel blockade by oximes and recovery of diaphragm muscle from soman poisoning in vitro. Br J Pharmacol 108(4):1006–1015
Thomsen R, Christensen MH (2006) MolDock: a new technique for high-accuracy molecular docking. J Med Chem 49(11):3315–3321. https://doi.org/10.1021/jm051197e
Walker RC, Crowley IF, Case DA (2008) The implementation of a fast and accurate QM/MM potential method in Amber. J Comput Chem. https://doi.org/10.1002/jcc.20857
Acknowledgements
The authors are grateful to the Military Institute of Engineering (IME), Institute of Chemical, Biological, Radiological and Nuclear Defense (IDQBRN), Brazilian Army Technological Center (CTEx), Federal University of Lavras (for software licenses), University of Hradec Kralové and the CQDM SynergiQc grant.
Funding
This research was funded by the Brazilian agencies Conselho Nacional de Pesquisa (CNPq), grant no. 308225/2018–0; Fundação de Amparo a Pesquisa do Estado do Rio de Janeiro (FAPERJ), grant no. E-02/202.961/2017; IFES—PRPPG, grant number 10/2019 (Productivity Researcher Program PPP); and FAPES, grant number 03/2020-2020-WMT5F. The authors are also grateful to the Excelence project PrF UHK 2217/2022-2023 for financial support.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Santos, M.C., Botelho, F.D., Gonçalves, A.S. et al. Are the current commercially available oximes capable of reactivating acetylcholinesterase inhibited by the nerve agents of the A-series?. Arch Toxicol 96, 2559–2572 (2022). https://doi.org/10.1007/s00204-022-03316-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-022-03316-z