
Abstract
2-Phenoxyethanol (PE), ethylene glycol monophenyl ether, is widely used as a preservative in cosmetic products as well as in non-cosmetics. Since PE has been used in many types of products, it can be absorbed via dermal or inhaled route for systemic exposures. In this study, the pharmacokinetic (PK) studies of PE and its major metabolite, phenoxyacetic acid (PAA), after dermal (30 mg and 100 mg) and inhaled administration (77 mg) of PE in rats were performed. PE was administered daily for 4 days and blood samples were collected at day 1 and day 4 for PK analysis. PE was rapidly absorbed and extensively metabolized to form PAA. After multiple dosing, the exposures of PE and PAA were decreased presumably due to the induction of metabolizing enzymes of PE and PAA. In dermal mass balance study using [14C]-phenoxyethanol ([14C]PE) as a microtracer, most of the PE and its derivatives were excreted in urine (73.03%) and rarely found in feces (0.66%). Based on these PK results, a whole-body physiologically-based pharmacokinetic (PBPK) model of PE and PAA after dermal application and inhalation in rats was successfully developed. Most of parameters were obtained from the literatures and experiments, and intrinsic clearance at steady-state (CLint,ss) were optimized based on the observed multiple PK data. With the developed model, systemic exposures of PE and PAA after dermal application and inhalation were simulated following no-observed-adverse-effect level (NOAEL) of 500 mg/kg/day for dermal application and that of 12.7 mg/kg/day for inhalation provided by the Environmental Protection Agency. The area under the concentration–time curve at steady state (AUCss) in kidney and liver (and lung for inhalations), which are known target organs of exhibiting toxicity of PE, as well as AUCss in plasma of PE and PAA were obtained from the model.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
2-Phenoxyethanol (PE), ethylene glycol monophenyl ether, is a colorless oily and viscous liquid at room temperature and widely used as a preservative in cosmetic products as well as in non-cosmetics (SCCS 2016; Scognamiglio et al. 2012). PE is used as a preservative in many types of cosmetics, including perfumes, soaps, body lotions, face creams, shampoo, and shower gel, among others, at a maximum concentration of 1.0%, as it exhibits antiseptic effects from Gram negative to Gram positive bacteria, mold, and yeast. PE is also used in lubricants, greases, metal working fluids, coating products, inks, and toners (ECHA 2019). Since PE has been used in many products, there are many documents reporting toxicological evaluation of PE.
The target organs for toxicity of PE include the hematopoietic system, liver and kidneys (Troutman et al. 2015). PE has hemolytic effects, inducing severe intravascular hemolysis with regenerative anemia. The hemolytic effects occurred after oral and dermal sub-chronic exposure (Breslin et al. 1991; Scortichini et al. 1987), and these effects are caused by a major metabolite of PE, phenoxyacetic acid (PAA) (Starek-Swiechowicz et al. 2012; Starek et al. 2008).
Unlike the acute outbreak hemolytic effect typically observed with high doses of PE, liver and kidney toxicities appeared at low dose levels of PE in rats. PE increased liver enzymes (such as AST and ALT) and bilirubin levels and decreased triglycerides in female rats at 10,000 ppm. Kidney toxicity included increased urinary protein, urothelial hyperplasia, renal papillary necrosis and mineralization (Dreno et al. 2019; Troutman et al. 2015). In addition to these toxicities, local irritation and effects at the site of exposures have also been reported; however, we considered only systemic toxicities in this study.
After oral and dermal administration of PE in rats, PE is rapidly absorbed and most of the PE is metabolized by cytochrome P450 (CYP), alcohol dehydrogenase (ADH), and aldehyde dehydrogenase (ALDH) to form PAA in liver and blood; PAA is then mainly excreted in urine (SCCS 2016; Troutman et al. 2015). Other minor metabolites were found with either ring sulfonation after hydroxylation or conjugation with glucuronic acid at the side chain (SCCS 2016). Although many pharmacokinetic (PK) studies of PE after oral and intravenous administration in rats have been reported, reports in rats after dermal and inhaled administration of PE are limited (Kim et al. 2015; SCCS 2016). Several cases of safety evaluation after dermal and inhaled exposures of PE have been reported and these were summarized in OPINION ON Phenoxyethanol adopted by the Scientific Committee on Consumer Safety (SCCS 2016). However, to the best of our knowledge, PK and TK studies after dermal applications have only been conducted by Kim et al. (2015) and studies on PE inhalation have never been reported. Kim and colleagues (2015) reported PKs of PE after dermal application and the authors compared the PK according to formulation. Since PE is present in various products such as cosmetics and perfumes, humans are highly likely to be exposed to PE in daily life via dermal and inhaled routes of exposures. Therefore, the PK evaluations after percutaneous and inhaled absorption are critical for assessing exposure-safety response analysis.
The aims of this study were to (1) evaluate PK characteristics of PE and PAA after dermal and inhaled exposures of PE in rats, and (2) establish a rat physiologically-based PK (PBPK) model. We first evaluated PK characteristics of PE and PAA after dermal and inhaled administration of PE in rats at high doses and in dermal applications, a mass balance study using [14C]-phenoxyethanol ([14C]PE) was performed to elucidate the excretion pathway of PE. Based on these PK results, a PBPK model was developed, which involved inhaled and dermal routes of exposures. Using this model, steady-state PKs of PE and PAA were simulated and not only plasma concentrations but also target organ concentrations of PE and PAA were predicted at known no-observed-adverse-effect-levels (NOAEL). Since a PBPK model of PE in rats and humans was developed by Troutman and his colleagues (Troutman et al. 2015), we focused on developing PBPK model after dermal and inhaled routes of exposures in rats.
Materials and methods
Materials and reagents
PE (purity 99.8%), chlorpropamide (used as an internal standard, IS), dimethylsulfoxide, lanolin, homosalate, petrolatum, steric acid, propylparaben, methyl paraben, disodium edentate, propylene glycol, and triethanolamine were purchased from Sigma-Aldrich Corporation (St. Louis, MO, USA), and PAA (purity 98.0%) was purchased from Tokyo Chemical Industries (Tokyo, Japan). Propylene glycol was from Tokyo Chemical Industries (Tokyo, Japan) and disodium edentate was from United States Pharmacopeia (Rockville, MD, USA). [14C]PE (specific activity 95 mCi/mmol) was synthesized by American Radiolabeled Chemicals, Inc. (St. Louis, MO, USA). Other chemicals and solvents were HPLC grade and obtained from Burdick & Jackson Company (Morristown, NJ, USA).
Animals
Male Sprague–Dawley rats (8–9 weeks old and weighing 290–310 g) were purchased from Young Bio (Sungnam, South Korea). The procedures used for housing and handling of the rats were similar to those described in previous studies (Kim et al. 2013). Rats were housed and handled at a temperature of 20 ± 2 °C with a 12-h light–dark cycle and relative humidity of 55 ± 5%; rats were accommodated for at least 3 days before the experiments.
PK studies in rats
Dermal applications
For dermal application of PE, a lotion formulation was prepared based on the previous reference with modifications (Kim et al. 2015). Briefly, the formulation was composed of two phases, a water phase and fatty phase. The fatty phase consisted of lanolin (4.8%), homosalate (7.7%), petrolatum (2.4%), steric acid (3.8%), and propylparaben (0.048%), and the water phase was composed of water (70.35%), propylene glycol (4.80%), triethanolamine (0.96%), methylparaben (0.096%), and disodium edentate (0.048%). The fatty phase and water phase were melted and heated separately in a water bath at 80 °C until completely solubilized. The fatty phase was added to the water phase while stirring with a homogenizer (T-18 digital ULTRA-TURRAX, IKA, Staufen Germany). The mixture was cooled to 50 °C while being stirred and then a PE stock of 20-fold higher than the final doses used for experiments (30 and 100 mg/rat) (5%) was added to the mixture and cooled at room temperature. The PE lotion formulation was kept in light-resistant containers at room temperature until use.
The dermal study was conducted according to the OECD guideline for in vivo skin absorption tests (2004). At 24 h before the experiment, an area of dorsal skin of rats of 11 × 8 cm2 was shaved with an electric clipper; a shaving cream was applied and gently wiped off with saline gauze 3–5 min later. For collecting plasma samples, the carotid artery was cannulated using previously reported procedures (Kim et al. 2013). Lotion (1 mL) was applied to a 3.5 × 5 cm2 patch and the patch was attached to the middle of the dorsal skin of rats for 4 h. PKs of PE and PAA were evaluated after single and multiple exposures, and for the multiple exposure study, patches were applied for 4 h daily for 4 days. Applied PE doses were 30 mg or 100 mg. Approximately 0.22 mL of blood was collected from each rat at 0, 90, and 240 min after patch attachment and 3, 5, 15, 30, 45, 60, 120, 180, 240, 360, and 1440 min after patch removal. The blood samples were immediately centrifuged (14 000 g, 3 min) and 100 μL plasma was collected in a polyethylene tube and stored at − 20 °C until the LC–MS/MS analysis. After dermal application, the area was rinsed with normal saline to remove any remaining lotion on the skin. The removed patches were immersed in 20 mL methanol for 24 h to measure the PE amount remaining in the patch and 100 μL aliquots of methanol were stored at − 20 °C until the LC–MS/MS analysis.
Inhalation study
The inhalation study was conducted using nose-only equipment, VT-HOOD-12, manufactured by Vitals Inc. (Daejeon, Korea). The carotid artery was cannulated to rats for collecting blood samples and then rats were placed in a 12-port rodent nose-inhalation tower for inhalation of PE for 3 h. Blood samples were collected at 0, 10, 30, and 120 min during inhalation and 3, 5, 10, 20, 30, 45, 60, 90, 120, 180, and 240 min after inhalation. Other procedures for blood sampling and sample treatment were similar to those for the dermal study mentioned above. The total airflow through the tower was maintained at 1 L/min, which was equally divided into the ports holding rats for inhalation, and 1.107 g/mL of PE solution was constantly infused into a vacuum container. The PE dose to each rat was calculated based on Eq. (1):
Where AC is the aerosol concentration of PE in the chamber, RMV represents the respiratory minute volume, which is the respiratory rate multiplied by the tidal volume, and D is the duration of exposure to the aerosol. The values of respiratory rate and the tidal volume were from Fioni et al. (Fioni et al. 2018); the calculated RMV was 0.263 L/min and D was 180 min in this study. The calculated PE dose was 77 mg/rat. PKs of PE and PAA were evaluated after single and multiple exposures; for the multiple study, PE was inhaled for 3 h at 24 h intervals for 4 days.
Mass balance study of PE after dermal application
The mass balance study of PE was performed to elucidate the excretion of PE after dermal application (30 mg) in rats (n = 5). [14C]PE was used as a microtracer and its specific activity was measured the day before the experiment. The dose of [14C]PE used in the experiment was 2.59 nCi. The method for applying patches to rats was the same as described above. During the experiment period, each rat was kept in a metabolic cage (Tecniplast™, Varese, Italy), which was designed to completely separate the samples of urine and feces. After dermal applications, urine and feces were collected from 0–4, 4–8, 8–12, 12–24, 24–48, and 48–72 h and after 72 h; each metabolic cage was swabbed and swabs were also kept for analysis. Collected samples were weighed and kept at − 20 °C until accelerator mass spectrometer (AMS) analysis.
PK and statistical analysis
The total area under the plasma concentration–time curve from time zero to infinity (AUC) and that from time zero to the last measured time t (AUClast) were calculated using the trapezoidal rule extrapolation method (Chiou 1978). Standard methods (Gibaldi and Perrier 1982) using a non-compartmental analysis (Pheonix WinNonlin ver. 8.1, Pharsight Corporation; Mountain View, CA, USA) were used to calculate the following PK parameters: terminal half-life, apparent steady-state volume of distribution (Vss/F), and total body clearances (CL/F). Percentages of the dose excreted in urine or feces sample at each time were also calculated. The peak plasma concentration (Cmax) and time to reach Cmax (Tmax) were read directly from the experimental data.
Differences between two means for the unpaired data were analyzed using Student’s t tests; a p value < 0.05 was considered to indicate statistical significance. All data are presented as means ± standard deviation (SD) unless otherwise specified.
Quantitative analysis of PE and PAA in biological samples
LC–MS/MS analysis
The previously developed LC–MS/MS method (Kim et al. 2015) was modified for the quantitative analysis of PE and PAA in biological samples. The AB SCIEX 6500 triple quadrupole (AB Sciex, Toronto, Canada) with an Agilent 1290 UPLC system (Agilent, CA, USA) was used for measuring PE and PAA concentrations. Instrument control, data acquisition, and process were performed using Analyst® software (version 1.7, AB SCIEX). The electrospray ionization (ESI) source was used in a positive ion mode (PE) and a negative ion mode (PAA) with multiple reaction monitoring. The instrument parameters were set as follows: gas temperature of 200 °C, ion source gas 1 of 45 psi, ion source gas 2 of 40 psi, ionspray voltage of 5500 V (PE) or − 4000 V (PAA), curtain gas 20 psi, and collision gas of 9 psi. The fragmentor voltages were set as 60 V (PE, PAA, and IS positive) and 25 V (IS negative). The collision energy was 35 V (PE), 31 V (PAA and IS positive), and 34 V (IS negative), respectively. The precursor to product ion transitions used for quantification were m/z 139.1 → 77.1 for PE, m/z 151 → 93.0 for PAA, m/z 276.9 → 174.9 for IS positive, and 274.9 → 189.8 for IS negative.
Sample preparation was conducted as follows. Aliquots of 100-μL methanol containing 20 ng/mL of chlorpropamide (IS) was added to 100-μL plasma samples. After mixing and centrifugation (14 000 g, 10 min), the supernatant was collected and injected directly onto a reversed-phase Poroshell® 120 C18 column (3.0 mm i.d. × 50 mm, 2.7 μm, Agilent) connected to Security Guard Pre-Column (C18, 4 × 2 mm; phenomenex). The gradient elution of mobile phase, 1% formic acid in distilled water (A) and 1% formic acid in methanol (B), was run through the column at a flow rate of 0.4 mL/min as follows: [B%; 0–1 min, 40%; 1–1.1 min, 40–70%; 1.1–1.8 min, 70%; 1.8–1.9 min, 70–40%; 1.9–3 min, 40%]. The column and autosampler temperature were set at 40 °C and 4 °C, respectively. The injection volume was 3 μL for PAA and 10 μL for PE and total run time was 3 min.
Liquid scintillation counting (LSC) analysis
Before mass balance experiments, a LSC system (Tri-carb® 5110 TR; PerkinElmer, MD, USA) was used to confirm the specific activity of [14C]PE. Sample preparation procedures were conducted in accordance with the manufacturer’s instructions (PerkinElmer Wallac, Gaithersburg, MD, USA). Briefly, a 1 mL aliquot of the collected sample was mixed with 14 mL of a scintillation cocktail (Utima Gold, PerkinElmer, MA, USA) by vortexing for 2 min. Each sample was detected for 20 min with seven cycles. The total radioactivity of [14C]PE was measured in triplicate.
AMS analysis
The samples obtained from mass balance study were analyzed by AMS. To measure a sample with AMS, the sample must first be converted to elemental carbon (fullerene often referred to a graphite). The sample graphitization was performed according to the septa-tube method described by Ognibene et al. (Ognibene et al. 2003). Briefly, a 20 μL aliquot of homogenized or well-mixed feces, urine, or swab samples was added to 2 mL diluted tributyrin solution (10% w:v tributyrin:methanol) and mixed by vortexing for 2 min. The carbon diluent dominated the total carbon content of the samples and eliminated any need for elemental analysis of the carbon content of the sample (Vogel et al. 2010). A 20 μL aliquot of the diluent (possessing 1.23 mg carbon) was then transferred to a small quartz insert and dried by vacuum centrifugation for 2 h at 35 °C. The quartz insert containing the dried sample was filled with copper oxide (approximately 200 mg) and placed into a larger quartz combustion tube. The combustion tubes were torched–sealed under vacuum and placed in a muffle furnace set to operate for 3 h at 900 °C. The products of oxidation, carbon dioxide, water and other gases were then transferred into a reduction tube submersed in liquid nitrogen. The tube contained zinc powder (for generation of hydrogen from water) and iron (4 mg) inside a pyrex insert inside the vial. The vial was placed in a custom-made open-top reduction block set at 525 °C for 3 h. The vials were allowed to cool to room temperature and the graphite that condensed on the iron was homogenized by mechanical-stirring action. The entire graphite sample was then pressed into aluminum targets. The [14C]s containing the graphite sample were measured by 500 kV tandem AMS (National Electrostatics Corp, WI, USA), which allows the measurement of the isotope ratio, 14C/12C, with a sensitivity of 2 × 10–15 and an instrument precision of 0.3–3%.
PBPK model development of PE
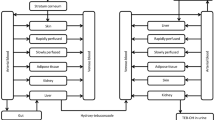
A comprehensive whole-body PBPK model of PE in rats was developed based on a previously reported model with modifications (Elmokadem et al. 2019; Troutman et al. 2015). The schematic of the PBPK model for PE and PAA after dermal and inhaled exposures of PE is depicted in Fig. 1. The model covers 29 compartments: 14 compartments for PE (gut, adipose, brain, heart, bone, kidney, liver, lung, muscle, spleen, arterial, vein, skin, rest of body), 14 compartments for PAA (same as PE) and a depot for dosing. The compartments are linked by blood flow, and distribution in all tissues is assumed to be well-stirred or perfusion-limited. Ordinary differential equations (ODEs) were adapted from previously described equations as follows (Elmokadem et al. 2019; Jones and Rowland-Yeo 2013; Pawaskar et al. 2013; Thompson and Beard 2011):

Structure of the PBPK model for PE and its major metabolite, PAA in rats. QAd, QBo, QBr, QGu, QHa, QHe, QKi, QLi, QLu, QSk, QSp, and QRe are blood flow rate in adipose, bone, brain, gut, hepatic artery, heart, kidney, liver, lung, skin, spleen, and rest of body, respectively. CLki, CLki,paa CLLi and CLLi,paa represent kidney clearance of PE, kidney clearance of PAA, metabolic clearance of PE to form PAA, and metabolic clearance of PAA in liver, respectively
Equations (2, 3, 4, 5) represent ODEs for tissue, lung, arterial, and venous compartments, respectively, in which CT is the concentration of drug in tissue T; QT is the blood flow to that tissue; Carterial and Cvenous are the drug concentrations in the arterial and venous blood compartments; Kp,T is the tissue:plasma partition coefficient; and BP is the blood-to-plasma concentration ratio. In Eq. (5), lung, spleen, and gut compartments were not included in T.
For absorptions, the permeation rate after dermal applications is given by the following equations (Troutman et al. 2015; Wong 2007):
where Kper and SA represents a permeability coefficient through skin and surface area over which the PE formulation is applied, respectively.
The metabolic intrinsic clearance (CLint) of PE to form PAA was scaled using hepatocellularity per gram of liver (HPGL, 163 × 106 cells/g) and liver weight (10.98 g) (Smith et al. 2008). Physiological parameters used in the model were obtained from previously reported literature, and compound-related parameters of PE and PAA were obtained from the experiments and published articles. The physiological parameters of rats used in the PBPK model are summarized in Table 1. The PBPK model that incorporated the final parameters was generated in RStudio (version 1.2.5033) with R (version 4.0.0) using mrgsolve, mainly utilized to build R codes for model development, and other packages such as dplyr, ggplot2, tidyverse, gridExtra, and grid.
The simulated kinetic profiles of PE and PAA were confirmed by observations and sensitivity analysis was performed. The sensitivity of the final model to single parameters was calculated (local sensitivity analysis) using Eq. (7) (Hanke et al. 2018):
with S is sensitivity of the AUC to the examined model parameter, and ∆AUC, AUC, p, and ∆p represents change of the AUC, simulated AUC with the original parameter value, original model parameter value, and change of the examined model parameter value ranged ± 50% of p, respectively. A sensitivity value of + 1.0 signifies that a 10% increase of the examined parameter causes a 10% increase of the simulated AUC (Hanke et al. 2018).
Simulation of PE and PAA concentrations
For model validation, the predicted plasma concentrations of PE and PAA and AUCs were compared with those from observed data. With the final PBPK model of PE, systemic exposures of PE and PAA after dermal application and inhalation were simulated following NOAEL of 500 mg/kg/day for dermal application and that of 12.7 mg/kg/day for inhalation provided by the Environmental Protection Agency (EPA) (EPA 2019). Concentrations in kidney and liver (and lung for inhalations), which are known target organs of exhibiting toxicity of PE, as well as plasma concentrations of PE and PAA were simulated.
Results
PK studies in rats
Dermal applications
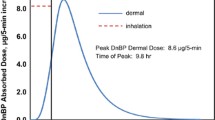
After dermal applications of PE to rats, PE is rapidly absorbed and extensively metabolized to form PAA. The mean plasma concentration–time profiles of PE and PAA after single and multiple dermal applications of PE (doses of 30 and 100 mg) are shown in Fig. 2 and the relevant PK parameters are listed in Table 2. Since high doses of PE were applied to each rat, the remaining PE amounts in patches were measured and the actually absorbed doses of PE were calculated as 8.84 ± 1.95 for 30 mg single application, 17.2 ± 1.56 for 30 mg multiple applications, 34.9 ± 55.6 for 100 mg single application, and 51.1 ± 4.10 for 100 mg multiple applications. In the PK results of PE, the terminal half-lives of PE after multiple applications (28.7 min for 30 mg and 15.6 mg for 100 mg) were significantly (p < 0.01) shorter than those after a single application (1352 min for 30 mg and 838 mg for 100 mg). The AUClast and AUCinf after multiple applications were also lower than those after a single application of PE, which may be due to induction of metabolic enzymes by PE. In PAA results, after the 30 mg dose, the Cmax, terminal half-life, AUClast, and AUCinf values of PAA in the single application group were significantly higher (by 191%, 705%, 196% and 202%) compared with multiple dosing group. However, at 100 mg dosing of PE, there were no significant differences in PK parameters between the single and multiple applications. This result was presumed to be due to the saturation of metabolic processes of PAA in high-dosing groups. The calculated AUCPAA/AUCPE ratios were 1.44 (30 mg, single), 5.50 (100 mg, single), 1.42 (30 mg, multiple), and 3.79 (100 mg, multiple).

Mean arterial plasma concentration–time profiles of PE a, c and PAA b, d following single (●) and multiple (○) dermal application of PE at a dose of 30 mg a, b and 100 mg c, d to rats. Data are expressed as mean ± SD
Inhalation study
As in the case of dermal application of PE, after inhalation of PE to rats, PE is rapidly absorbed and extensively metabolized to form PAA. The mean plasma concentration–time profiles of PE and PAA after single and multiple (for 4 days) inhalation administration of PE (25.7 mg/h for 3 h inhalation) are shown in Fig. 3 and the relevant PK parameters are listed in Table 3. Because the respiratory rate of each rat and the actual amount of PE absorbed by the rat could not be measured during the experiments, the absorbed dose for each rat was calculated using Eq. (1). In the PK results of PE, the AUClast and AUCinf values after a single dosing were significantly greater (165% and 169%) than those after multiple dosing, which may be due to induction of metabolic enzymes of PE. However, Cmax and terminal half-life of PE were comparable between single and multiple inhalations. In the PAA results, the average Cmax of PAA after a single inhalation was significantly lower (by 144%) than that following multiple inhalations; whereas, the terminal half-life of PAA was comparable between the two groups. The calculated AUCPAA/AUCPE ratio in the multiple inhalation group (4.09) was significantly higher than that in the single group (1.35). This result may be due to not only the induction of metabolic enzymes to form PAA in the multiple inhalations but also the saturation of metabolic processes of PAA.

Mean arterial plasma concentration–time profiles of PE a and PAA b following single (n = 6, ●) and multiple (n = 4, ○) inhaled administration of PE at a dose of 77 mg to rats. Data are expressed as mean ± SD
Mass balance study of PE after dermal application
The fractions of cumulative radioactive doses recovered in urine, feces, and swab after dermal applications of 30 mg PE containing 2.59 nCi of [14C]PE as a microtracer are shown in Fig. 4 and the excretion of total radioactivity is listed in Table 4. The total recovery of the radioactive dose was 75.5% within 72 h after dermal application and the reaming amount of PE in the patch was 1.81 nCi, indicating that 30.1% of PE was actually absorbed into the skin. Urine and feces were collected at specific time intervals and swabs were collected once after cage washing at 72 h after dermal application. Most of PE and its derivatives were excreted in urine (73.03%) and rarely found in feces (0.66%). These results indicate that after dermal applications, absorbed PE is metabolized to PAA and mainly excreted in urine, not bile.

Cumulative recovery of total [14C]-radioactivity in excreta following a single dose of 30 mg (containing 2.59 nCi of [14C]PE) for dermal application (n = 5) to rats. Total recovery was included each collection samples and swab at 72 hr. Each value represents the mean ± SD
PBPK model development of PE and simulations
A whole-body PBPK model of PE and PAA after dermal application and inhalation was successfully developed and the model structure is shown in Fig. 1. Rat physiological parameters including blood flow and tissue volume were obtained from the study by Law et al. (Law et al. 2017) and are listed in Table 1. Both the volume and blood flow of each tissue were calculated based on the body weight of rat as 0.3 kg. Most biochemical-specific values used as input parameters including fu, fu,hep, BP ratio, CLint, CLki, and Kper were directly obtained from in-house experimental data or calculated using experimental data; whereas, distribution-related parameters were estimated using commercially available PBPK software, Simcyp® (Table 5). Specifically, the values of CLki of both PE and PAA were obtained from intravenous administration of PE to rats and the final values of CLint of PE and PAA were modified from experimental values. The values of fu,hep were calculated as fu,hep = fu0.5 (for non-basic compounds) (Winiwarter et al. 2019) and the Kper of PE was directly measured from in vitro skin absorption study using minipig skin. The values of in vitro CLint of PE and PAA were scaled using HPGL (163 × 106 cells/g) and liver weight (10.98 g) (Smith et al. 2008); we assumed that metabolism of PE to form PAA occurred in the liver and all PE metabolized in the liver is biotransformed to PAA. Moreover, based on the observed data in which a significant decrease of exposure after multiple dosing of PE may be due to the induction of metabolic enzymes, the steady state metabolic clearance, CLint,ss, was estimated based on the observed multiple dosing data. The values of CLint,ss of PE and PAA were firstly estimated using Parameter Estimation module in Simcyp® and these values were applied as initial values to the PBPK model developed using R. Then, the CLint,sss were manually adjusted by fitting the observed in vivo PK concentrations of PE and PAA after multiple applications of PE.
To compare the simulated plasma concentrations of PE and PAA with the observed data, PE and PAA concentrations in plasma were simulated under the same exposure scenarios as in the experiments. Plasma concentrations of PE and PAA after both single and multiple applications of PE were simulated. For dermal applications, 8.84 mg for a single dose and 17.2 mg for multiple doses were used for the simulation. For inhalations of PE, a dose of 25.7 mg/h for 3 h was applied. Figures 5 and 6 shows predicted and observed plasma concentrations of PE and PAA after PE applications and the predicted AUCs were also compared with the observed AUCs summarized in Table 6. The predicted AUC/observed AUC ratios of PE and PAA were 0.86 and 1.03 in a single dermal application (1.44 and 1.13 in multiple dermal applications) and those were 0.98 and 1.24 in a single inhalation (0.68 and 1.23 in multiple inhalations), respectively.

Predicted and observed plasma PK profiles of PE a, c and PAA b, d after single (8.8 mg, a, b) and multiple (17.2 mg, c, d) dermal applications of PE. Dots represent observed concentration of PE and PAA; whereas, black lines are simulated concentrations of PE and PAA with the developed PBPK model

Predicted and observed plasma PK profiles of PE a, c and PAA b, d after single a, b and multiple c, d inhalations (77 mg) of PE. Dots represent observed concentration of PE and PAA whereas black lines are simulated concentrations of PE and PAA with the developed PBPK model
The sensitivity analysis was conducted including the parameters if they were optimized, if they were obtained from experiments, or if they had significant impact in the model. Sensitivity of the models was calculated as changes of AUC according to the ± 50% changes of the parameters and the results are shown in Table 7. The BP ratios of PE and PAA were the most sensitive parameters influencing the concentration of PE and PAA in plasma.
With the final PBPK model, the concentration–time profiles of PE and PAA after dermal application and inhalation were simulated following NOAEL of 500 mg/kg/day for dermal application and that of 12.7 mg/kg/day for inhalation. The duration of exposures of 6 h was applied to the exposure scenarios and kidney and liver (and lung for inhalations) concentrations as well as plasma concentrations of PE and PAA were simulated and shown in Figs. 7 and 8. The predicted internal exposures (AUC and steady-state concentrations, Css) of PE and PAA in plasma and target tissues are listed in Table 8. In dermal applications, the AUCss values of PE in plasma, liver, and kidney were 133, 48.2, and 130 μg·h/mL and the AUCss values of PAA in plasma, liver, and kidney were 150, 443, and 545 μg·h/mL, respectively. In inhalations, the AUCss values of PE in plasma, liver, kidney, and lung were 1.62, 0.590, 1.58, and 1.28 μg·h/mL and the AUCss values of PAA in plasma, liver, kidney, and lung were 11.5, 34.1, 42.0, and 13.8 μg·h/mL, respectively.

Simulated steady-state plasma a, liver b, and kidney c concentration—time profiles of PE (line) and PAA (dashed) after dermal applications of 500 mg/kg/day. From day 1, PE and PAA were assumed to have reached steady state exposures and PE was applied for 6 h/day for 4 days

Simulated steady-state plasma a, liver b, kidney c, and lung d concentration—time profiles of PE (line) and PAA (dashed) after inhalations of 12.7 mg/kg for 4 days. From day 1, PE and PAA were assumed to have reached steady state exposures and PE was inhaled for 6 h/day for 4 days
Discussion
PE is one of the widely used biocides, and the details of the toxicity and PK are well-documented in the final version of ‘Opinion on phenoxyethanol’ (SCCS 2016). Nevertheless, there is not much information about the PK of PE and PAA after multiple dermal applications and even less information about the PK of PE and PAA after inhalation of PE. Therefore, in this study, we performed PK studies of PE. We evaluated PK characteristics of PE and PAA after dermal and inhaled exposures of PE in rats and also developed a rat PBPK model of PE and PAA.
After dermal and inhaled applications of PE, PE was rapidly absorbed, which is consistent with findings in previous studies (Dreno et al. 2019; Roper et al. 1997). The half-life of PE after intravenous administration was 11.6 min (data not shown), which was sufficient time for PE to reach steady-state absorption. Therefore, exposure durations of 4 h for the skin and 3 h for inhalation were chosen. In dermal applications, the absorbed dose of PE, calculated as \(\left( {{\text{the}}\;{\text{amount}}\;{\text{applied}}\;{\text{to}}\;{\text{the}}\;{\text{patch}}} \right)\; - \;\left( {{\text{the}}\;{\text{amount}}\;{\text{remaining}}\;{\text{in}}\;{\text{the}}\;{\text{patch}}} \right)\), was higher in the multiple dosing group than in the single dosing group. We speculate this may be from skin irritations caused by repeated patch attachment in the same area, and the amount of PE that penetrated skin during multiple dosing was greater than that of a single dose (1.95-fold at 30 mg and 1.58-fold at 100 mg). Nevertheless, the systemic exposures (i.e., Cmax and AUC) of both PE and PAA in the multiple dosing group were lower than those in the single dosing group. In particular, the AUCPAA/AUCPE ratio was significantly higher in multiple dosing groups and since the half-life of PE was significantly longer in a single dose, the low systemic exposures of PE in multiple dosing were considered to be due to induction of metabolic enzymes of PE in liver and blood. In inhalations, PE was also rapidly absorbed and metabolized to form PAA. The value of AUC after multiple inhalations of PE was significantly lower than that after a single inhalation, whereas the value of AUCPAA was increased in the multiple inhalation groups (AUCPAA/AUCPE ratio was significantly higher in multiple dosing groups) and these were considered to be from the induction of metabolic enzymes of PE, as in the dermal application study.
PE is metabolized by alcohol dehydrogenase and aldehyde dehydrogenase in liver (Lockley et al. 2005; Roper et al. 1997). Rat skin also has the ability to metabolize PE, but PE metabolism in the skin would be minimal because the residence time of PE in the skin is short (Lockley et al. 2005; Roper et al. 1997). After multiple dosing of glycol ether groups, specific metabolic enzymes are induced. After multiple dermal administrations of ethanol and 2-butoxyethanol to rats, the metabolism of ethanol and 2-butoxyethanol in rat whole skin cytosol was increased (Lockley et al. 2005) and ethanol induced CYP3A and CYP2E1 (Feierman et al. 2003). The results of this study, which showed a significant decrease in PE concentration after multiple doses, support the possibility of enzyme induction by PE.
A mass balance study of PE after dermal application was also conducted and most of the PE and its derivatives were excreted in urine rather than via bile and feces. The total recovery was 75.51% for 72 h after dermal applications. Based on the total recovery after oral and intravenous administration of PE (100.0% after oral and 107.1% after intravenous; data not shown) and the short residence time of PE in the skin, few of the PEs that did not recovered for 72 h would be accumulated or remained in the layers of the skin. This result was consistent with previously published results (Howes 1991) that reported that 60% of the dose was recovered in the urine at 48 h, mainly as free or conjugated PAA, and total recovery was 64–76% of the original dose. These findings indicated that the relatively low recovery after dermal application was because of evaporation of the applied PE (Howes 1991; Roper et al. 1997). Most PE was excreted in urine as PAA, and this may be related with the kidney toxicity of PE (Dreno et al. 2019; Troutman et al. 2015); therefore, examining PAA concentration in kidney would be critical to evaluate exposure-toxicity relationships.
A whole-body PBPK model of PE and PAA after dermal application and inhalation was successfully developed. The PBPK model facilitates the quantitative analysis of exposure–toxicity relationships. Results and findings from toxicology studies can be better understood when relevant and reliable internal estimates of toxicant exposure in blood or target tissues are available (McLanahan et al. 2012). In particular, when metabolites as well as the parent compound exhibit significant toxic effects, such as in the case of PE and PAA, the use of a PBPK model for risk assessment can help in an intensive and quantitative understanding of the toxicity of the compound.
The model was first developed and evaluated based on the PK data of PE and PAA after intravenous administration. The final clearance (CL) and volume of distribution (Vd) parameters had been determined in advance and then absorption parameters were applied to the model using in vitro and in vivo dermal and inhalation data. Biochemical reactions are dependent on the free concentration of the compound in plasma or target organs and metabolism in hepatocytes can also occur only unbound form of compound; therefore, plasma-to-tissue partition coefficients (Kp), fu, and fu,hep were used in the model. The Kp in each tissue was estimated using Method 2 incorporated within Simcyp® or obtained from published values (Kim et al. 2015). Method 2 is the Rogers and Rowland (R&R) method to calculate Kp in each tissue and takes into account the values of tissue components including neutral lipids, neutral phospholipid, extracellular water, and intracellular water; the advantage of this method is that ionization and lipophilicity of compounds are incorporated (Rodgers and Rowland 2006). The values of fu were obtained in in vitro experiments and fu,hep values were calculated using fu. The challenge to develop a PBPK model of PE was (1) to determine absorption parameters and (2) to determine metabolism parameters after multiple dosing. For dermal applications, the model validation was conducted using observed data and the actually absorbed dose was calculated using the concentration of PE in the product (formulation), in vitro Kper, applying surface area, and applying time. A dosing compartment (depot) was used for the absorption of PE. For inhalation, the inhaled amount of PE was directly dosed into the lung compartment and the bioavailability as 0.48 was applied to the final model based on the observed data. After multiple inhaled dosing of PE, the systemic exposures of PE decreased while those of PAA increased compared with those after a single dose. These were presumably due to the saturation of metabolism of PAA, therefore, the CLint of PAA was used instead of CLint,ss for multiple inhalation simulations. Since in vitro induction parameters (e.g., EC50 and Emax) for the PBPK model were not available, CLint,ss was optimized based on the observed PK profiles.
With the developed model, simulations following NOAEL of 500 mg/kg/day for dermal application and that of 12.7 mg/kg/day for inhalation were performed. For the considerations of chronic exposures of PE, CLint,ss rather than CLint were used for the simulations. Both PE and PAA concentrations in plasma, liver, and kidney (and lung for inhalations) were predicted and AUC and Css of PE and PAA were also calculated. At 500 mg/kg/day for dermal applications, the AUCs of PE and PAA in systemic exposure were much higher than those at 12.7 mg/kg/day for inhalation.
This study has several limitations. In the inhalation PK study, the actual absorbed inhalation dose could not be directly measured during the experiments. The inhaled amount was calculated using the respiratory rate and tidal volume of rats; therefore, respiratory rate is a critical factor in determining inhalation dose. However, because it was not possible to directly measure the actual respiratory rate of rats used in this experiment, the respiratory rate of rats was obtained from a published article. The respiratory rate of rats used during the experiment may differ from the literature value, but it could not be reflected in the dose calculation. The CLint parameter obtained from in vitro experiments and the literature also could not accurately describe the observed data. The final CLint parameters incorporated in the model were optimized based on the observed data. In the development of the in vitro-in vivo extrapolation (IVIVE)–based PBPK model, discrepancies between the PK parameters obtained from in vivo data and those obtained from in vitro data are common. In this case, the in vitro parameter used as input parameters of the model can be modified according to the observed data, but if there are no in vivo data, an incorrect model may be developed. Therefore, the in vitro parameters used in model should be carefully selected, and for the same reason, sensitivity analysis of parameters should be conducted for model verification.
In conclusion, this study evaluated the PK profiles of PE and PAA after dermal and inhaled route of exposures of multiple dosing of PE and also developed a PBPK model of PE in rats. In accordance with the physicochemical properties of PE, which is highly permeable across the membrane, PE was rapidly absorbed regardless of the route of exposure. After modifying details to describe human physiology, this model structure can be applied to develop a human PBPK model of PE, and the internal dose metric derived by this model can be used for human risk assessment of PE after dermal and inhaled routes of exposures.
Data availability
The data are available upon the reasonable request to the corresponding author.
Abbreviations
- AC:
-
Aerosol concentration
- AMS:
-
Accelerator mass spectrometer
- AUC:
-
Area under the plasma concentration–time curve
- AUCinf :
-
Area under the plasma concentration–time curve from time zero to infinity
- AUClast :
-
Area under the plasma concentration–time curve within time span zero to last
- AUCPAA/AUCPE :
-
Metabolic ratio calculated by phenoxyacetic acid AUC/phenoxyethanol AUC ratio
- Carterial :
-
Drug concentrations in arterial blood compartments
- CL:
-
Clearance
- CLint :
-
Intrinsic clearance
- Cmax :
-
Maximum observed drug concentration in plasma
- Css :
-
Concentration at steady-state
- CT :
-
Concentration of drug in tissue
- CV:
-
Coefficient of variation
- CYP:
-
Cytochrome P450s
- D:
-
Duration of exposure to the aerosol
- ESI:
-
Electrospray ionization
- fu :
-
Unbound fraction in plasma
- fu ,hep :
-
Unbound fraction in hepatocytes
- IS:
-
Internal standard
- IVIVE:
-
In vitro-in vivo extrapolation
- Kper :
-
Permeability coefficient through skin
- Kp ,T :
-
Tissue:plasma partition coefficient
- LC–MS/MS:
-
Liquid chromatography tandem mass spectrometry
- LLOQ:
-
Lower limit of quantification
- LSC:
-
Liquid scintillation counting
- MRM:
-
Multiple reaction monitoring
- NOAEL:
-
No-observed-adverse-effect level
- p :
-
Original model parameter value
- PBPK:
-
Physiologically-based pharmacokinetic
- PK:
-
Pharmacokinetic
- QC:
-
Quality control
- QT :
-
Tissue blood flow
- RMV:
-
Represents respiratory minute volume, which is the respiratory rate multiplied by the tidal volume
- RSD:
-
Relative standard deviation
- SA:
-
Surface area of applied PE formulation
- t ½ :
-
Terminal elimination half-life
- Tmax :
-
Time to reach maximum (peak) plasma concentration following drug administration
- Vd :
-
Volume of distribution
- VT :
-
Volume in tissue compartment
References
Breslin WJ, Phillips JE, Lomax LG et al (1991) Hemolytic activity of ethylene glycol phenyl ether (EGPE) in rabbits. Fundam Appl Toxicol 17(3):466–481. https://doi.org/10.1016/0272-0590(91)90198-d
Chiou WL (1978) Critical evaluation of the potential error in pharmacokinetic studies of using the linear trapezoidal rule method for the calculation of the area under the plasma level–time curve. J Pharmacokinet Biopharm 6(6):539–546. https://doi.org/10.1007/BF01062108
Dreno B, Zuberbier T, Gelmetti C, Gontijo G, Marinovich M (2019) Safety review of phenoxyethanol when used as a preservative in cosmetics. J Eur Acad Dermatol Venereol 33(Suppl 7):15–24. https://doi.org/10.1111/jdv.15944
ECHA (2019) European Chemicals Agency (ECHA). Substance Infocard: 2-phenoxyethanol. https://echa.europa.eu/substance-information/-/substanceinfo/100.004.173
Elmokadem A, Riggs MM, Baron KT (2019) Quantitative systems pharmacology and physiologically-based pharmacokinetic modeling with mrgsolve: a hands-on tutorial. CPT Pharmacometrics Syst Pharmacol 8(12):883–893. https://doi.org/10.1002/psp4.12467
EPA (2019) Environmental Protection Agency (EPA). 2-phenoxyethanol; exemption from the requirement of a tolerance. Federal Register https://www.federalregister.gov/documents/2019/09/27/2019-20529/2-phenoxyethanol-exemption-from-the-requirement-of-a-tolerance
Feierman DE, Melinkov Z, Nanji AA (2003) Induction of CYP3A by ethanol in multiple in vitro and in vivo models. Alcohol Clin Exp Res 27(6):981–988. https://doi.org/10.1097/01.ALC.0000071738.53337.F4
Fioni A, Selg E, Cenacchi V et al (2018) Investigation of lung pharmacokinetic of the Novel PDE4 inhibitor CHF6001 in preclinical models: evaluation of the preciseinhale technology. J Aerosol Med Pulm Drug Deliv 31(1):61–70. https://doi.org/10.1089/jamp.2017.1369
Gibaldi M, Perrier D (1982) Pharmacokinetics, 2nd edn. Marcel-Dekker, New York
Hanke N, Frechen S, Moj D et al (2018) PBPK models for CYP3A4 and P-gp DDI prediction: a modeling network of rifampicin, itraconazole, clarithromycin, midazolam, alfentanil, and digoxin. CPT Pharmacometrics Syst Pharmacol 7(10):647–659. https://doi.org/10.1002/psp4.12343
Howes D (1991) Absoprtion and metabolism of 2-phenoxyethanol in rat and man. 15th IFSCC Int Congr Cosmet Sci 3:415–434
Jones H, Rowland-Yeo K (2013) Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacometrics Syst Pharmacol 2:e63. https://doi.org/10.1038/psp.2013.41
Kim JM, Yoon JN, Jung JW et al (2013) Pharmacokinetics of hederacoside C, an active ingredient in AG NPP709, in rats. Xenobiotica 43(11):985–992. https://doi.org/10.3109/00498254.2013.788231
Kim TH, Kim MG, Kim MG et al (2015) Simultaneous determination of phenoxyethanol and its major metabolite, phenoxyacetic acid, in rat biological matrices by LC-MS/MS with polarity switching: application to ADME studies. Talanta 144:29–38. https://doi.org/10.1016/j.talanta.2015.05.075
Law FCP, Yao M, Bi HC, Lam S (2017) Physiologically based pharmacokinetic modeling of tea catechin mixture in rats and humans. Pharmacol Res Perspect 5(3):e00305. https://doi.org/10.1002/prp2.305
Lockley DJ, Howes D, Williams FM (2005) Cutaneous metabolism of glycol ethers. Arch Toxicol 79(3):160–168. https://doi.org/10.1007/s00204-004-0619-3
McLanahan ED, El-Masri HA, Sweeney LM, Kopylev LY, Clewell HJ, Wambaugh JF, Schlosser PM (2012) Physiologically based pharmacokinetic model use in risk assessment—why being published is not enough. Toxicol Sci 126(1):5–15
Ognibene TJ, Bench G, Vogel JS, Peaslee GF, Murov S (2003) A high-throughput method for the conversion of CO2 obtained from biochemical samples to graphite in septa-sealed vials for quantification of 14C via accelerator mass spectrometry. Anal Chem 75(9):2192–2196. https://doi.org/10.1021/ac026334j
Pawaskar DK, Straubinger RM, Fetterly GJ et al (2013) Physiologically based pharmacokinetic models for everolimus and sorafenib in mice. Cancer Chemother Pharmacol 71(5):1219–1229. https://doi.org/10.1007/s00280-013-2116-y
Rodgers T, Rowland M (2006) Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci 95(6):1238–1257. https://doi.org/10.1002/jps.20502
Roper CSHD, Blain PG, Williams FM (1997) Percutaneous penetration of 2-phenoxyethanol through rat and human skin. Food Chem Toxicol 35:1009–1016
SCCS (2016) Opinion on phenoxyethanol. Scientific Committee on Consumer Safety https://ec.europa.eu/health/scientific_committees/consumer_safety/docs/sccs_o_195.pdf
Scognamiglio J, Jones L, Letizia CS, Api AM (2012) Fragrance material review on 2-phenoxyethanol. Food Chem Toxicol 50(Suppl 2):S244–S255. https://doi.org/10.1016/j.fct.2011.10.030
Scortichini BH, Quast JF, Rao KS (1987) Teratologic evaluation of 2-phenoxyethanol in New Zealand White rabbits following dermal exposure. Fundam Appl Toxicol 8(2):272–279. https://doi.org/10.1016/0272-0590(87)90126-6
Smith R, Jones RD, Ballard PG, Griffiths HH (2008) Determination of microsome and hepatocyte scaling factors for in vitro/in vivo extrapolation in the rat and dog. Xenobiotica 38(11):1386–1398. https://doi.org/10.1080/00498250802491662
Starek A, Szabla J, Kiec-Kononowicz K, Szymczak W (2008) Comparison of the in vitro hemolytic effects produced by alkoxyacetic acids on human and rat erythrocytes. Int J Occup Med Environ Health 21(2):147–155. https://doi.org/10.2478/v10001-008-0009-9
Starek-Swiechowicz B, Miranowicz-Dzierzawska K, Szymczak W, Budziszewska B, Starek A (2012) Hematological effects of exposure to mixtures of selected ethylene glycol alkyl ethers in rats. Pharmacol Rep 64(1):166–178. https://doi.org/10.1016/s1734-1140(12)70743-0
Thompson MD, Beard DA (2011) Development of appropriate equations for physiologically based pharmacokinetic modeling of permeability-limited and flow-limited transport. J Pharmacokinet Pharmacodyn 38(4):405–421. https://doi.org/10.1007/s10928-011-9200-x
Troutman JA, Rick DL, Stuard SB, Fisher J, Bartels MJ (2015) Development of a physiologically-based pharmacokinetic model of 2-phenoxyethanol and its metabolite phenoxyacetic acid in rats and humans to address toxicokinetic uncertainty in risk assessment. Regul Toxicol Pharmacol 73(2):530–543. https://doi.org/10.1016/j.yrtph.2015.07.012
Vogel JS, Giacomo JA, Schulze-Konig T, Keck BD, Lohstroh P, Dueker S (2010) Accelerator mass spectrometry best practices for accuracy and precision in bioanalytical (14)C measurements. Bioanalysis 2(3):455–468. https://doi.org/10.4155/bio.10.13
Winiwarter S, Chang G, Desai P et al (2019) Prediction of fraction unbound in microsomal and hepatocyte incubations: a comparison of methods across industry datasets. Mol Pharm 16(9):4077–4085. https://doi.org/10.1021/acs.molpharmaceut.9b00525
Wong BA (2007) Inhalation exposure systems: design, methods and operation. Toxicol Pathol 35(1):3–14. https://doi.org/10.1080/01926230601060017
Acknowledgements
We especially thank the support from Professor Kyung-Min Lim in Ewha Womans University, Professor Ok-Nam Bae in Hanyang University, and Professor Soo Kyung Bae in the Catholic University of Korea, Republic of Korea.
Funding
This study was funded by the Korea Ministry of Environment (MOE) as “the Environmental Health Action Program” and “Technology Program for establishing biocide safety management” (2019002490005 1485016231 and 2019002490004 1485016253) and also supported by a grant of the Korea Institute of Radiological and Medical Sciences (KIRAMS), funded by Ministry of Science and ICT(MSIT), Republic of Korea (No. 50539-2020).
Author information
Authors and Affiliations
Contributions
All authors were involved in the conception and design of the study and data interpretation. MK and SHB drafted the paper, conducted PK studies, and performed PBPK modeling. JBP and MK conducted PK studies and JS and CSY conducted sample analysis. All authors critically revised the paper and approved it for submission.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest to be disclosed.
Ethics approval
The animal experiment was approved by the Institutional Animal Care and Use Committee (IACUC) of the Korea Institute of Radiological & Medical Sciences (KIRAMS), Republic of Korea (approval number: Kirams2019-45). In addition, all animal experimental procedures were carried out in accordance with the revised Guidelines for Ethical Conduct in the care and Use of Animals and the rules of Good Laboratory Practice.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kwon, M., Park, J.B., Kwon, M. et al. Pharmacokinetics of 2-phenoxyethanol and its major metabolite, phenoxyacetic acid, after dermal and inhaled routes of exposure: application to development PBPK model in rats. Arch Toxicol 95, 2019–2036 (2021). https://doi.org/10.1007/s00204-021-03041-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-021-03041-z