
Abstract
The metabolic activity of hepatocytes is a central prerequisite for drug activity and a key element in drug–drug interaction. This central role in metabolism largely depends on the activity of the cytochrome P450 (CYP450) enzyme family, which is not only dependent on liver cell maturation but is also controlled in response to drug and chemical exposure. Here, we report the use of VividDye fluorogenic CYP450 substrates to directly measure and continuously monitor metabolic activity in living hepatocytes. We observed time- and dose-dependent correlation in response to established and putative CYP450 inducers acting through the aryl hydrocarbon receptor and drug combinations. Using repetitive addition of VividDye fluorogenic substrate on a daily basis, we demonstrated the new application of VividDye for monitoring the maturation and dedifferentiation of hepatic cells. Despite a lack of high specificity for individual CYP450 isoenzymes, our approach enables continuous monitoring of metabolic activity in living cells with no need to disrupt cultivation. Our assay can be integrated in in vitro liver-mimetic models for on-line monitoring and thus should enhance the reliability of these tissue model systems.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Monitoring metabolic activity is an important prerequisite to proof reliability of in vitro liver models to be used for drug testing and drug–drug interaction studies. Liver and in particular hepatocytes are central for the metabolism of drugs and xenobiotics, which is performed by enzymes of the cytochrome P450 (CYP450) superfamily (Montellano 2005; Hewitt et al. 2007). There are several subclasses of CYP450, notably CYP1A, CYP2B, CYP2C9, CYP2C19, CYP2E1 CYP3A and CYP4A, which are induced after exposure to xenobiotics or drugs. CYP1A1/1A2 and 1B1 are induced through the aryl hydrocarbon receptor (AhR) in concert with its co-regulator aryl hydrocarbon receptor nuclear translocator (ARNT), which regulates transcription of target genes upon ligand binding and subsequent translocation to the nucleus (Ma 2001; Denison and Heath-Pagliuso 1998; Hankinson 1995), where it binds to drug or xenobiotic response elements (DRE/XRE) in promoter or enhancer regions of target genes, including the various CYP450. Drugs are mostly substrates of one specific CYP450 enzyme. Direct monitoring of CYP450 activity is of high interest to enable early detection of drug toxicity and drug–drug interactions (DDI) (Friedman et al. 1999; Lasser et al. 2002). Various in vitro tests are in use to assess CYP450 activity in drug discovery. These methods commonly use cell lysates or liver microsomes enriched in CYP450 enzymes in combination with a range of various downstream analytical approaches to identify metabolic activity and metabolites, including HPLC, and radioactive-labeled substrates, as well as fluorescence- and luminescence-based assays. So far, human liver microsomes have been used as the gold standard to determine drug metabolism and metabolic interaction during drug development in vitro. All cell lysate- and microsome-based approaches have in common that they only analyze actual metabolic turnover but exclude other effects inherent in living cells and organs such as gene expression induction and toxicity (Kelly and Sussman 2000; Reiners et al. 1990). To detect effects directly in living systems including liver-on-chip systems, we investigated on the use of the VividDye substrates for direct monitoring of CYP450 activities in living cells. By now, VividDye substrates are used in combination with liver BACULOSOMES®, a special type of liver microsomes overexpressing specific CYP450 isoforms, to analyze CYP450-dependent drug interaction (Trubetskoy et al. 2005). VividDye substrates are metabolized by CYP450 enzymes, thereby forming highly fluorescent leaving groups, which can be detected in standard plate readers. We show that the assay can be used for several applications including, but not limited to, CYP450 response after drug treatment, co-drug treatment, comparison of cellular environment for mono- and co-culture systems, and changes in CYP450 activity during cellular maturation and dedifferentiation. We tested our system on several CYP450-expressing cell types including primary murine, HepaRG, HepG2, upcyte® Hepatocytes, murine iHep and HepG2 and liver endothelial cells lacking CYP450 expression.
In the drug-treatment experiments, we focused on induction and inhibition via the AhR receptor. We, therefore, investigated on 2,3,7,8 Tetrachlorodibenzo-p-dioxin (TCDD) as reference substance and the indirubin derivative E804 as potential AhR ligand (Göttel et al. 2014), as well as on resveratrol as AhR inhibitor. Indirubin is the major active compound of a herbal recipe in traditional Chinese medicine (Cheng et al. 2010), which is used for treatment of various chronic diseases in China (Cheng et al. 2016, 2014). Recent results have shown the potential applicability of indirubin derivatives in regenerative medicine due to diverse mechanisms of action (Sato et al. 2004; Cheng et al. 2015a, b). Moreover, the detection of indirubin in human urine led to the identification of indirubin as a potent endogenous AhR ligand (Adachi et al. 2004, 2001; Guengerich et al. 2004).
In our differentiation assay, we made use of HepaRG cells, which undergo different stages during the maturation process (Andersson et al. 2012; Gerets et al. 2012; Kanebratt and Andersson 2008), leading to increased levels of VividDye metabolism. We finally optimized the assay to be used in microscope setups on single cell level and for integration into microfluidic-based in vitro systems.
Materials and methods
Chemicals
E804 was synthesized as previously reported and characterized by NMR, HPLC and MS. (https://doi.org/10.1021/acs.jmedchem.7b00324). The purity was at least more than 96%. TCDD and Resveratrol were purchased from Sigma-Aldrich (Germany). VividDyes were purchased from Life Technologies (Thermofisher, Germany) and reconstituted in acetonitrile.
Cell isolation and culture
Primary mouse hepatocytes (pmH) were isolated from male C57BL/6 mice (8–13 weeks old) using collagenase perfusion as described (Levy et al. 2015). The work and procedure was approved by the government of Baden-Württemberg’s Animal Care Committee, Regierungspräsidium Karlsruhe, Germany, and all animals received humane care in compliance with the German Animal Protection Act (reference number 35-9185.81/G-145-14 valid until 30.09.2019). The typical viability of HCs was 95%. PmH were cultured in three kinds of media: Medium 1: Williams’ medium E supplemented with 10% fetal calf serum (FCS) 2 mM l-glutamine, 1% penicillin/streptomycin and 100 nM dexamethasone. Medium 2: medium 1 without FCS. Medium 3: Williams’ medium E supplemented only with 2 mM l-glutamine and 1% penicillin/streptomycin. Freshly isolated HCs were plated on collagen-coated 3.5-cm plates at a density of 4 × 105 cells/dish in medium 1 and incubated in 5% CO2 at 37 °C for 4 h. Then medium 1 was replaced with medium 2 for serum starvation and cell cycle synchronization. On the second day, medium 2 was changed to medium 3.
HepG2 hepatocellular carcinoma cells (ATCC, Germany) were cultured in DMEM Glutamax medium containing 10% FCS, 1% Penicillin/Streptomycin, and were maintained under standard cell culture conditions. Cells were passaged upon reaching 70–80% confluency, and cells of low passage number (< 10) were used in our experiments.
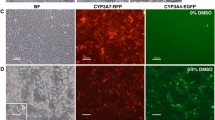
HepaRG cells were obtained from BIOPREDIC International at passage number 0. Cells of low passage number (< 6) were used in the experiments. For proliferation, HepaRG cells were cultured in William’s E medium containing 10% FCS, 2 mM l-glutamine 1% penicillin/streptomycin, 5 μg/ml insulin and 50 μM hydrocortisone-hemisuccinate. To induce differentiation, 1.5% DMSO was added to proliferation medium. Cells were cultured in proliferation medium for 14 days and the medium was changed weekly. Subsequently maturation of cells was induced by adding 1.5% DMSO to the medium for 14 days, where medium was changed every 3 days.
iHep
Mouse-induced hepatocytes were obtained directly from lineage reprogramming of mouse myofibroblasts with ectopic expression of Foxa3, Gata4, Hnf1a and Hnf4a as described before (Song et al. 2016). The expandable iHeps were cultivated in a mixture of hepatocyte culture medium (CC-3198, Lonza, Germany) and DMEM (1:1) in a 6-well plate coated with collagen (Roche, Germany) and passaged every 3–4 days (confluent 70–90%) with the help of cell culture scraper. The medium was refreshed every other day.
Upcyte® cells
Upcyte® Hepatocytes and upcyte® LSECs were purchased from upcyte® technologies, Germany, and cultured according to the manufactures protocols. In brief, flasks were collagen-coated, cells were thawed in hepatocyte thawing medium and maintained in hepatocyte high-performance medium and endothelial cell culture medium, respectively.
HepG2 Crispr/Cas9 knockdown AhR cell line
CRISPR/CAS9 AhR plasmid (SC-400297-NIC-NIC) was purchased from Santa Cruz, Germany. Lipofectamine 3000 (Life Technologies, Germany) was used for transfection according to the manufacturer’s instruction. HepG2 cells were seeded in a 24-well plate at a density of 400,000 cells/well. For the selection 0.5 µg/mL puromycin was used. The established HepG2 AhRkd cell line was cultivated in DMEM with 10% FCS, 1% PS and 0.5 µg/mL puromycin. The medium was changed two or three times per week.
qRT-PCR
As described before (Cheng et al. 2016), quantitative reverse transcription real-time PCR (RT-qPCR) was performed according to the manufacturer’s instruction (Lightcycle 96, Roche, Germany). cDNA was generated by reverse transcription of equivalent quantities of total RNA (ProtoScript® First Strand cDNA synthesis kit, NEB, Germany), isolated from cells using the RNeasy kit (Qiagen, Germany). RT-qPCR was performed using SYBR Green PCR master mix (qPCRBIO SyGreen Mix Lo-Rox, Nippon Genetics, Germany). PCR primers were obtained from Eurofins (Germany); respective primer sequences used are listed in supplementary information. Actin was used as endogenous control.
Immunoblotting
As we described before (Cheng et al. 2016), cell extracts were homogenized in urea lysis buffer (1 mM EDTA, 0.5% Triton X-100, 5 mM NaF, 6 M Urea, 1 mM Na3VO4, 10 µg/mL Pepstatin, 100 µM PMSF and 3 µg/mL Aprotinin in PBS). The immunoblot was detected by ECL solution. 40 µg of total protein was resolved on 8% SDS–PAGE gels and immunoblotted with specific antibodies. Primary antibodies, CYP1A1 (Adgen, Biomol, Germany) and AhR (Biomol, Germany) were incubated at a 1:1000 dilution in blocking buffer (5% BSA) with gentle agitation overnight at 4 °C. The secondary antibodies (Dianova, Germany) were used in 1:10,000 dilution.
Human albumin ELISA
Secretion of albumin was measured by enzyme-linked immunosorbent assay (ELISA) kit (Bethyl laboratories, Montgomery, TX). ELISA assay was performed in clear flat-bottom 96-well plates. Supernatant of cells was collected and kept frozen at − 20 °C prior to performing the assays. Prior to this, the assay samples were thawed to room temperature and prepared in accordance to the manufacturer protocol. Protein content was normalized using Bradford assay (Sigma-Aldrich, Germany). Albumin ELISA assay was optimized using different dilution of the investigated samples. Samples were diluted in the supplied buffers. The assay protocol was followed stepwise. Plate’s absorbance at 450 nm wavelength was measured at room temperature using a microplate reader (Ultra TECAN plate reader, Germany).
Urea synthesis assay
Urea assay was performed in accordance to the urea assay kit (Sigma-Aldrich, Germany). In brief, the supernatant was collected on the same days as the albumin assay and stored at − 20 °C. Samples were diluted with the supplied buffer to adjust concentrations to the linear range of the assay. Assays were run in clear flat-bottom 96-well plates and measured at 570 nm using a microplate reader (Ultra TECAN plate reader, Germany).
Fluorescent kinetic assays in HepG2 and primary murine hepatocytes
Cells were plated at a density of 3.5 × 104 for HepG2 and 2 × 104 cells for pmH on transparent 96-well microplates. After 6 h, cells were washed carefully with HBSS to remove dead cells. Medium was replaced and cells were treated with TCDD, E804 or a combination of both with resveratrol for 24 h. VividDye substrates BOMCC, EOMCC, BOMR and OOMR (Lifetechnologies, Germany) were reconstituted in acetonitrile and stored at − 20 °C. Substrates are linked to different subgroups of CYP450 isoenzymes, also specificity in vivo is unclear. VividDye substrates become fluorescent upon metabolic cleavage, BOMCC and EOMCC utilize 7-hydroxycoumarin as fluorescent product, whereas cleaved BOMR and OOMR contain fluorescent resorufin. In 96-well plate format, cells were pre-treated with the drugs for 24 h before medium was replaced with 100 µl VividDye 1 µM assay solution of the respective VividDye.
Kinetic assay measurements were started immediately after addition of the VividDye mixture. Cells were placed into a TECAN Sapphire reader with sample temperature set to 37 °C. Measurements were taken every 3 min. For EOMCC and BOMCC, excitation and emission wavelengths were set according to the manufacturers’ protocol (Excitation: 415 Emission: 460 for BOMCC and EOMCC and Excitation: 550 nm Emission 590 nm for BOMR and OOMR) with a window of 10 nm; to minimize variations caused by different cell seeding density, absorbance at 580 nm was also measured. Fluorescence signals were divided by absorbance to normalize values relative to cell number/density.
Culture of hepatocytes in optimized tissue culture condition
The influence of different culture methods was compared: conventional 2D cell culture, collagen/matrigel sandwich culture, and co-culture systems using hepatocytes and liver endothelial cells. HepG2 and upctyte Hepatocytes were seeded into 24-well plates at 1.5 × 105 cells per well. For upcyte® hepatocytes, wells were pre-coated with collagen as described by the manufacturer. For sandwich culture, HepG2 cells were overlaid with collagen, and upcyte cells were overlaid with matrigel. For our co-culture experiments, following 24 h incubation, 0.5 × 105 human liver endothelial cells (HLEC) were added to hepatocytes, (both HepG2 and upcyte® hepatocytes). Experiments were performed in two groups: non-treated cells and cells where CYP450 activity was induced using 24 h pre-incubation with 20 nM TCDD. To analyze the CYP450 activity, BOMR VividDye was added on day 3 and 5 (HepG2) and day 5 and 8 (upcyte) to the cells for 2 h and the supernatant was analyzed.
Long-term monitoring of CYP450 activity during hepatocyte maturation and dedifferentiation
Functionality of hepatocytes is crucially associated with the quality of hepatocellular maturation and differentiated phenotype. CYP450 activity of HepaRG cells was investigated over 21 days differentiation and maturation. Dedifferentiation of pmH during 9 days of cultivation. Cells were seeded into a 24-well plate at 1 × 105 cells. One group of cells was continuously exposed to 10 nM TCDD, the other group was treated with media only. Both groups were exposed to BOMR fluorescent substrate for 1 h, supernatant was taken and measured as described above, using 96-well plate and a TECAN Sapphire reader.
Microscopic monitoring of the hepatocyte differentiation and in cell recording of CYP450 activity
Direct determination of CYP450 activity in cells by microscopy was established to detect changes in CYP activity during differentiation of HepaRG cells. HepaRG cells were seeded at a concentration of 1 × 105 cells in four-well µ-Slides, (ibiTreat, ibidi, Germany). To induce CYP activity, HepaRG cells were pre-incubated with 10 nM TCDD for 24 h. Cells were washed with PBS and VividDye substrates were added to the cells; pictures were taken by time laps microscopy for a period of 20 min and differentiated cells were compared to non-treated cells.
Pictures were taken using the following filter combination purchased from Chroma (see Table 1):
Background on microscopic setup
High-resolution fluorescence images were acquired using a Ti-TuCam microscope (Nikon Imaging Centre, Heidelberg). We used a Nikon Apo LWD 40x NA 1.15 λS water immersion objective optimized for coverslips. Pictures were analyzed using Nikon Dx software; background area was defined at cell-free places in each µslide chamber. Double negative controls were performed with a second background reference. In case of time laps, “light gating” was fixed on the last picture of each series. Intensity increase as fold change in fluorescence was compared to t 0 time point.
On-chip cell cultivation and fluorescence detection
Experiments were performed using our in-house microfluidic system. The microfluidic setup and fluid conditions used are described in our previous manuscript (Theobald et al. 2017). The eight-chamber rhomboid microfluidic chips with two inlet/outlet ports are designed and manufactured by microfluidic ChipShop (Jena, Germany). For on-chip cultivation, HepG2 cells were seeded at 35,000 cells/chamber followed by overnight incubation to establish cell layers. Then, cells were pre-treated with 10 nM TCDD as either a single treatment of 24 h or repetitive treatments for 4 h on consecutive days. For on-chip measurements of CYP450 activity, medium containing the respective VividDye substrates were passed through chambers over 1 h. Subsequently, medium was collected and stored at − 20 °C. Sample collection was performed in the same manner for all time points. Samples were measured as described above using a 96-well plate in a TECAN Sapphire reader.
Results
Direct measurement of CYP450 activity in living hepatic cells using VividDye substrates
In need of a straightforward protocol for online determination of CYP450 activity, we tested the repurposing of VividDye substrates. As outlined in Fig. 1, we aimed to have an assay, that can be used to address the urgent need to observe CYP450 activity in living cells and its specific response, induction or inhibition to drug treatment and changes during hepatocyte maturation of dedifferentiation.

General overview on the application of the VividDye assay. Use of VividDye substrates for monitoring CYP450 activity in living cells and its application for drug testing (top) and for monitoring cell differentiation (bottom) in liver cell cultures and organoids. VividDye fluorogenic CYP450 probes are available with different CYP450 specificity linked with a selection of fluorescent dyes, which can be detected simultaneously in one assay. BOMR and EOMCC are examples of VividDye substrates used during this study
To ensure broad application of the present live cell CYP450 activity imaging presented here, we tested a wide range of experimental parameters, including the use of different cell lines, determination of optimal VividDye substrate concentrations, as well as other experimental conditions such as cell number, volume of medium as well as medium and incubation conditions.
The assay was tested in cells expressing or not expressing CYP450 genes, namely, HLEC, as CYP450 negative liver endothelial cells and a range of hepatocytes, namely, HepG2WT cells, HepaRG, immature iHep, upcyte® hepatocytes cells and primary murine hepatocytes. In these initial experiments, we investigated on basal and induced CYP450 activity, using BOMR dye. Results are summarized in Table 1 (more detailed kinetics measurements are provided in supplementary Fig. 1). For basal CYP450 activity, the results are presented as fold difference of cells incubated with Dye for 4 h to cells maintained in parallel without adding the dye. To further induce CYP450 activity, cells were pre-incubated with 20 nM TCDD for 24 h, before the dye was added and fold change was calculated as above. Highest basal levels were seen in pmH, while HepG2 and differentiated HepaRG showed the highest induction. Undifferentiated cells (iHep day 3, HepaRG day 3) showed only low activity levels and limited induction.
We further analyzed CYP450 expression using qPCR (see supplementary Fig. 2, please note that delta CT values are presented). In good accordance with the CYP450 activity level, pmH showed the highest expression over all investigated CYP450 isoenzymes, followed by iHep cells, HepG2 WT, upcyte® hepatocytes and differentiated HepaRG cell which showed similar levels for CYP1A1, CYP1A2 and CYP2E1, only for CYP3A4 the expression of was found to be lower in HepG2 WT.
To further optimize the protocol, we then used HepG2 WT and pmH. In addition to live cell recordings, CYP450 activity was also measured in cell lysates (see supplementary Fig. 3a). The VividDye fluorescence levels varied depending on cell type, cell number and cell density (supplementary Fig. 3). Interestingly, evaluating the impact of cell density, no linear correlation between cell number and fluorescent signal was observed, while different volumes of culture media showed that with lower volumes a more rapid signal saturation occurred. For the analysis of long-term stability of the dye, we performed an experiment with 300 measurement cycles (900 min) and observed a stable signal after saturation was reached. After cycle 110 (330 min), fluorescence signals declined, which we attributed to evaporation and bleaching (see supplementary Fig. 3c). We advise the adjustment of the total volume depending on the length of the experiment. Furthermore, we observed that dye concentrations ranging from 1 to 100 µM had no cytotoxic effects on cells (see supplementary Fig. 3d). The results obtained indicate that a concentration of 1 µM of VividDye substrates is sufficient in our experimental setup, showing at least a ten-fold difference in signal intensity compared to cells treated with the dye only.
A time-dependent correlation between intracellular fluorescence signals and fluorescence in the supernatant was observed. Initially, the signal within the cells appears to be higher compared to the signal in the supernatant. This indicates that VividDye substrates diffuse into cells; then the dyes are metabolized inside the cells; subsequently, the fluorescent metabolites diffuse out and are detectable in the supernatant (see supplementary Fig. 3e). We investigated on the stability of the VividDye in the supernatant. Therefore, samples were taken at various time points from 10 to 240 min. Samples were measured at each of the following time points, whereby no significant differences were observed (supplementary Fig. 3f).
We also used liver endothelial cells which do not express CYP450 as control. In these cells, no induction of fluorescence was observed upon pre-treatment with TCDD and E804 and subsequent VividDye measurements (see supplementary Fig. 4), further indicating that CYP450 activity is required to obtain induction of fluorescence signal and that fluorescence is not due to a chemical interaction of VividDye substrate and inducers.
To determine assay quality, we used the so-called Z′ values (see Eq. 1), a statistical parameter to determine the confidence level of an assay as a screening tool.
where \(\sigma\) is the standard deviation and µ the mean of the signal s and the background b (non-treated cells). A Z′ value of 1 describes a perfect assay, values above 0.5 are sufficient for screening. For the determination of our Z′ values, we used the last measuring point of each kinetic. Z′-values were calculated for the treatment showing the highest induction upon TCDD respective E804 for EOMCC, BOMCC and BOMR dyes. For OOMR, no Z′ value was calculated. For our VividDye assay, we determined Z′ values greater than 0.72 (Table 2).
Monitoring CYP450 induction in response to small molecule inducers: TCDD and E804
A key feature of hepatocyte metabolism is rapid and specific induction of CYP450 isoenzymes in response to drugs. In our drug-treatment experiments, we focused on two factors important in CYP-mediated drug interaction, induction and inhibition of CYP450 expression. 2,3,7,8 tetrachlorodibenzo-p-dioxin (TCDD) and dioxin-like compounds are known to act via specific binding to AhR present in many cell types including hepatocytes (Poellinger 2000; Mandal 2005). We also analyzed the so-far unknown indirubin derivative E804 assuming that the basic structure of indirubins and in particular of some derivatives could be potent AhR ligands and thus provide alternative CYP450 inducers (Adachi et al. 2001a, b).
It should be noted that TCDD is effective at much lower concentrations than E804 (10 nM vs. 1 µM). TCDD was analyzed at three different concentrations (5, 10, 20 nM) and showed a dose-dependent response with all VividDye substrates (see Fig. 2a–d). The range of induction observed with E804 was relatively narrow, which can be explained with (1) the higher concentration needed for CYP-induction followed and (2) the rapid transition of E804 to severe toxicity (IC50 5 µM in HepG2). Thus, no fluorescence signal was observed with 5 µM E804 (see Fig. 2f–h). In untreated cells and cells exposed to VividDye substrates without CYP450 inducing substances, only low fluorescence signals were observed, which can be attributed to basal levels of CYP450 expression in HepG2 cells. Fold induction changes were calculated upon normalization to non-treated cells.

Kinetic response of VividDye and associated CYP450 enzyme presumably responsible for VividDye metabolism. Online detection of VividDye metabolism by CYP450 enzymes using a selection of VividDye substrates after CYP450 induction. HepG2 cells were pre-treated with various concentration of TCDD (5–20 nM) (a–d) and E804 (10–5 µM) for 24 h, before VividDye substrates were added. VividDye were added as follows: BOMCC (a + e), EOMCC (b + f), BOMR (c + g) and OOMR (d + h). The kinetic recording of substrate turnover shows a dependence on the type and the concentration of the inducer used. Induction was observed for BOMCC, EOMCC, BOMR dye, but only a slight induction is visible for OOMR. The turnover rate is given as fluorescence signal divided by absorbance measured at λ580 nm to normalize for relative cell density. Fold induction (insert table) was calculated as increase in fluorescence signal relative to untreated control. Note that small symbols are a result of multiple time points, non-treated condition and Dye only condition are overlaying in panel a,e-h. Induction of CYP1A1 and expression of AhR in response to stimulation by TCDD (10, 20 nM) and E804 (1, 2 µM) on protein level were analyzed by Western blot (Original Western blot pictures can be found in supplementary Figs. 8 + 9). Pictures are cropped, full imagines are provided in supplementary information’s (l). RT-qPCR was performed to analyze induction on transcriptional (mRNA) upon treatment with TCDD 10 nM (j) and 1 µM E804 (k) for CYP1A1, CYP1A2 and CYP1B1 compared to mock treatment after 1 and 24 h. Experiments a–h are performed n = 5 times and for experiments j–k n = 3 repetitions were performed, error bars show the standard deviation
Kinetic measurements with different VividDye substrates show differences in the response. For example, EOMCC and BOMCC reach significant and stable fluorescence levels already after about 45 min, while with BOMR stable fluorescence is only reached after about 75 min, indicating faster metabolism of EOMCC and BOMCC compared to BOMR. In contrast, with OOMR only a weak CYP450 induction with TCDD or E804 was observed. Relative induction of CYP450 activity as a function of fluorescence signal fold change was highest with BOMR dye upon pre-treatment with E804 (see Fig. 2g), whereas with TCDD pre-treatment, the highest induction was observed with EOMCC dye (see Fig. 2b). VividDyes are commonly used as a rapid method to detect interaction between specific cytochromes and drugs using BACULOSOME® liver microsomes. These assays are based on overexpressed recombinant cytochromes as enzyme component to study drug interference. In the protocol presented here, assignment to a specific CYP 450 isoenzyme is not possible, due to cross reactivity of various CYP450 isoenzymes with the available substrates. Thus, fluorescence recorded from cells expressing different CYP450 enzymes reflects the combined activity of enzymes metabolizing the VividDye substrates used.
We next asked if differences in BOMCC, EOMCC and BOMR substrates dependent on fluorescence signals can be attributed to increased activity of either CYP1A1, CYP1A2 or CYP1B1. We, therefore, prepared RNA and proteins after induction and analyzed CYP450 isoenzyme expression on mRNA and protein levels (see Fig. 2i–k). On the mRNA level, a clear induction of CYP1A1, CYP1A2 and CYP1B1 is shown by RT-qPCR after 1 and 24 h treatments with both, TCDD or E804. In case of TCDD, induction was significantly stronger after 24 h. Interestingly, E804 pre-treatment led to a strong induction of CYP1A1 and CYP1B1 only but not of CYP1A2 (see Fig. 2k). Induction of CYP1A1 by E804 and TCDD after 24 h was also confirmed by Western blots (see Fig. 2i). At the same time, protein levels of AhR remained unchanged during treatment with both molecules (see Fig. 2i). So far, a clear association with of specific CYP450 activities with individual VividDyes was not obvious, and also slight differences in metabolic turnover were observed.
To demonstrate general applicability to monitor CYP450 activity independent of the cellular background, we performed similar experiments, using other hepatocyte model cell lines considered to be more similar to primary human hepatocytes, namely, HepaRG (Andersson et al. 2012) and upcyte® hepatocytes (Levy et al. 2015), and also included Omeprazole as CYP450 inducer (see Table 3 and supplementary Fig. 5).
Conformation of AhR-dependent induction of CYP450
TCDD is a ligand of the AhR receptor, we assumed that E804 is also a ligand of AhR. We, therefore, included an AhR inhibitor, resveratrol, and an AhR-deficient cell line (HepG2 AHRkd) to analyze the dependence of the observed CYP450 induction on AhR. Resveratrol is known as a competitive antagonist of dioxin and other AhR ligands. Similar to TCDD, it promotes translocation of the AhR complex to the nucleus and binding to dioxin-responsive element, however lacking transactivation (Casper et al. 1999; Ciolino et al. 1998; Ciolino and Yeh 1999). Consequently, resveratrol inhibits activation of dioxin-inducible genes including, but not limited to members of the CYP1 family.
In the presence of resveratrol (0.5, 5, 50 µM), we observed a dose-dependent decrease in fluorescence response to stimulation with TCDD and E804 (Fig. 3a + b), reaching background levels of non-treated cells with the highest resveratrol concentration for E804. Further, 50 µM resveratrol was not toxic to the cells (data not shown). Inhibition of AhR transactivation also decreased mRNA levels of CYP1A1, CYP1A2 and CYP1B1 upon compound induction (Fig. 3c).

Co-drug treatment of AhR-associated inducer and inhibitor. Detection of CYP450 activity with EOMCC VividDye substrate in co-treatment with inducer and inhibitor. Cells were pre-treated with mixtures of 1 µM E804 and various concentrations of resveratrol (0.5–50 µM) (a) and 20 nM TCDD and various concentrations of resveratrol (0.5–50 µM) (b) for a period of 24 h. The turnover rate is given as fluorescence divided by absorbance (λ580 nm) to normalize for relative cell density. Fold induction was calculated as increase in fluorescent signal compared to non-treated cells. Heat map showing relative mRNA levels of AhR, CYP1A1, CYP1A2, CYP1B1 upon treatment with TCDD, resveratrol and their combination (c). Experiments a, b are performed n = 5 times and for experiments c n = 3 repetitions were performed, error bars show the standard deviation. Comparison of the relative mRNA expression of AhR was compared, HepG2 AhrKD showed significant lower expression, experiment were performed for n = 3, (d). Comparison of protein expression level of AhR between HepG2 WT and HepG2 Ahr KD are shown, significant lower expression were observed for the knock-down cells, n = 5 (e)
In line, a CRISP/Cas9 AhR-knockdown cell line, HepG2 AHRkd showed significantly lower fluorescence levels to BOMCC, EOMCC and BOMR dye in comparison with the parental HepG2 WT cells, upon induction with 10 nM TCDD (Fig. 4a–c). It should be noted that our rapid protocol using batch selection did not lead to a complete depletion of AhR in the knockdown cells, however a significant decrease for both mRNA expression (60% reduction) protein level (48% reduction) between HepG2 WT and HepG2 AHRkd was observed (see Fig. 4d + e, please note that original picture of the western blot are provided in the supplementary Fig. 7).

Comparison of CYP450 induction in HepG2 and HepG2 AhRkD cells. Cells were pre-treated with 10 nM TCDD for 24 h and CYP450 activity was measured either by BOMCC (a), EOMCC (b) or BOMR (c) for 225 min. The turnover rate is given as fluorescence divided by absorbance (λ580 nm) to normalize for relative cell density. Fold induction was calculated as increase in fluorescent signal compared to non-treated cells. Experiments a–c are performed n = 5 times and error bars show the standard deviation
Visualization of CYP450 activity in individual cells after differentiation of HepaRG
To visualize CYP450 in hepatocyte-like cells, we used HepaRG cells, which pass through several stages of differentiation before reaching a mature status with higher CYP450 activity, compared to HepG2 cells. HepaRG cells were differentiated for 21 days according to manufacturer’s protocol. We further analyzed hepatocyte-specific inducibility of CYP450 expression. Differentiated HepaRG (day 24) cells were pre-treated with 10 nM TCDD for 24 h, before EOMCC and BOMR dyes were added. Pictures were taken every minute for a period of 20 min (Fig. 5a). Pictures were analyzed using double background normalization, and gating was set on the last measurement. Results indicate at least a sixfold induction of fluorescence for both dyes (Fig. 5c). Without TCDD induction, no increase in fluorescent signal upon addition of the dyes was observed (Fig. 5b).

Detection of CYP450 activity with VividDye in living cells by fluorescence microscopy and during cellular differentiation. Pictures were taken every minute and time point 0, 10, 20 min are shown as example (a). For quantitative analysis fluorescent signals were background corrected. Fold change in cellular fluorescence were calculated with EOMCC and BOMR dye staining and fluorescence intensities of cells at time point t = 0 min was set to 1. Without CYP450 induction fluorescence remained constant throughout the measurement (b). After pre-treatment with 10 nM TCDD for 24 h an increase in fluorescence can be seen at later time points (c)
Long-term differentiation of HepaRG and dedifferentiation of primary murine hepatocytes
Since the functionality of hepatocytes strongly depends on their differentiation status, we investigated on the potential application of the VividDye assay for assessment of maturation status. We, therefore, monitored albumin and urea, since those are among the most important biomarkers of a functional liver, for 27 days (see Fig. 6a, b). A significant increase in albumin secretion was observed starting day 9, urea synthesis stayed constant over the culturing time. Furthermore, the relative mRNA levels of several CYP450 enzymes during the differentiation of HepaRG were monitored, as observed for albumin an increased expression of different isoenzymes was observed starting from day 10 (see Fig. 6c). For the VividDye assay, a non-treated and an induced group were examined. For the first, cells were continuously treated with TCDD and 5.5-fold induction of BOMR fluorescent level was already observed on day 3. For the latter, fluorescent signals started increasing at day 9, reaching 8.8-fold signal on day 21. Note that day 21 measurement of 10 nm TCDD treatment is missing due to cells death, presumably due to long-term exposure to TCDD (see Fig. 6d). Both increase in albumin expression on day 9 and relative CYP450 mRNA level correlated with the increase of VividDye signal in non-treated HepaRG cells.

Monitoring maturation of HepaRG cells using Urea synthesis, Albumin secretion and CYPP450 activity level. Albumin secretion level of HepaRG cells during 27 days of maturation. Significant increase of albumin secretion was observed after day 9, n = 3, error bars show the standard deviation (a). Urea synthesis of HepaRG cells during 27 days of maturation. Comparison of mRNA expression of HepaRG cells during differentiation, and CYP450 enzymes associated with VividDye metabolism were continuously monitored for 25 days; an increase was observed starting day 10 (c). Fluorescent signal of metabolized VividDye Substrate BOMR is plotted against days of differentiation cells. Cells were either treated, non-treated or continuous treated with TCDD, for both cases negative control using no dye was performed. In case of TCDD initial induction of 5.5 was observed, while for non-treated cells induction is only seen starting day 11 of differentiation. Day 21 is missing fluorescent signal from TCDD-treated cells due to complete cell death, the experiment was performed with n = 5 independent cells populations, error bars show the standard deviation (d)
Freshly isolated hepatocytes do not maintain a differentiated phenotype and lose functionality when cultured in vitro (Guguen-Guillouzo and Guillouzo 2010; Godoy et al. 2013). We asked if we could follow the dedifferentiation of primary murine cells, including the loss of CYP450 activity, upon ex vivo culture. Following initial increase in metabolic turnover of the VividDye on day 2–4, signals started decreasing over the culture period of 9 days (see supplementary Fig. 5).
Influence of different culture methods on hepatocyte functionality
It had been shown, that more complex tissue culture approaches, such as co-culture endothelial liver cells and ECM sandwich cultures, resembling more closely the in vivo condition, lead to an improve hepatocyte phenotype. We, therefore, investigated if the VividDye assay can be used to determine improved functionality of hepatocytes in response to cellular environment in the tissue culture. We compared hepatocytes cultured in conventional 2D, in co-culture with endothelial cells and in sandwich culture. Effects of the applied culture system were measured on day 5 (see Fig. 7a + b). A significant increase was observed in both groups non-treated and 20 nM TCDD induced. Additional immunohistochemical analysis of the different culture conditions also showed improved expression for albumin and e-cadherin in co-culture and sandwich (see supplementary Fig. 10).

Effects of culture methods on CYP450 activity. HepG2 and upcyte® hepatocytes were culture under different environments including 2D mono cell culture, co-culture (with human liver endothelial cells) and 3D-like sandwich culture. Cells were exposed to BOMR dye for 1 h subsequent supernatant was analyzed. a HepG2 WT after 5 days of culture, b upcyte® hepatocytes after 5 days in culture with significant difference for both non-treated and induced cells. Insert tables show fold change of fluorescent signal compared to cells treated with no dye. Error bars show standard deviation of the mean n = 5
VividDye substrates in a microfluidic liver-on-chip system
Mimicking liver metabolism in liver organoids is a great challenge for in vitro drug testing and requires tools to monitor system functionality. Since the development of such systems is closely linked with microfluidic nutrition, we investigated if VividDye substrates can be used for activity monitoring in a microfluidic culture system. Using our in-house-developed microfluidic setup with microfluidic rhombic chambers (microfluidic ChipShop Jena) (Fig. 8a), we could see transiently induced fluorescent signals upon single-pulse treatments with TCDD, and could also follow multiple treatments with TCDD on subsequent days in longer term experiments. In this setup, cells were exposed to VividDye containing media for 1 h, and fluorescent signal was determined in the flow-through supernatant. Upon extended pre-incubation (24 h) with TCDD fluorescence signals, a decrease over time, possibly due to readjustment and normalization of CYP450 expression levels, was observed (Fig. 8b). We further investigated multiple treatments and cells were treated with TCDD for 4 h. The response to TCDD treatment was observed on two consecutive days. A decline in fluorescent response was observed on the pre-induction measurement on day 2, this is attributed due to overnight incubation with normal media lacking induction of AhR receptor (see Fig. 8c).

Determination of CYP450 activity in continuous microfluidic systems. Illustration of the applied microfluidic chip consisting of eight individual rhombic chambers with two ports (inlet/outlet). Each chamber measures 14.28 mm in length and 2.6 mm in width with a total volume of 20 µL (a). Live VividDye assay was performed with HepG2 cells at concentration of 50,000 cells/chamber. Samples pre-treated with TCDD 20 nM for 24 h are shown as solid bars, cells without pre-treatment (control) are shown as wide bars. Cells were exposed to VividDye BOMR for 1 h and fluorescence signals were determined in flow-through at time points indicated. Between VividDye exposure, chambers were maintained with normal cell culture medium (b). Repeated induction of HepG2 cells at concentration of 50,000 cells/chamber with TCDD for 4 h on two successive days. VividDye exposure and measurement as described in b was done in 1 day intervals (c). Experiments were performed with n = 4 cell chambers, error bars show the standard deviation
Discussion
CYP450 enzymes are strongly linked to drug metabolism and elimination; therefore, in early stages of drug development, it is of interest to monitor the cytochrome activity in response to drug treatment. Assays currently used to monitor the reciprocal influence of drugs and of CYP450 activity, include chromatography-, luminescence- and fluorescent-based assays using cell extracts and liver microsomes.
We here report the development of a fluorescent CYP450 activity assay for hepatocytes and hepatocyte-like cells using VividDye substrates. A major advantage of our protocol is the possibility to measure CYP450 activity in living cells as opposed to cellular extracts, e.g., microsomes or cell lysates, enabling repeated real-time measurements over extended time spans, thus meeting the requirements of in vitro cell-based systems. In comparison with luminescence-based assays, our setup is also compatible with microscopy, allowing monitoring on cell level. Since VividDye substrates exhibit only very low background fluorescence, no further steps were required to achieve high signal-to-background ratio. Different concentrations of VividDye substrates were screened for readout optimization. While all tested concentrations were non-toxic to cells, the final concentration used in the study was 1 µM, aimed at maintaining acetonitrile concentration in the culture medium low (0.1% v/v) to avoid toxicity and alterations of CYP activity. We are aware of the fact that for BACULOSOME®-based in vitro assay VividDye substrates are promoted to be used at different concentration to optimize specify towards individual CYP450 isoenzymes. In the living cell environment lacking the specificity for individual CYP450 enzymes, increase of dye concentration did not result in more selective response to drug treatment. This lack of specificity may also be the reason that in the cell-based protocol, different well-known CYP450 inducers (Rifampicin, Acetaminophen, Tryptamine, Ketoconazole) did not result in a selective response with individual dyes. We established our VividDye assay with several cell types including primary murine hepatocytes, iHep, HepaRG, HepG2 cells and upcyte® hepatocytes, which are reported to have similar functionality as primary human hepatocytes with expanded lifespan (Levy et al. 2015). Due to their adequate response and ease in culture, we primarily used HepG2 cells to optimize the use of VividDyes to monitor CYP450 alteration upon drug treatment in living cells. In case of differentiation and maturation HepaRG, iHep and upcyte® cells were used.
Cell density was optimized for HepG2 cells at 35,000 cells/well, resulting in a confluent monolayer of cells after 24 h. High standard deviation between the wells was normalized by applying absorbance at λ580 nm as a correction factor. We further assessed the applied assay volume for periods up to 5 h, and found 100 µL to be sufficient. For long-term assays, volumes should be increased to counter steer evaporation effects. Our results indicate that VividDye is metabolized inside the cells and subsequently secreted into the supernatant, since (1) fluorescent signals can be detected in both cells and supernatant; (2) metabolites are stable within the supernatant for at least 12 h; (3) no further increase in fluorescence signal was present once supernatant was removed from the cells and measured at later time points; (4) ratio between fluorescence signal within the and in supernatant shifts in a time dependent manner; (5) cells not expressing CYP450 lack induction. For drug induction and inhibition studies HepG2 cells were used as assay evaluation a model. Even though HepG2 express low basal cytochrome levels, induction upon drug treatment of certain cytochromes, such as CYP1A1, CYP1A2, CYP1B1 and CYP3A4, has been reported (Huang et al. 2006; Li et al. 1998; Chung and Bresnick 1994).
We further optimized the assay for microscopic setup to investigate the CYP450 activity on the single cell level. Using double background subtraction, a time-dependent increase of the fluorescent signal was observed, with distinguishable differences between individual cells. The culturing method of hepatocytes is crucial for their enzymatic expression and functionality (Godoy 2013). Optimized culturing protocols increase the interaction of hepatocyte with the extracellular environment (ECM) and with neighboring cells providing a more natural environment (Putnam and Mooney 1996). Several approaches were made to embed hepatocytes into non-adhesive hydrogels including MatrigelTM (BD Biosciences) (Kleinman et al. 1986), ExtracelTM (Ranucci et al. 2000; Prestwich et al. 2007), AlgimatrixTM (Rowley et al. 1999) and HGF/heparin-immobilized collagen scaffolds (Hou et al. 2010; Kim et al. 2010a). It is further known that co-culture of hepatocytes among other non-parenchymal cell lines induces liver function (Bhatia et al. 1998). We, therefore, investigated if our VividDye assay can be applied to analysis changes upon different culture methods. Our results show that after cells established their network levels of CYP450, both basal and induced, are significantly higher in sandwich and co-culture systems compared to conventional 2D culture (see Fig. 6).
We proofed the assay to be capable of following maturation in HepaRG cells for a period of 21 days, which shows higher CYP450 activity levels even without drug induction after differentiation starting at Day 9. Even though we investigated on the expression of several CYP450 enzymes associated with VividDye metabolism, a general increase was observed. In this experiment, the isoenzyme composition responsible for the metabolism of VividDye BOMR substrate could not be solved at the moment.
Freshly isolated primary hepatocytes are the gold standard to replicated in vivo hepatic function (Hengstler et al. 2000). However, more than 3000 genes (Zellmer et al. 2010) and several signaling pathways (Godoy et al. 2009) alter once primary cells are cultured ex vivo. Further cells quickly dedifferentiate (Paine and Andreakos 2004; Elaut et al. 2006; Vinken et al. 2006) and loose their cytochrome activity (Tong et al. 1990; LeCluyse 2001; Khetani et al. 2015), when placed in culture. Therefore, we checked if we can monitor loss of functionality in ex vivo liver system. We followed the CYP450 activity of primary isolated murine hepatocytes for a period of 9 days and as expected observed a transient activity profile, reflecting adhesion and reorganization in the tissue culture leading to increased activity levels at day 2–3 followed by a loss of activity in extended culture.
We further integrated the assay into our in-house microfluidic system, allowing for controlled addition of drugs and VividDye.
Taking the multiple application possibilities together, we propose that the VividDye assay can be integrated in an easy manner to monitor CYP450 activity in the upcoming field of in vitro liver-mimetic systems, as organoid-(Baptista et al. 2011; Funatsu et al. 2001; Ramachandran et al. 2015), spheroid-(Agastin et al. 2011; Lee et al. 2012; Nupura et al. 2016) and 3D static scaffold-based (Chevallay and Herbage 2000) cell culture systems, as well as microfluidic-based liver-on-chip systems (Kang et al. 2015; Lee et al. 2007). Thus, we expect that the assay can be used as a screening tool for functionality of single liver mimetic compartments before interconnecting them into more complex systems (Loskill et al. 2015).
References
Adachi J, Mori Y, Matsui S, Takigami H, Fujino J et al (2001a) Indirubin and indigo are potent aryl hydrocarbon receptor ligands present in human urine. J Biol Chem 276:31475–31478
Adachi J, Mori Y, Matsui S, Takigami H, Fujino J et al (2001b) Indirubin and indigo are potent aryl hydrocarbon receptor ligands present in human urine. J Biol Chem 276:31475–31478
Adachi J, Mori Y, Matsui S, Matsuda T (2004) Comparison of gene expression patterns between 2,3,7,8-tetrachlorodibenzo-p-dioxin and a natural arylhydrocarbon receptor ligand, indirubin. Toxicol Sci 80:161–169
Agastin S, Giang U-BT, Geng Y, DeLouise LA, King MR (2011) Continuously perfused microbubble array for 3D tumor spheroid model. Biomicrofluidics 5:024110
Andersson TB, Kanebratt KP, Kenna JG (2012) The HepaRG cell line: a unique in vitro tool for understanding drug metabolism and toxicology in human. Expert Opin Drug Metab Toxicol 8:909–920
Baptista PM, Siddiqui MM, Lozier G, Rodriguez SR, Atala A et al (2011) The use of whole organ decellularization for the generation of a vascularized liver organoid. Hepatology 53:604–617
Bhatia S, Balis U, Yarmush M, Toner M (1998) Microfabrication of hepatocyte/fibroblast co-cultures: role of homotypic cell interactions. Biotechnol Prog 14:378–387
Casper RF, Quesne M, Rogers IM, Shirota T, Jolivet A et al (1999) Resveratrol has antagonist activity on the aryl hydrocarbon receptor: implications for prevention of dioxin toxicity. Mol Pharmacol 56:784–790
Cheng X, Rasque P, Vatter S, Merz KH, Eisenbrand G (2010) Synthesis and cytotoxicity of novel indirubin-5-carboxamides. Bioorg Med Chem 18:4509–4515
Cheng X, Merz KH, Vatter S, Christ J, Wolfl S et al (2014) 7,7′-Diazaindirubin—a small molecule inhibitor of casein kinase 2 in vitro and in cells. Bioorg Med Chem 22:247–255
Cheng X, Yoshida H, Raoofi D, Saleh S, Alborzinia H et al (2015a) Ethyl 2-((4-Chlorophenyl)amino)thiazole-4-carboxylate and Derivatives Are Potent Inducers of Oct3/4. J Med Chem 58:5742–5750
Cheng X, Dimou E, Alborzinia H, Wenke F, Gohring A et al (2015b) Identification of 2-[4-[(4-Methoxyphenyl)methoxy]-phenyl]acetonitrile and Derivatives as Potent Oct3/4 Inducers. J Med Chem 58:4976–4983
Cheng X, Kim JY, Ghafoory S, Duvaci T, Rafiee R et al (2016) Methylisoindigo preferentially kills cancer stem cells by interfering cell metabolism via inhibition of LKB1 and activation of AMPK in PDACs. Mol Oncol 10:806–824
Chevallay B, Herbage D (2000) Collagen-based biomaterials as 3D scaffold for cell cultures: applications for tissue engineering and gene therapy. Med Biol Eng Comput 38:211–218
Chung I, Bresnick E (1994) 3-Methylcholanthrene-mediated induction of cytochrome P4501A2 in human hepatoma HepG2 cells as quantified by the reverse transcription-polymerase chain reaction. Arch Biochem Biophys 314:75–81
Ciolino HP, Yeh GC (1999) Inhibition of aryl hydrocarbon-induced cytochrome P-450 1A1 enzyme activity and CYP1A1 expression by resveratrol. Mol Pharmacol 56:760–767
Ciolino HP, Daschner PJ, Yeh GC (1998) Resveratrol inhibits transcription of CYP1A1 in vitro by preventing activation of the aryl hydrocarbon receptor. Cancer Res 58:5707–5712
De Montellano PRO (2005) Cytochrome P450: structure, mechanism, and biochemistry: Springer Science & Business Media, Berlin
Denison M, Heath-Pagliuso S (1998) The Ah receptor: a regulator of the biochemical and toxicological actions of structurally diverse chemicals. Bull Environ Contam Toxicol 61:557–568
Elaut G, Henkens T, Papeleu P, Snykers S, Vinken M, Vanhaecke T, Rogiers V (2006) Molecular mechanisms underlying the dedifferentiation process of isolated hepatocytes and their cultures. Curr Drug Metab 7:629–660. https://doi.org/10.2174/138920006778017759
Friedman MA, Woodcock J, Lumpkin MM, Shuren JE, Hass AE et al (1999) The safety of newly approved medicines: do recent market removals mean there is a problem? JAMA 281:1728–1734
Funatsu K, Ijima H, Nakazawa K, Yamashita Y, Shimada M et al (2001) Hybrid artificial liver using hepatocyte organoid culture. Artif Organs 25:194–200
Gerets HHJ, Tilmant K, Gerin B, Chanteux H, Depelchin BO et al (2012) Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biol Toxicol 28:69–87
Godoy P, Hengstler JG, Ilkavets I, Meyer C, Bachmann A, Muller A, Tuschl G, Mueller SO, Dooley S (2009) Extracellular matrix modulates sensitivity of hepatocytes to fibroblastoid dedifferentiation and transforming growth factor beta-induced apoptosis. Hepatology 49:2031–2043. https://doi.org/10.1002/hep.22880
Godoy P, Hewitt N, Albrecht U, Andersen M, Ansari N et al (2013) Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch Toxicol 87:1315–1530
Göttel M, Le Corre L, Dumont C, Schrenk D, Chagnon M-C (2014) Estrogen receptor α and aryl hydrocarbon receptor cross-talk in a transfected hepatoma cell line (HepG2) exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Rep 1:1029–1036
Guengerich FP, Sorrells JL, Schmitt S, Krauser JA, Aryal P et al (2004) Generation of new protein kinase inhibitors utilizing cytochrome P450 mutant enzymes for indigoid synthesis. J Med Chem 47:3236–3241
Guguen-Guillouzo C, Guillouzo A (2010) General review on in vitro hepatocyte models and their applications. In: Maurel P (ed) Hepatocytes. Methods in molecular biology (Methods and protocols), vol 640. Humana Press, pp 1–40
Hankinson O (1995) The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol 35:307–340
Hengstler JG, Utesch D, Steinberg P, Platt K, Diener B et al (2000) Cryopreserved primary hepatocytes as a constantly available in vitro model for the evaluation of human and animal drug metabolism and enzyme induction. Drug Metab Rev 32:81–118
Hewitt NJ, Gómez Lechón MJ, Houston JB, Hallifax D, Brown HS et al (2007) Primary hepatocytes: current understanding of the regulation of metabolic enzymes and transporter proteins, and pharmaceutical practice for the use of hepatocytes in metabolism, enzyme induction, transporter, clearance, and hepatotoxicity studies. Drug Metab Rev 39:159–234
Hou YT, Ijima H, Matsumoto S, Kubo T, Takei T, Sakai S, Kawakami K (2010) Effect of a hepatocyte growth factor/heparin-immobilized collagen system on albumin synthesis and spheroid formation by hepatocytes. J Biosci Bioeng 110:208–216. https://doi.org/10.1016/j.jbiosc.2010.01.016
Huang H, Wang H, Sinz M, Zoeckler M, Staudinger J et al (2006) Inhibition of drug metabolism by blocking the activation of nuclear receptors by ketoconazole. Oncogene 26:258–268
Kanebratt KP, Andersson TB (2008) Evaluation of HepaRG cells as an in vitro model for human drug metabolism studies. Drug Metab Dispos 36:1444–1452
Kang YB, Sodunke TR, Lamontagne J, Cirillo J, Rajiv C et al. (2015) Liver sinusoid on a chip: Long-term layered co-culture of primary rat hepatocytes and endothelial cells in microfluidic platforms. Biotechnol Bioeng
Kelly JH, Sussman NL (2000) A fluorescent cell-based assay for cytochrome P-450 isozyme 1A2 induction and inhibition. J Biomol Screen 5:249–253
Khetani SR, Berger DR, Ballinger KR, Davidson MD, Lin C et al (2015) Microengineered liver tissues for drug testing. J Lab Autom 20:216–250
Kim M, Lee JY, Jones CN, Revzin A, Tae G (2010) Heparin-based hydrogel as a matrix for encapsulation and cultivation of primary hepatocytes. Biomaterials 31:3596–3603. https://doi.org/10.1016/j.biomaterials.2010.01.068
Kleinman HK, McGarvey ML, Hassell JR, Star VL, Cannon FB, Laurie GW, Martin GR (1986) Basement membrane complexes with biological activity. Biochemistry 25:312–318. https://doi.org/10.1021/bi00350a005
Lasser KE, Allen PD, Woolhandler SJ, Himmelstein DU, Wolfe SM et al (2002) Timing of new black box warnings and withdrawals for prescription medications. JAMA 287:2215–2220
LeCluyse EL (2001) Human hepatocyte culture systems for the in vitro evaluation of cytochrome P450 expression and regulation. Eur J Pharm Sci 13:343–368. https://doi.org/10.1016/S0928-0987(01)00135-X
Lee PJ, Hung PJ, Lee LP (2007) An artificial liver sinusoid with a microfluidic endothelial-like barrier for primary hepatocyte culture. Biotechnol Bioeng 97:1340–1346
Lee S-A, Choi YY, Park D, Jang JY, Kim D-S et al (2012) Functional 3D human primary hepatocyte spheroids made by co-culturing hepatocytes from partial hepatectomy specimens and human adipose-derived stem cells. PLoS One 7:e50723
Levy G, Bomze D, Heinz S, Ramachandran SD, Noerenberg A et al (2015) Long-term culture and expansion of primary human hepatocytes. Nat Biotechnol 33:1264–1271
Li W, Harper PA, Tang B-K, Okey AB (1998) Regulation of cytochrome P450 enzymes by aryl hydrocarbon receptor in human cells: CYP1A2 expression in the LS180 colon carcinoma cell line after treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin or 3-methylcholanthrene. Biochem Pharmacol 56:599–612
Loskill P, Marcus SG, Mathur A, Reese WM, Healy KE (2015) μOrgano: A Lego®-like plug and play system for modular multi-organ-chips. PLoS One 10:e0139587
Ma Q (2001) Induction of CYP1A1. The AhR/DRE paradigm transcription, receptor regulation, and expanding biological roles. Curr Drug Metab 2:149–164
Mandal PK (2005) Dioxin: a review of its environmental effects and its aryl hydrocarbon receptor biology. J Comp Physiol B 175:221–230
Nupura SB, Vijayan M, Solange M, Ali T, Masoumeh G et al (2016) A liver-on-a-chip platform with bioprinted hepatic spheroids. Biofabrication 8:014101
Paine AJ, Andreakos E (2004) Activation of signalling pathways during hepatocyte isolation: relevance to toxicology in vitro. Toxicol In Vitro 18:187–193. https://doi.org/10.1016/S0887-2333(03)00146-2
Poellinger L (2000) Mechanistic aspects the dioxin (aryl hydrocarbon) receptor. Food Addit Contam 17:261–266
Prestwich GD, Liu Y, Yu B, Shu XZ, Scott A (2007) 3-D culture in synthetic extracellular matrices: new tissue models for drug toxicology and cancer drug discovery. Adv Enzyme Regul 47:196–207
Putnam AJ, Mooney DJ (1996) Tissue engineering using synthetic extracellular matrices. Nat Med 2(7):824–826
Ramachandran SD, Schirmer K, Münst B, Heinz S, Ghafoory S et al (2015) In vitro generation of functional liver organoid-like structures using adult human cells. PLoS One 10:e0139345
Ranucci CS, Kumar A, Batra SP, Moghe PV (2000) Control of hepatocyte function on collagen foams: sizing matrix pores toward selective induction of 2-D and 3-D cellular morphogenesis. Biomaterials 21:783–793. https://doi.org/10.1016/S0142-9612(99)00238-0
Reiners JJ, Cantu AR, Pavone A, Smith SC, Gardner CR et al (1990) Fluorescence assay for per-cell estimation of cytochrome P-450-dependent monooxygenase activities in keratinocyte suspensions and cultures. Anal Biochem 188:317–324
Rowley JA, Madlambayan G, Mooney DJ (1999) Alginate hydrogels as synthetic extracellular matrix materials. Biomaterials 20:45–53. https://doi.org/10.1016/S0142-9612(98)00107-0
Sato N, Meijer L, Skaltsounis L, Greengard P, Brivanlou AH (2004) Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat Med 10:55–63
Song G, Pacher M, Balakrishnan A, Yuan Q, Tsay H-C et al (2016) Direct reprogramming of hepatic myofibroblasts into hepatocytes in vivo attenuates liver fibrosis. Cell Stem Cell 18:797–808
Theobald J, Ghanem A, Wallisch P, Banaeiyan AA, Andrade-Navarro MA, Taskova K, Haltmeier M, Kurtz A, Becker H, Reuter S, Mrowka R, Cheng X, Wölfl S (2017) Liver-kidney-on-chip to study toxicity of drug metabolites. ACS Biomater Sci Eng. https://doi.org/10.1021/acsbiomaterials.7b00417
Tong JZ, Bernard O, Alvarez F (1990) Long-term culture of rat liver cell spheroids in hormonally defined media. Exp Cell Res 189:87–92. https://doi.org/10.1016/0014-4827(90)90260-H
Trubetskoy OV, Gibson JR, Marks BD (2005) Highly miniaturized formats for in vitro drug metabolism assays using Vivid® fluorescent substrates and recombinant human cytochrome P450 enzymes. J Biomol Screen 10:56–66
Vinken M, Papeleu P, Snykers S, de Rop E, Henkens T, Chipman JK, Rogiers V, Vanhaecke T (2006) Involvement of cell junctions in hepatocyte culture functionality. Crit Rev Toxicol 36:299–318. https://doi.org/10.1080/10408440600599273
Zellmer S, Schmidt-Heck W, Godoy P, Weng H, Meyer C, Lehmann T, Sparna T, Schormann W, Hammad S, Kreutz C, Timmer J, von Weizsacker F, Thurmann PA, Merfort I, Guthke R, Dooley S, Hengstler JG, Gebhardt R (2010) Transcription factors ETF, E2F, and SP-1 are involved in cytokine-independent proliferation of murine hepatocytes. Hepatology 52:2127–2236. https://doi.org/10.1002/hep.23930
Acknowledgements
The authors would like to acknowledge the Nikon Imaging Centre, University of Heidelberg, for providing the possibility to use their microscopically setup. Special thanks to Dr. Ulrike Engel for her support in optimizing the microscopically setup and her expert assistance and technical support. The authors would like to thank Saskia Schmitteckert for the substantial help in performing the experiments.
Author information
Authors and Affiliations
Contributions
JT, Experimental design, performed experiments, performed data analysis, wrote the manuscript. XC, Performed experiments, experimental design, performed data analysis. AG, Experimental design, performed experiments. HG Isolated primary murine hepatocytes, performed experiments. GS Developed and provided iHep cells. EK Participated in experimental design. JW Participated in experimental design. HB, Developed and manufactured the Chip-design. RM, Participated in experimental design. KB-H Experimental design, corrected the manuscript. SD, Supervised experiments, corrected the manuscript. SW, Supervised experiments, experimental design, corrected the manuscript.
Corresponding author
Ethics declarations
Founding sources
BMBF SysTox: FKZ 031A303E iPS-Profiler: FKZ 01EK1612C, DFG Grant program (CH 1690/2-1).
Conflict of interest
The authors declare that they have no competing financial interest.
Data availability statement
All data are is included in the manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Theobald, J., Cheng, X., Ghanem, A. et al. Monitoring cytochrome P450 activity in living hepatocytes by chromogenic substrates in response to drug treatment or during cell maturation. Arch Toxicol 92, 1133–1149 (2018). https://doi.org/10.1007/s00204-017-2128-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-017-2128-1