
Abstract
Hepatocellular carcinoma (HCC) is one of the most fatal malignancies with high mortality worldwide. Here the underlying antitumor mechanism of gallotannin was elucidated in HCC cells. Gallotannin suppressed viability and colony formation, increased subG1 portion and also induced senescence via upregulation of p21, G0/G1 arrest and higher SA-β-gal activity in HepG2 and SK-Hep1 cells. However, pan-caspase inhibitor Z-VAD-FMK reversed the ability of gallotannin to activate caspase 3 at 48 h after treatment in two HCC cells. Of note, gallotannin also induced autophagic features by increasing LC3 punctae, LC3B-II conversion, autophagic vacuoles and decreasing the expression of Beclin1 in two HCC cells. Furthermore, autophagy flux assay using GFP–mRFP–LC3 plasmid revealed increased yellowish color and late autophagy inhibitor CQ or NH4Cl enhanced cytotoxicity, LC3B-II conversion, and LC3 punctae in gallotannin-treated HepG2 and SK-Hep1 cells compared to early autophagy inhibitor 3-MA or wortmannin. Interestingly, gallotannin attenuated the expression of SIRT1 and mTOR and activated phosphorylation of AMPK in two HCC cells. Furthermore, AMPK activator AICAR significantly enhanced SA-β-gal activity and antiproliferation induced by gallotannin, while AMPK inhibitor compound C did not in two HCC cells. Consistently, LC3B-II conversion by gallotannin was not shown in AMPKα1 −/− MEF cells compared to WT AMPK +/+ MEF cells. Consistently, gallotannin reduced in vivo growth of HepG2 cells implanted in NCr nude mice along with decreased expression of PCNA and SIRT1 and increased AMPKα1 and TUNEL. Overall, these findings highlight evidence that regulation of SIRT1/AMPK is critically involved in gallotannin-induced senescence and impaired autophagy leading to cell death in HCC cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC) is one of the most intractable tumors with high mortality worldwide. Five-year overall survival rate of HCC is significantly lower than that of the other cancers (Baffy et al. 2012; Flores and Marrero 2014). Furthermore, recently anticancer phytochemicals including sulforaphane (Chi et al. 2015), luteolin (Ding et al. 2014), resveratrol (Tang et al. 2013), quercetin (Casella et al. 2014), berberine (Yip and Ho 2013), decursin (Kim et al. 2016), 1,2,3,4,6-penta-O-galloyl-beta-d-glucose (PGG) (Oh et al. 2001), curcumin (Marquardt et al. 2015), genistein (Dai et al. 2015), and cryptotanshinone (Park et al. 2014) are attractive with less toxicity, since several anticancer drugs, which have been used for HCC therapy, showed side effects of toxicity, recurrence, and resistance(Berk et al. 2013; Wei et al. 2009).
Cellular senescence is categorized into replicative senescence and stress-induced premature senescence (SIPS) with features of p21 upregulation, enlarged and flattened cell morphology, and increased senescence-associated β-galactosidase (SA-β-gal) activity (Pietrocola et al. 2013; Raghuram and Mishra 2014; Zhao and Darzynkiewicz 2014). In addition, the senescence induction is another target for cancer therapy, since therapy-induced cellular senescence suppresses the proliferation of cancer cells as an important tumor suppressor mechanism (Asfour et al. 1990; Gewirtz 2013; Ghosh and Zhou 2015; Sabin and Anderson 2011; Wang et al. 2011; Yang et al. 2013; Zu et al. 2010).
Autophagy is the catabolic process for the recycling of macromolecules and the degradation of long-lived proteins, pathogens, damaged DNA elements, and damaged organelles activated especially during metabolic stress conditions such as nutrient deprivation or chemotherapy (Gewirtz 2014; Klionsky et al. 2012; Sharma et al. 2014). Growing evidences have demonstrated that autophagy can exert either survival or death in cancer cells. In terms of survival mechanism of autophagy, once autophagy response is induced, the entire autophagic flux is completed in progress and then the homeostasis of the cell is maintained (Dong et al. 2014; Puissant et al. 2010; Vucicevic et al. 2011; Zhao et al. 2013; Zhou et al. 2012). In contrast, impaired autophagy by blocking the fusion of autophagosome and lysosome rather induces cell death, because the energy consumption is too high to survive in the process (Feng et al. 2014; Gali-Muhtasib et al. 2015; Lao et al. 2014; Marino et al. 2014; Mizushima et al. 2010; Singh et al. 2014; Tang et al. 2013; Wang et al. 2013).
It is well documented that AMPK plays a pivotal role in senescence, autophagy, and apoptosis in cancer cells (Guo et al. 2012; Lee et al. 2012; Park et al. 2014; Puissant et al. 2010; Vucicevic et al. 2011; Wang et al. 2011; Wu et al. 2011). In addition, SIRT1, a type of protein deacetylase, is closely associated with the survival, metabolic homeostasis, aging, cancer metabolism, and inflammation. Since SIRT1 is overexpressed in prostate, ovary, stomach, colon, and liver cancers, suppression of SIRT1 is a key target to inhibit tumor growth, induce cell cycle arrest and apoptosis in various cancer cells (Bae et al. 2014; Canto et al. 2010; Ghosh and Zhou 2015; Hou et al. 2008; Zu et al. 2010).
Gallotannin, one of hydrolysable tannic acids derived from Rhus javanica, was reported to have anti-inflammatory, antioxidant, and anticancer effects in colon, breast, and prostate cancers (Al-Ayyoubi and Gali-Muhtasib 2007; Al-Halabi et al. 2015; Park et al. 2015; Zhao et al. 2014). Nevertheless, its underlying antitumor mechanism is not fully understood in liver cancer. Thus, in the current study, the anticancer mechanism of gallotannin was elucidated in HepG2 and SK-Hep1 HCC cells in association with senescence, proliferation, autophagy, and apoptosis via regulation of SIRT1/AMPK signaling in vitro and in vivo.
Materials and methods
Chemicals and reagents
Gallotannin (tannic acid, T0200, Fig. 1a) was purchased from Sigma-Aldrich (St Louis, MO, USA). 3-Methyladenine (3-MA, M9281), wortmannin (W1628), chloroquine (CQ, 50-63-5), ammonium chloride (NH4Cl, A9434) ribonuclease A (R6513) and MG132 (C2211) were purchased from Sigma-Aldrich. X-tremeGENE HP DNA transfection reagent (06 366 236 001) was purchased from Roche (Mannheim, Germany). DC Protein Assay Kit II (500-0113) was purchased from Bio-Rad (Hercules, CA, USA). ECL Western blotting detection reagent (RPN2209) was purchased from GE Healthcare (Buckinghamshire, UK). Senescence β-galactosidase staining kit (9860) and AICAR (9944) were purchased from Cell Signaling Technology (Beverly, MA, USA). Histological mounting medium (Histomount, 1330-20-7) was purchased from National Diagnostics (Atlanta, USA). Compound C (171261) and pan-caspase inhibitor (627610) were purchased from Calbiochem (Hessen, Germany). Other chemical reagents were obtained from Sigma-Aldrich.

Gallotannin exhibits anti-proliferative activity and induces senescence in HepG2 and SK-Hep1 cells. a Chemical structure of gallotannin (Empirical Formula: C76H52O46, Molecular weight: 1701.2). b Cytotoxicity of gallotannin in HepG2 and SK-Hep1 cells. Cells were treated with gallotannin at the indicated concentrations for 24, 48, and 72 h and cytotoxicity by MTT assay was presented as means ± SD. c Effect of gallotannin on colony formation of HepG2 and SK-Hep1 cells. Following treatment of 40 µM gallotannin for 24 h, the cells were incubated in new fresh complete medium for 12–14 days and stained with 0.5% crystal violet. Stained cells were solubilized with 1% SDS buffer to measure the optical density. Relative percentages of clonogenic cells were represented as means ± SD of three independent experiments, ***p < 0.001 vs control by Student’s t test. d G0/G1 arrest was increased by gallotannin in HepG2 and SK-Hep1 cells. Cells were treated with indicated concentrations of gallotannin for indicated times. After fixation with 75% ethanol, cells were stained with PI (propidium iodide, 50 µg/ml) and then cell cycle was analyzed using flow cytometry. Data were represented as means ± SD of three independent experiments. **p < 0.01, ***p < 0.001 vs. untreated control by Student’s t test (for two group comparison) or one-way ANOVA. e Effect of gallotannin on the expression of p21 in HepG2 and SK-Hep1 cells in a time course. The cells were treated with 40 µM gallotannin for indicated times. The cell lysates were separated on 12% SDS-PAGE gels, transferred to Hybond ECL membrane, and probed with anti-p21 antibody. Membranes were probed with an anti-β-actin antibody as a loading control. Image J densitometric analysis of the p21/β-actin ratio was shown. f SA-β-galactosidase-positive cells were increased in gallotannin-treated HepG2 and SK-Hep1 cells. The SA-β-galactosidase-positive cells were counted and presented as relative values of the total cells in HepG2 and SK-Hep1 cells. (means ± SD of three independent experiments, *p < 0.05, **p < 0.01, ***p < 0.001 vs. untreated control by Student’s t test). g Gallotannin-induced weak apoptosis through a caspase-dependent pathway in HepG2 and SK-Hep1 cells. Cells were treated with 40 μM gallotannin for 24, 48, and 72 h. After treatment, the cell lysates were subjected to Western blotting with anti-PARP, anti-procaspase 3, and anti-procaspase 8 as apoptosis-related proteins. Protein contents were normalized by probing the same membrane with an anti-β-actin antibody. h Effect of pan-caspase inhibitor on caspase 3/8 in gallotannin-treated HepG2 cells. Cells were pretreated with pan-caspase inhibitor (10 and 20 μM) for 2 h and then exposed to 40 μM gallotannin for 48 h. After treatment, the cell lysates were subjected to Western blotting with indicated proteins
Antibodies
Antibodies for Beclin 1 (3495, 1:2000 dilution), p62/SQSTM1 (5114, 1:2000 dilution), LC3B (3868, 1:2000 dilution), p21(2947, 1:2000 dilution), p-mTOR (2971, 1:2000 dilution), mTOR (2972, 1:2000 dilution), phospho-AMPKα1 (2535, Thr172, 1:500 dilution), AMPKα1 (2532, 1:2000 dilution), cleaved caspase 3 (9664, 1:1000 dilution), and β-actin (4967, 1:5000 dilution) were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies for PARP (SC-7150, 1:2000 dilution), Caspase 3 (SC-7148, 1:2000 dilution), and Caspase 8 (SC-7890, 1:2000 dilution) were purchased from Santa Cruz Biotechnology (CA, USA). SIRT1 (2273971, 1:2000 dilution) was purchased from Millipore (MA, USA).
Plasmids
HisB–SIRT1 expression vector was kindly provided by Prof. SW. Nam (Catholic University, Seoul, Korea) (Bae et al. 2012). GFP–mRFP–LC3 plasmid were kindly provided by Prof. H. Hu (China Agricultural University, Beijing, China) (Song et al. 2015).
Cell culture
HepG2 (ATCC® HB-8065™) and SK-Hep1 (ATCC® HTB-52™) cells were obtained from ATCC (American Type Culture Collection) and maintained in DMEM supplemented with 10% fetal bovine serum. AMPKα1 +/+ and AMPKα1 −/− mouse embryonic fibroblast (MEF) cells were provided by Prof. JH Ha (Kyung Hee University, Seoul, Korea) and maintained in DMEM supplemented with 10% FBS (fetal bovine serum). Dulbecco’s modified Eagle’s medium (DMEM, LM 001-05) and fetal bovine serum (FBS, PK004-07) were purchased from Welgene (GD, Korea). The cells were kept in the medium at 37 °C in a humidified atmosphere containing 5% CO2 incubator.
Cytotoxicity assay
HepG2 or SK-Hep1 cells were seeded onto 96-well microplates at a density of 1 × 104 cells/well and incubated in the absence or presence of various concentrations of gallotannin for 24–72 h with or without 3-MA (3 mM), wortmannin (1 μM), chloroquine (CQ, 30 μM), and ammonium chloride (NH4Cl, 10 mM) for 24 h. Cytotoxicity of gallotannin in HCC cells was evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich., St Louis, MO, USA) assay. After gallotannin treatment, culture medium was discarded and 50 μl of MTT solution (1 mg/ml) was added to each well and incubated at 37 °C for 2 h. After incubation the optical density (O. D.) was measured by microplate reader (Sunrise, TECAN, Switzerland) at 570 nm. The cell viability was calculated by the following equation: cell viability (%) = [O.D. (gallotannin) − O.D. (blank)]/[O.D. (control) − O.D. (blank)] × 100.
Colony formation assay
To determine colony formation, HepG2 and SK-Hep1 cells at the exponential growth phase were harvested, seeded at about 1 ~ 2 × 103 cells per well of a six-well plate and cultured in complete medium supplemented with 10% FBS at 37 °C in 5% CO2 incubator for 24 h. The cells were exposed to 40 μM gallotannin in the absence or presence of 3-MA (3 mM), wortmannin (1 μM), AICAR (1 mM), and compound C (5 μM) for 24 h. Then the cells were cultured in new complete medium for 12–14 days and finally rinsed with PBS twice, fixed with 100% methanol for 30 min at root temperature, and stained with 0.5% crystal violet for 1 h at room temperature. Stained cells were solubilized with 1 ml of 1% SDS buffer at room temperature for 30 min and 200 μl of the lysates were transferred onto the 96-well plate. Then the optical density was measured using microplate reader (Sunrise, TECAN, Mannedorf, Switzerland) at 590 nm wave length.
Cell cycle analysis
HepG2 and SK-Hep1 cells were treated with gallotannin for 6–72 h and fixed with 75% ethanol at −20 °C. The fixed cells were washed twice with PBS and resuspended in PBS-containing RNase A (1 mg/ml) and incubated for 1 h at 37 °C. Then the cells were stained with 500 μl of propidium iodide (50 μg/ml) for 30 min at room temperature in the dark. Finally, the DNA contents of the stained cells were analyzed using CellQuest Software with the FACSCalibur flow cytometer (Becton–Dickinson, Franklin Lakes, NJ, USA).
Senescence-associated β-galactosidase (SA-β-gal) staining
SA-β-gal staining was performed using senescence β-galactosidase staining kit according to the manufacturer’s protocol. Briefly, HepG2 and SK-Hep1 cells were treated with or without gallotannin in the absence or presence of AICAR or compound C. The cells were washed with PBS twice, fixed for 15 min and incubated in staining mixture overnight at 37 °C in the dark. After incubation, the cells were observed and counted under a light microscope (Axio Observer A1, Carl Zeiss, Inc., USA).
Western blot analysis
HepG2 and SK-Hep1 cells were washed twice with cold PBS on ice and harvested. The cells were pelleted by centrifugation at 4 °C and resuspended directly into a lysis buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 1 mM EDTA, 1 mM Na3VO4, 1 mM NaF, protease inhibitor cocktail). Cell lysates were subjected to SDS-PAGE and transferred to a nitrocellulose membrane. After blocking in 5% skim milk and washing with Tris-buffered saline–0.1% Tween-20 (TBS-T), the membranes were immunoblotted with antibodies against p21, LC3B, p62/SQSTM1, Beclin1, SIRT1, p-mTOR, mTOR, p-AMPKα1 (Thr172), AMPKα1, PARP, caspase 8, and caspase 3. β-Actin protein was immunoblotted to normalize the quantity of sample protein. Signals were detected by Konica photoconductor (SRX-101A, Konica Minolta) and densitometric analysis was performed using ImageJ software.
Transfection assay
HepG2 and SK-Hep1 cells were transfected with HisB–SIRT1 overexpression vector or GFP–mRFP–LC3 construct using the X-tremeGENE HP DNA transfection reagent according to the manufacturer’s protocol. One day after transfection, the cells were treated with or without gallotannin for 24 h.
Immunofluorescence assay
HepG2 and SK-Hep1 cells were grown on Lab-Tek chamber slide (Nunc, Rochesta) to 70% confluency. The cells were fixed in 3.7% formaldehyde in PBS for 15 min at room temperature and permeabilized with 0.2% Triton X-100 in PBS for 10 min and blocked with 1% BSA in PBS for 1 h. The fixed cells were incubated overnight with anti-LC3 primary antibody, and then washed in PBS and incubated with Alexa Fluor® 568-conjugated anti-rabbit IgG antibody (Molecular Probes, Oregon) for 1 h. The cells were then stained with 0.5 mg/ml of DAPI to visualize nuclei, mounted on glass slides and observed with an LSM510 confocal laser microscope (Carl Zeiss, Oberkochen, Germany).
Autophagic flux assay
HepG2 and SK-Hep1 cells were transfected with GFP–mRFP–LC3 construct using X-treme GENE HP DNA transfection reagent. After gallotannin treatment, the cells were washed twice in ice-cold PBS, fixed, mounted with Histological Mounting Medium (Histomount, USA) and observed with LSM510 confocal laser microscope (Carl Zeiss, Germany).
Transmission electron microscopy (TEM) observation
HepG2 cells were harvested by trypsinization, fixed in 2% paraformaldehyde–glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) and washed in 0.1 M phosphate buffer (PB). Then the cells were fixed with 1% OsO4 dissolved in 0.1 M PB for 2 h, dehydrated in ascending gradual series (50–100%) of ethanol and infiltrated with propylene oxide. Specimens were embedded by Poly/Bed 812 kit (Polysciences). After pure fresh resin embedding and polymerization at 65 °C electron microscope oven (TD-700, DOSAKA, Japan) for 24 h. Sections at about 200–250 nm thick were stained with toluidine blue (sigma, T3260) and double stained with 6% uranyl acetate (EMS, for 20 min) and lead citrate (Fisher, for 10 min) for contrast staining. The sections were cut by LEICA EM UC-7 (Leica Microsystems, Austria) with a diamond knife (Diatome) and transferred on copper and nickel grids. All the thin sections were observed by TEM (JEM-1011, Japan) at the acceleration voltage of 80 kV and camera-Megaview III (Soft imaging system-Germany).
Xenograft tumor model
Six-week-old male NCr (CrTac:NCr-Foxn1 nu) nude mice (25 ± 3 g) were purchased from Nara Biotech Co. (Seoul, Korea) and housed in animal facility at 23 ± 2 °C and 60 ± 10% humidity under light-controlled (12 h, 07:00–19:00) environment. All materials including bedding and feed were sterilely cleaned by UV rays for 15 min before treatment to the mice. The experiments were conducted in accordance with the guidelines approved by Institutional Animal Care and Use Committee, Kyung Hee University (KHUASP(SE)-11-024). After adaptation, HepG2 cells (5 × 106) were subcutaneously injected into right flank of NCR nude mice. Twenty days later, mice were subdivided into three groups: control (PBS; n = 8), T1 (gallotannin, 10 mg/kg; n = 7), and T2 (gallotannin, 20 mg/kg; n = 7) groups. The mice were intraperitoneally injected by PBS or gallotannin 3 times a week for 15 days and the mice were killed and tumors were collected at 36 days after HepG2 cell inoculation. Tumor size was measured 2–3 times a week using a caliper and the tumor volume was monitored by the formulation of AB 2/2 (A is the longer dimension and B is the smaller dimension).
Immunohistochemistry and TUNEL assay using tumor tissues
Immunohistochemistry (IHC) was performed using the indirect avidin–biotin-enhanced horseradish-peroxide method as previously described (Jung et al. 2015). Briefly, paraffin sections were deparaffinized with xylene and dehydrated with an increasing series of ethanol. After deparaffinization, dehydration, endogenous peroxidase blocking and antigen retrieval, specimens were incubated overnight at 4 °C with antibodies of PCNA (1:150), SIRT1 (1:150), and p-AMPKα1 (Thr172, 1:150) in a humidity chamber. The sections were washed with PBS and incubated with biotinylated goat anti-rabbit (1:200, Vector Laboratories, Burlingame, CA, USA) or biotinylated rabbit anti-rat IgG (1:200, Abcam, Boston, MA, USA) for 40 min in a humidity chamber. After further washes, the target antibodies including PCNA, pAMPKα1, and SIRT1 were detected with the vector ABC complex/horseradish peroxidase (HRP) kit (Vector Laboratories, Burlingame, CA, USA), developed with 3,3′-diaminobenzidine (DAB substrate kit for peroxidase, SK-4100, Vector Laboratories, Burlingame, CA, USA) and observed with microscope (LEICA DM 2500, Germany). To avoid false-negative and false-positive results, all processes such as fixation, processing, and molecular assays were identically conducted for the slides tested and IHC results were scored blinded to the source of the HRP conjugate. For semiquantitation, the positive cells for PCNA, SIRT1, and p-AMPKα1 were counted from at least total 165 cells in a photo (200×) obtained with a CCD camera. The quantification of immunostained cells was presented as a percentage of [No. of positive cells/No. of total cells] × 100. In addition, TUNEL assay was performed using tumor tissues from the mice and apoptosis was detected to count TUNEL-positive green fluorescent cells from at least total 280 cells per photo using in situ Cell Death Detection kit according to the manufacturer’s instructions (11 684 795 910, Roche, Mannheim, Germany) by confocal microscope (FLUOVIEW, FV10C-W, Tokyo, Japan).
Statistical analysis
The results were expressed as mean ± SD or median ± SEM from at least three independent experiments. Student’s t test was performed for two group comparisons. In addition, the one-way analysis of variance (ANOVA) followed by a Turkey post hoc test was conducted for multi-group comparison using GraphPad Prism software (Version 5.0, California, USA). Significant difference was considered if the p value was less than 0.05.
Results
Gallotannin exhibited anti-proliferative activity and induced senescence through p21 upregulation and G0/G1 arrest in HepG2 and SK-Hep1 cells
To assess the effect of gallotannin (Fig. 1a) on the viability and proliferation of HCC cells, MTT assay and colony formation assay were performed. As shown in Fig. 1b, gallotannin exerted cytotoxicity in a time-dependent (24–72 h) and concentration-dependent manner in HepG2 and SK-Hep1 cells with IC50 values of over 150 and 70 µM, respectively. In addition, 40 µM of gallotannin treatment for 12–14 days potently suppressed long-term proliferative activity of HepG2 and SK-Hep1 cells by colony formation assay (Fig. 1c). Consistently, gallotannin significantly increased G0/G1 arrest in HepG2 and SK-Hep1 cells (Fig. 1d) in a time- and concentration-dependent fashion. However, p21 was upregulated at early time points (6–12 h), but rapidly declined at later time points (24–72 h) in gallotannin-treated HepG2 and SK-Hep1 cells (Fig. 1e). Furthermore, the number of SA-β galactosidase-positive cells was significantly increased in gallotannin-treated HepG2 and SK-Hep1 cells by SA-β galactosidase (SA-β-gal) assay widely used for senescence experiment (Barth et al. 2010) (Fig. 1f). In addition, gallotannin cleaved PARP and caspase 3, but attenuated the expression of procaspase 8 from 24-h culture in HepG2 cells and from 48-h culture in SK-Hep1 cells (Fig. 1g). In contrast, gallotannin increased subG1 population in a time- and concentration-dependent fashion (Fig. S1). Furthermore, pan-caspase inhibitor Z-VAD-FMK blocked the ability of gallotannin to cleave PARP and caspase 3 in HepG2 cells (Fig. 1h).
Gallotannin induced impaired autophagy via inhibition of autophagy flux in HepG2 and SK-Hep1 cells
Autophagy is also closely associated with senescence in cancers, since tumor cells tend to secrete factors that promote senescence (Gewirtz 2014). The amount of LC3B-II conversion is positively related to the formation of autophagosomes during autophagy (Dupont et al. 2014). As shown in Fig. 2a, gallotannin increased LC3B-II conversion in a concentration-dependent manner and also decreased Beclin1 in HepG2 and SK-Hep1 cells (Fig. 2b). Consistently, immunofluorescence assay showed the increased distribution of LC3 punctae compared to untreated control in gallotannin-treated HepG2 and SK-Hep1 cells (Fig. 2c). Furthermore, gallotannin increased the number of autophagosomes in HepG2 cells by TEM observation, which was blocked by early-stage autophagy inhibitor 3 methyladenine (3-MA) (Fig. 2d). In addition, 3-MA reduced LC3B-II conversion in gallotannin-treated HepG2 and SK-Hep1 cells (Fig. 2e). Consistently, early-stage autophagy inhibitors including 3-MA and wortmannin did not affect colony formation and weakly enhanced cytotoxicity in gallotannin-treated HepG2 and SK-Hep1 cells (Fig. 3a). In contrast, late-stage autophagy inhibitors such as CQ and NH4Cl (ammonium chloride) enhanced cytotoxicity (Fig. 3b), LC3B-II accumulation and downregulation of SIRT1 (Fig. 3c, d) in gallotannin-treated HepG2 and SK-Hep1 cells.

Autophagic response was initiated by gallotannin in HepG2 and SK-Hep1 cells. a, b Gallotannin promotes conversion of LC3-I to LC3B-II in a concentration- and time-dependent fashion in HepG2 and SK-Hep1 cells. Cells were treated at indicated concentrations of gallotannin for indicated times and the expression of LC3B and Beclin1 was detected by Western blotting. Quantification of LC3B-II or Beclin 1 relative to β-actin in gallotannin-treated HepG2 and SK-Hep1 cells was presented by Image J densitometric analysis. c Effect of gallotannin on LC3 punctation of HepG2 and SK-Hep1 cells. For immunostaining, cells were grown on chamber slide and fixed. The fixed cells were incubated overnight with anti-LC3 B primary antibody and then incubated with Alexa Fluor®568-conjugated anti-rabbit IgG antibody. The cells were stained with 0.5 mg/ml of DAPI to visualize nuclei. LC3 puncta was observed under confocal laser microscope (×400 and ×600). d TEM picture of gallotannin-treated HepG2 cells. The number of autophagic vesicles (red arrows) was increased by gallotannin treatment and the formation of autophagosome was inhibited by 3-MA treatment. Lower panel is the enlarged image in the black frame. Red arrows indicate autophagic vesicles. Scale bar regular image: 5000 nm, enlarge image: 2000 nm. e Effect of 3-MA on gallotannin-mediated early autophagic response in HepG2 and SK-Hep1 cells. Cells were incubated with 40 µM gallotannin and/or 3-MA for 24 h. LC3B expression was detected by Western blotting. β-Actin was used as a loading control. Quantification of LC3B-II relative to β-actin was presented by Image J densitometric analysis (color figure online)

Gallotannin induces impaired autophagy in HepG2 and SK-Hep1 cells. a, b Effect of autophagy inhibitors on cytotoxicity in gallotannin-treated HepG2 and SK-Hep1 cells. Cytotoxicity of gallotannin was evaluated in HepG2 and SK-Hep1 cells in the presence or absence of autophagy inhibitors (3-MA, wortmannin, CQ or NH4Cl) by MTT assay. Data were represented as means ± SD of three independent experiments. **p < 0.01 and ***p < 0.001 vs. gallotannin treatment by one-way ANOVA). c and d Effect of CQ or NH4Cl on the expression of SIRT1 and LC3-BII in gallotannin-treated HepG2 and SK-Hep1 cells. Cells were treated with gallotannin and/or late-stage autophagy inhibitors (CQ or NH4Cl) for 24 h. The expression of SIRT1 and LC3-B was visualized by Western blotting with the antibodies SIRT1 and LC3B-II. e Effect of CQ on autophagosomal LC3 puncta distribution in gallotannin-treated HepG2 and SK-Hep1 cells. HepG2 and SK-Hep1 cells were transfected with GFP–mRFP–LC3 construct and then exposed to 40 µM gallotannin and/or CQ for 24 h. Then the merged color was observed in gallotannin- and/or CQ-treated cells under the confocal laser microscope (×400 and ×600). Quantitative analysis of yellowish LC3 puncta was represented as mean ± SD. ***p < 0.01 vs gallotannin treatment by one-way ANOVA
Consistently, to confirm whether or not autophagic flux is completed during gallotannin-induced autophagy, autophagy flux assay was conducted in gallotannin-treated HepG2 and SK-Hep1 cells transfected by a tandem fluorescent-tagged LC3 reporter plasmid (GFP–mRFP–LC3). The yellow fluorescence for GFP–mRFP–LC3 puncta by merging both green and red fluorescence in autophagosomes indicates impaired autophagy, while red fluorescence for only the mRFP signal after fusion with lysosomes implies complete autophagic flux (Klose et al. 2014; Pietrocola et al. 2013; Zhang et al. 2013). Here, as shown in Fig. 3e, the number of yellow-colored LC3 puncta was increased in gallotannin-treated HepG2 and SK-Hep1 cells compared to untreated control, which was enhanced by CQ.
Gallotannin suppressed the expression of SIRT1 and activated phosphorylation of AMPKα1 in HepG2 and SK-Hep1 cells
To examine the roles of AMPK and SIRT1 in gallotannin-induced senescence and cell death, Western blotting was performed in gallotannin-treated HepG2 and SK-Hep1 cells. In the current study, gallotannin increased phosphorylation of AMPKα1(Thr172) and decreased the expression level of SIRT1 and p-mTOR in a concentration-dependent fashion in HepG2 and SK-Hep1 cells (Fig. 4a). In addition, gallotannin attenuated the expression of SIRT1 in HepG2 and SK-Hep1 cells, but overexpression of SIRT1 blocked LC3B-II conversion in gallotannin-treated HepG2 and SK-Hep1 cells. Interestingly, proteosomal inhibitor MG132 reversed the ability of gallotannin to reduce the expression of SIRT1 and accumulation of LC3B-II in gallotannin-treated HepG2 and SK-Hep1 cells (Fig. 4b, c). To find out the correlation of SIRT1 and AMPKα1 in gallotannin-treated cells, HepG2 cells were co-transfected with HisB–SIRT1 and HA–AMPKα1 and then immunoprecipitation was performed. As shown in Fig. 4d, the ectopically expressed HA–AMPKα1 was immunoprecipitated with HisB-tagged SIRT1, indicating the binding of SIRT1 and AMPKα1 in HepG2 cells regardless of gallotannin treatment. Furthermore, overexpression of SIRT1 blocked the G0/G1 arrest (Fig. 4e) and anti-proliferative effect by colony formation assay induced by gallotannin in HepG2 cells (Fig. 4f).

The pivotal role of SIRT1 in gallotannin-induced autophagy and anti-proliferative activity in HepG2 and SK-Hep1 cells. a Effect of gallotannin on the expression of SIRT1 and phosphorylation of AMPKα1 in HepG2 and SK-Hep1 cells. Cells were treated with indicated concentrations of gallotannin for 24 h. Then the whole lysates were subjected to Western blotting with antibodies of SIRT1, p-mTOR, mTOR, pAMPKα1(Thr172), and AMPKα1. b Effect of SIRT1 overexpression on gallotannin-induced LC3B-II accumulation in HepG2 and SK-Hep1 cells. Cells were transfected with HisB–SIRT1 plasmid and exposed to 40 μM gallotannin for 24 h. Then the cell lysates were subjected to Western blotting with indicated antibodies. c Effect of proteosomal inhibitor MG132 on gallotannin-induced LC3-II accumulation and SIRT1 stability in HepG2 and SK-Hep1 cells. Cells were transfected with HisB–SIRT1 plasmid and exposed to 40 μM gallotannin for 24 h and/or MG132 (10 μM) for 4 h before harvest. Then the cell lysates were subjected to Western blotting with indicated antibodies. d Effect of gallotannin on the binding between SIRT1 and AMPKα1 in HepG2 cells by immunoprecipitation. HepG2 cells were transfected with HisB–SIRT1 (2 μM) and HA–AMPKα1 (2 μM), and then exposed to gallotannin (40 μM) for 24 h. The transfected cells were immunoprecipitated with HisB–SIRT1 antibody for HisB–SIRT1 and HA–AMPKα1 and then were subjected to Western blotting. e Effect of SIRT1 overexpression on gallotannin increased G0/G1 population in HepG2 cells. Cell cycle analysis was conducted in HepG2 cells transfected with HisB–SIRT1 for 24 h, and exposed to 40 μM gallotannin for 24 h. G0/G1 arrest was represented as means ± SD of three independent experiments. **p < 0.01 vs. control by one-way ANOVA. f Effect of SIRT1 overexpression on gallotannin-induced anti-proliferative activity by clonogenic assay. HepG2 cells transfected with HisB–SIRT1 were cultured with or without 40 μM gallotannin for 24 h. Then the cells were continuously incubated in new fresh complete medium for 12–14 days. Relative percentage of clonogenic cells were represented as means ± SD of three independent experiments, ***p < 0.001 vs gallotannin treatment alone by one-way ANOVA
Critical roles of AMPK and SIRT1 in gallotannin-induced senescence, inhibition of proliferation and impaired autophagy in HepG2 and SK-Hep1 cells
To test the role of AMPK in gallotannin-induced autophagy and senescence in HCC cells, AMPK activator 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) and AMPK inhibitor compound C were used in HepG2 and SK-Hep1 cells. As shown in Fig. 5a, AICAR significantly increased the number of SA-β galactosidase-positive cells in gallotannin-treated HepG2 and SK-Hep1 cells, while compound C significantly reduced SA-β-gal-positive cells in gallotannin-treated HepG2 and SK-Hep1 cells. Likewise, AICAR significantly reduced the number of clonogenic cells, while compound C significantly increased the number of clonogenic cells in gallotannin-treated HepG2 and SK-Hep1 cells (Fig. 5b). As shown in Fig. 5c, the number of autophagic vesicles induced by gallotannin was inhibited by compound C using TEM observation in HepG2 cells. Consistently, compound C reversed AMPKα1 activation, LC3B-II accumulation and downregulation of SIRT1 induced by gallotannin in HepG2 and SK-Hep1 cells (Fig. 5d). Of note, decreased expression of SIRT1 and LC3-II conversion by gallotannin was shown in AMPKα1 +/+ wild MEF cells, while there was no change by gallotannin in AMPKα1 −/− MEF cells (Fig. 5e).

The important role of AMPK in gallotannin-induced senescence and impaired autophagy in HCC cells. a Effect of AMPK activator AICAR or AMPK inhibitor compound C on senescence in HepG2 and SK-Hep1 cells. Cells were pretreated with AICAR or compound C for 1 h, and exposed to 40 μM gallotannin for 24 h. Then, SA-β-galactosidase staining was performed. SA-β galactosidase-positive cells were presented as relative values of the total cells (means ± SD of three independent experiments, *p < 0.05, **p < 0.01 vs. gallotannin treatment alone by one-way ANOVA). b Effect of AICAR or compound C on colony formation in HepG2 and SK-Hep1 cells. Cells were pretreated with AICAR or compound C for 1 h, and exposed to 40 μM gallotannin for 24 h. Then cells were continuously incubated in new fresh complete medium for 12–14 days. Relative percentage of clonogenic cells were represented as means ± SD of three independent experiments. *p < 0.05 and ***p < 0.001 vs. gallotannin treatment alone by one-way ANOVA). c Compound C decreased gallotannin-induced autophagic vesicles in HepG2 cells. After pre-treatment of compound C for 1 h, HepG2 cells were exposed to gallotannin for 24 h and then TEM observation was performed. Lower panel is the enlarged image in the black frame. Red arrows indicate autophagic vesicles. Scale bar regular image: 5000 nm, enlarge image: 2000 nm. d Effect of compound C on the reduced expression of SIRT1 and LC3B-II accumulation by gallotannin in HepG2 and SK-Hep1 cells. The cells were exposed to 40 μM gallotannin for 24 h with or without compound C. Then the cell lysates were subjected to Western blotting with indicated antibodies. e Important role of AMPK in gallotannin-induced LC3B-II accumulation in AMPKα1-WT and AMPKα1−/− mouse embryonic fibroblast (MEF) cells. The cells were treated with 40 μM gallotannin for 24 h and the expression of SIRT1 and AMPKα1 was detected by Western blotting
Gallotannin attenuated the in vivo growth of HepG2 in a xenograft tumor model
To confirm in vitro anticancer effects of gallotannin, animal study was conducted in HepG2 xenograft tumor model. Gallotannin at the doses of 10 mg/kg (T1) and 20 mg/kg (T2) was intraperitoneally injected into the mice three times a week for 15 days from 20 days after HepG2 cells were subcutaneously injected into the right flank of NCr nude mice. Gallotannin significantly suppressed tumor size (Fig. 6a, b) and reduced tumor volume by 1.9–2.3-fold compared to PBS control group (Fig. 6c) without any body weight loss in mice. However, tumor weight was significantly reduced only at 20 mg/kg (T2) compared to PBS-treated control (Fig. 6d). In addition, immunohistochemistry for tumor tissues revealed the decreased expression of PCNA for proliferation and SIRT1 for senescence and also increased expression of p-AMPKα1 and TUNEL for apoptosis compared to PBS-treated control (Fig. 6e, f). Consistently, Western blotting showed decreased expression level of PCNA and SIRT1 and increased expression level of p-AMPKα1 in gallotannin-treated group compared to PBS control group (Fig. 6g).

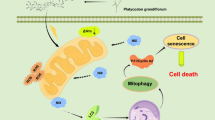
Antitumor effect of gallotannin in HepG2 xenograft mouse model. a Photos of mice bearing HepG2 cells from control (n = 8; PBS), T1 (n = 7; 10 mg/kg) and T2 (n = 7; 10 mg/kg) groups. b Representative tumors isolated from NCr nude mice from ***control, T1, and T2 groups. c Tumor volume of mice from three groups during experiment period. Tumor volumes were represented by median ± SEM. *p < 0.05 vs. control by one-way ANOVA. d Effect of gallotannin on tumor weights from three groups. The weight of tumor was measured from sacrificed mice. The data were represented by median ± SEM. *p < 0.05 vs. control by one-way ANOVA. e Effect of gallotannin on the expression of PCNA, SIRT1 and p-AMPKα1 by immunohistochemistry. Scale bar 50 μm. Quantification of IHC staining-positive cells in tumor tissues were represented by mean ± SD (n = 7–8). **p < 0.01 and ***p < 0.001 vs. control by one-way ANOVA. f Effect of gallotannin on the number of TUNEL-positive cells in tumor sections from mice of three groups (mean ± SEM, n = 5). **p < 0.01 and ***p < 0.001 vs. control by one-way ANOVA. Scale bar 50 μm. g Effect of gallotannin on the expression of PCNA, SIRT1, p-AMPKα1, and procaspase 3 in isolated tumors. Protein levels were quantified by Image J software and plotted relative to the control group. *p < 0.05, **p < 0.01 or ***p < 0.001 vs. control by one-way ANOVA. h Schematic diagram on antitumor mechanism of gallotannin via SIRT1/AMPK-mediated senescence and impaired autophagy leading to cell death
Discussion
In the present study, the underlying antitumor mechanism of gallotannin was elucidated in HepG2 and SK-Hep1 HCC cells and mice in association with SIRT1 and AMPK signaling pathways. Here gallotannin increased cytotoxicity and sub-G1 population and also reduced long-term colony formation in HepG2 and SK-Hep1 cells, indicating the possibility of programmed cell death by gallotannin in HCCs. However, pan-caspase inhibitor Z-VAD-FMK effectively blocked cleavages of PARP and caspase 3 induced by gallotannin 48 h after treatment in HepG2 cells, implying gallotannin can induce caspase-dependent apoptosis at the late stage of treatment. Likewise, Al-Ayyoubi and Gali-Muhtasib (2007) claimed that gallotannin induced p21-dependent disturbance of the cell cycle and apoptosis in p53-negative HCT116 colon cancer cells better than in p53 wild-type HCT colon cancer cells, indicating apoptotic potential of gallotannin.
It is well documented that cellular senescence is a known terminal differentiation condition for tumour suppression and aging (Althubiti et al. 2014; Campisi 2000). Interestingly, gallotannin induced senescence through G0/G1 cell cycle arrest, increased SA-β gal positive cells and upregulated p21 as a cyclin-dependent kinase inhibitor in HepG2 and SK-Hep1 cells, implying the senescence-mediated antitumor effect of gallotannin. However, it is noteworthy that gallotannin induced transient p21 upregulation and subsequently its rapid decline in two HCC cells in the current study. Here we can postulate this biphasic phenomena of p21 imply p21-dependent senescence at early time points and p21-independent apoptosis at later time points by gallotannin, since p21 is temporarily activated in senescence (Baker et al. 2013; Romanov et al. 2010), and then decreases after cell cycle arrest in many cell lines including senescent human diploid fibroblasts (Alcorta et al. 1996). However, Al-Halabi and his colleagues (Al-Halabi et al. 2015) reported that gallotannin induces senescence independently of p53 and p21 in HCT116 colon cancer cells.
Notably, 1,2,3,4,6-penta-O-galloyl-b-d-glucose (PGG) is a key intermediate and immediate precursor involved in the biosynthesis of hydrolyzable tannins including gallotannin in many plants and fruits (Zhang et al. 2009). Though PGG was well known to have antitumor activity through anti-angiogenic (Huh et al. 2005), apoptotic (Huh et al. 2005), autophagic (Dong et al. 2014), anti-inflammatory (Kiss et al. 2013), and antioxidant (Shaikh et al. 2016) effects in several cancers in breast (Lee et al. 2011), prostate (Park et al. 2010; Zhang et al. 2011), liver (Kant et al. 2016), lung (Kant et al. 2016), and blood (Kwon et al. 2012), the underlying autophagic mechanism of gallotannin was for the first time elucidated in the current study. Autophagy is a physiological dynamic process consisting of three sequential steps such as formation of autophagosomes, the fusion of autophagosomes with autolysosomes and lysosomal degradation (Barth et al. 2010; Dupont et al. 2014). Here gallotannin showed autophagic features by increasing the number of LC3 puncta and autophagic vesicles and increased the conversion of LC3B-I to LC3B-II, but induced decreased expression of Beclin 1 in HepG2 and SK-Hep1 cells, indicating autophagic potential of gallotannin. Since the accumulation of LC3-puncta, autophagic vesicles and LC3B-II represents either complete autophagy or impaired autophagy due to inefficient lysosomal fusion and decreased lysosomal degradation (Dupont et al. 2014; Zhang et al. 2013), autophagic flux was assessed using a tandem fluorescent-tagged LC3 reporter plasmid (GFP–mRFP–LC3) transfection in gallotannin-treated HepG2 and SK-Hep1 cells. Immunofluorescence revealed that gallotannin increased the number of yellow-colored LC3 puncta through merging of GFP and mRFP–LC3, which was enhanced late-stage autophagy inhibitor CQ in HepG2 and SK-Hep1 cells. Consistently, late autophagy inhibitors CQ and NH4Cl significantly enhanced cytotoxicity by MTT assay, while early autophagy inhibitors 3-MA and wortmannin did not in HepG2 and SK-Hep1 cells, demonstrating the impaired autophagy of gallotannin in two HCC cells.
There are accumulating evidences that SIRT1 is aberrantly overexpressed in a subset of human HCCs, and so the silencing of SIRT1 in cancer cells suppresses the growth of cancer cells leading to cell cycle arrest and apoptosis (Lau et al. 2014; Raghuram and Mishra 2014; Zhou et al. 2015; Zu et al. 2010). In addition, previous evidences reveal that AMPK controls proliferation, apoptosis, and autophagy in cancer cells as a potent tumor suppressor (Lee et al. 2012). Hence, SIRT1 and AMPK are the two energy sensor systems regulating proliferation, autophagy, and senescence of cells (Wang et al. 2011; Wu et al. 2011). In addition, LKB1/AMPK signaling was activated in SIRT1-dependent or -independent manner in several cancer cells (Canto et al. 2010; Guo et al. 2012; Hou et al. 2008; Lee et al. 2012; Vucicevic et al. 2011; Wu et al. 2011). In the current study, gallotannin attenuated the expression of SIRT1 and mTOR and also activated phosphorylation of AMPK on Thr172 in HepG2 and SK-Hep1 cells. Interestingly, gallotannin also reduced the expression of overexpressed SIRT1 in HepG2 and SK-Hep1 cells like the native alleles. However, proteosomal inhibitor MG132 blocked the ability of gallotannin to attenuate the expression of SIRT1 and LC3B-II accumulation, implying gallotannin alters SIRT1 protein stability via proteasome-mediated degradation. Furthermore, AMPK inhibitor compound C blocked LC3B-II accumulation and higher SA-β-gal activity by gallotannin in HepG2 and SK-Hep1 cells. Consistently, LC3-II conversion by gallotannin was not shown in AMPKα1 −/− MEF cells compared to WT AMPK +/+ MEF cells, demonstrating the pivotal role of AMPK in gallotannin-induced senescence and impaired autophagy leading to cell death. Likewise, SIRT1 overexpression using HisB–SIRT1 vector reversed LC3-II conversion, G0/G1 arrest and anti-proliferative activity by gallotannin in HepG2 cells, indicating the important role of SIRT1 in gallotannin-induced senescence and impaired autophagy leading to cell death. In HepG2 xenograft tumor model to confirm in vitro antitumor effect of gallotannin, additionally, gallotannin significantly suppressed tumor size and volume compared to PBS-injected control group without any body weight loss in mice. Furthermore, IHC for tumor tissues showed the decreased expression of PCNA for proliferation, SIRT1 for senescence and also increased expression of p-AMPKα1 and TUNEL for apoptosis compared to PBS-treated control. Consistently, gallotannin reduced the expression level of PCNA and SIRT1 and increased phosphorylation of AMPKα1 compared to PBS control, strongly demonstrating antitumor effect of gallotannin via inhibition of PCNA and SIRT1, activation of AMPK α1, and apoptosis.
Collectively, gallotannin induced senescence, suppressed proliferation via G0/G1 cell cycle arrest and p21 upregulation at early time points, induced impaired autophagy and finally caused cell death in HepG2 and SK-Hep1 cells through regulation of SIRT1/AMPK axis, and also suppressed the growth of HepG2 cells by inhibition of PCNA, SIRT1 and activation of p-AMPK α1 and TUNEL in HepG2 Xenograft mouse model (Fig. 6h). Overall, these findings support evidences that gallotannin exerts antitumor effect through senescence-mediated impaired autophagy leading to cell death via regulation of SIRT1 and AMPK signaling as a potent anticancer agent for HCC treatment and prevention.
Change history
16 July 2018
This article contains an error.
Abbreviations
- p62/SQSTM1:
-
p62 sequestosome 1
- Gal:
-
Gallotannin
- MTT:
-
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- HCC:
-
Human hepatocellular carcinoma
- HepG2:
-
Human epithelial hepatocellular carcinoma
- SK-Hep1:
-
Human epithelial hepatic adenocarcinoma
- MEF:
-
Mouse embryo fibroblast
- CQ:
-
Chloroquine
- NH4Cl:
-
Ammoniun chloride
- AICAR:
-
5-Aminoimidazole-4-carboxamide ribonucleotide
- Com. C:
-
Compound C
- LC3:
-
Microtubule-associated protein 1 light chain 3
- LC3-I:
-
Soluble unlipidated form of LC3
- LC3-II:
-
LC3–phospholipid conjugate
- AMPK:
-
AMP-activated protein kinase
- DAPI:
-
4′,6-Diamidino-2-phenylindole
- FITC:
-
Fluorescein isothiocyanate
- GFP/RFP:
-
Green fluorescent protein/red fluorescent protein
- SIRT1:
-
NAD-dependent deacetylase sirtuin-1
- Caspase:
-
Cysteine aspartyl-specific protease
- 3-MA:
-
3-Methyladenine
- SA-β-gal:
-
Senescence-associated β-galactosidase
- TEM:
-
Transmission electron microscopy
- TUNEL:
-
Terminal deoxynucleotidyl transferase dUTP nick end labeling
References
Al-Ayyoubi S, Gali-Muhtasib H (2007) Differential apoptosis by gallotannin in human colon cancer cells with distinct p53 status. Mol Carcinog 46(3):176–186. doi:10.1002/mc.20252
Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC (1996) Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci USA 93(24):13742–13747
Al-Halabi R, Abou Merhi R, Chakilam S et al (2015) Gallotannin is a DNA damaging compound that induces senescence independently of p53 and p21 in human colon cancer cells. Mol Carcinog 54(10):1037–1050. doi:10.1002/mc.22172
Althubiti M, Lezina L, Carrera S et al (2014) Characterization of novel markers of senescence and their prognostic potential in cancer. Cell Death Dis 5:e1528. doi:10.1038/cddis.2014.489
Asfour OM, el-Fiky HM, Nassar A (1990) Lamellar keratoplasty combined with extracapsular cataract extraction. Refract Corneal Surg 6(2):125–126
Bae HJ, Chang YG, Noh JH et al (2012) DBC1 does not function as a negative regulator of SIRT1 in liver cancer. Oncol Lett 4(5):873–877. doi:10.3892/ol.2012.875
Bae HJ, Noh JH, Kim JK et al (2014) MicroRNA-29c functions as a tumor suppressor by direct targeting oncogenic SIRT1 in hepatocellular carcinoma. Oncogene 33(20):2557–2567. doi:10.1038/onc.2013.216
Baffy G, Brunt EM, Caldwell SH (2012) Hepatocellular carcinoma in non-alcoholic fatty liver disease: an emerging menace. J Hepatol 56(6):1384–1391. doi:10.1016/j.jhep.2011.10.027
Baker DJ, Weaver RL, van Deursen JM (2013) p21 both attenuates and drives senescence and aging in BubR1 progeroid mice. Cell Rep 3(4):1164–1174. doi:10.1016/j.celrep.2013.03.028
Barth S, Glick D, Macleod KF (2010) Autophagy: assays and artifacts. J Pathol 221(2):117–124. doi:10.1002/path.2694
Berk V, Kaplan MA, Tonyali O et al (2013) Efficiency and side effects of sorafenib therapy for advanced hepatocellular carcinoma: a retrospective study by the anatolian society of medical oncology. Asian Pacific J Cancer Prev APJCP 14(12):7367–7369
Campisi J (2000) Cancer, aging and cellular senescence. In vivo 14(1):183–188 (Athens, Greece)
Canto C, Jiang LQ, Deshmukh AS et al (2010) Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab 11(3):213–219. doi:10.1016/j.cmet.2010.02.006
Casella ML, Parody JP, Ceballos MP et al (2014) Quercetin prevents liver carcinogenesis by inducing cell cycle arrest, decreasing cell proliferation and enhancing apoptosis. Mol Nutr Food Res 58(2):289–300. doi:10.1002/mnfr.201300362
Chi X, Zhang R, Shen N et al (2015) Sulforaphane reduces apoptosis and oncosis along with protecting liver injury-induced ischemic reperfusion by activating the Nrf2/ARE pathway. Hep Intl 9(2):321–329. doi:10.1007/s12072-014-9604-y
Dai W, Wang F, He L et al (2015) Genistein inhibits hepatocellular carcinoma cell migration by reversing the epithelial-mesenchymal transition: partial mediation by the transcription factor NFAT1. Mol Carcinog 54(4):301–311. doi:10.1002/mc.22100
Ding S, Hu A, Hu Y, Ma J, Weng P, Dai J (2014) Anti-hepatoma cells function of luteolin through inducing apoptosis and cell cycle arrest. Tumour Biol J Int Soc Oncodev Biol Med 35(4):3053–3060. doi:10.1007/s13277-013-1396-5
Dong Y, Yin S, Jiang C et al (2014) Involvement of autophagy induction in penta-1,2,3,4,6-O-galloyl-beta-d-glucose-induced senescence-like growth arrest in human cancer cells. Autophagy 10(2):296–310. doi:10.4161/auto.27210
Dupont N, Orhon I, Bauvy C, Codogno P (2014) Autophagy and autophagic flux in tumor cells. Methods Enzymol 543:73–88. doi:10.1016/B978-0-12-801329-8.00004-0
Feng Y, Ke C, Tang Q et al (2014) Metformin promotes autophagy and apoptosis in esophageal squamous cell carcinoma by downregulating Stat3 signaling. Cell Death Dis 5:e1088. doi:10.1038/cddis.2014.59
Flores A, Marrero JA (2014) Emerging trends in hepatocellular carcinoma: focus on diagnosis and therapeutics. Clin Med Insights Oncol 8:71–76. doi:10.4137/CMO.S9926
Gali-Muhtasib H, Hmadi R, Kareh M, Tohme R, Darwiche N (2015) Cell death mechanisms of plant-derived anticancer drugs: beyond apoptosis. Apoptosis 20(12):1531–1562. doi:10.1007/s10495-015-1169-2
Gewirtz DA (2013) Autophagy and senescence: a partnership in search of definition. Autophagy 9(5):808–812. doi:10.4161/auto.23922
Gewirtz DA (2014) Autophagy and senescence in cancer therapy. J Cell Physiol 229(1):6–9. doi:10.1002/jcp.24420
Ghosh S, Zhou Z (2015) SIRTain regulators of premature senescence and accelerated aging. Protein Cell 6(5):322–333. doi:10.1007/s13238-015-0149-1
Guo L, Xie B, Mao Z (2012) Autophagy in premature senescent cells is activated via AMPK pathway. Int J Mol Sci 13(3):3563–3582. doi:10.3390/ijms13033563
Hou X, Xu S, Maitland-Toolan KA et al (2008) SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J Biol Chem 283(29):20015–20026. doi:10.1074/jbc.M802187200
Huh JE, Lee EO, Kim MS et al (2005) Penta-O-galloyl-beta-d-glucose suppresses tumor growth via inhibition of angiogenesis and stimulation of apoptosis: roles of cyclooxygenase-2 and mitogen-activated protein kinase pathways. Carcinogenesis 26(8):1436–1445. doi:10.1093/carcin/bgi097
Jung DB, Yun M, Kim EO et al (2015) The heparan sulfate mimetic PG545 interferes with Wnt/beta-catenin signaling and significantly suppresses pancreatic tumorigenesis alone and in combination with gemcitabine. Oncotarget 6(7):4992–5004. doi:10.18632/oncotarget.3214
Kant R, Yen CH, Lu CK, Lin YC, Li JH, Chen YM (2016) Identification of 1,2,3,4,6-penta-O-galloyl-β-d-glucopyranoside as a glycine N-methyltransferase enhancer by high-throughput screening of natural products inhibits hepatocellular carcinoma. Int J Mol Sci 17(5):669. doi:10.3390/ijms17050669
Kim J, Yun M, Kim EO et al (2016) Decursin enhances TRAIL-induced apoptosis through oxidative stress mediated- endoplasmic reticulum stress signalling in non-small cell lung cancers. Br J Pharmacol 173(6):1033–1044. doi:10.1111/bph.13408
Kiss AK, Filipek A, Zyzynska-Granica B, Naruszewicz M (2013) Effects of penta-O-galloyl-beta-d-glucose on human neutrophil function: significant down-regulation of l-selectin expression. Phytother Res PTR 27(7):986–992. doi:10.1002/ptr.4822
Klionsky DJ, Abdalla FC, Abeliovich H et al (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8(4):445–544
Klose J, Stankov MV, Kleine M et al (2014) Inhibition of autophagic flux by salinomycin results in anti-cancer effect in hepatocellular carcinoma cells. PLoS One 9(5):e95970. doi:10.1371/journal.pone.0095970
Kwon TR, Jeong SJ, Lee HJ et al (2012) Reactive oxygen species-mediated activation of JNK and down-regulation of DAXX are critically involved in penta-O-galloyl-beta-d-glucose-induced apoptosis in chronic myeloid leukemia K562 cells. Biochem Biophys Res Commun 424(3):530–537. doi:10.1016/j.bbrc.2012.06.150
Lao Y, Wan G, Liu Z et al (2014) The natural compound oblongifolin C inhibits autophagic flux and enhances antitumor efficacy of nutrient deprivation. Autophagy 10(5):736–749. doi:10.4161/auto.28034
Lau AW, Liu P, Inuzuka H, Gao D (2014) SIRT1 phosphorylation by AMP-activated protein kinase regulates p53 acetylation. Am J Cancer Res 4(3):245–255
Lee HJ, Seo NJ, Jeong SJ et al (2011) Oral administration of penta-O-galloyl-beta-d-glucose suppresses triple-negative breast cancer xenograft growth and metastasis in strong association with JAK1-STAT3 inhibition. Carcinogenesis 32(6):804–811. doi:10.1093/carcin/bgr015
Lee CW, Wong LL, Tse EY et al (2012) AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Res 72(17):4394–4404. doi:10.1158/0008-5472.CAN-12-0429
Marino G, Niso-Santano M, Baehrecke EH, Kroemer G (2014) Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol 15(2):81–94. doi:10.1038/nrm3735
Marquardt JU, Gomez-Quiroz L, Arreguin Camacho LO et al (2015) Curcumin effectively inhibits oncogenic NF-kappaB signaling and restrains stemness features in liver cancer. J Hepatol 63(3):661–669. doi:10.1016/j.jhep.2015.04.018
Mizushima N, Yoshimori T, Levine B (2010) Methods in mammalian autophagy research. Cell 140(3):313–326. doi:10.1016/j.cell.2010.01.028
Oh GS, Pae HO, Oh H et al (2001) In vitro anti-proliferative effect of 1,2,3,4,6-penta-O-galloyl-beta-d-glucose on human hepatocellular carcinoma cell line, SK-HEP-1 cells. Cancer Lett 174(1):17–24
Park KY, Lee HJ, Jeong SJ et al (2010) 1,2,3,4,6-Penta-O-galloyl-beta-d-glucose suppresses hypoxia-induced accumulation of hypoxia-inducible factor-1alpha and signaling in LNCaP prostate cancer cells. Biol Pharm Bull 33(11):1835–1840
Park IJ, Yang WK, Nam SH et al (2014) Cryptotanshinone induces G1 cell cycle arrest and autophagic cell death by activating the AMP-activated protein kinase signal pathway in HepG2 hepatoma. Apoptosis Int J Program cell Death 19(4):615–628. doi:10.1007/s10495-013-0929-0
Park E, Kwon HY, Jung JH et al (2015) Inhibition of myeloid cell leukemia 1 and activation of caspases are critically involved in gallotannin-induced apoptosis in prostate cancer cells. Phytother Res 29(8):1225–1236. doi:10.1002/ptr.5371
Pietrocola F, Izzo V, Niso-Santano M et al (2013) Regulation of autophagy by stress-responsive transcription factors. Semin Cancer Biol 23(5):310–322. doi:10.1016/j.semcancer.2013.05.008
Puissant A, Robert G, Fenouille N et al (2010) Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res 70(3):1042–1052. doi:10.1158/0008-5472.CAN-09-3537
Raghuram GV, Mishra PK (2014) Stress induced premature senescence: a new culprit in ovarian tumorigenesis? Indian J Med Res 140(Suppl):S120–S129
Romanov VS, Abramova MV, Svetlikova SB et al (2010) p21(Waf1) is required for cellular senescence but not for cell cycle arrest induced by the HDAC inhibitor sodium butyrate (Georgetown, Tex). Cell Cycle 9(19):3945–3955. doi:10.4161/cc.9.19.13160
Sabin RJ, Anderson RM (2011) Cellular senescence—its role in cancer and the response to ionizing radiation. Genome Integr 2(1):7. doi:10.1186/2041-9414-2-7
Shaikh QU, Yang M, Memon KH et al (2016) 1,2,3,4,6-Pentakis[-O-(3,4,5-trihydroxybenzoyl)]-alpha, beta-d-glucopyranose (PGG) analogs: design, synthesis, anti-tumor and anti-oxidant activities. Carbohydr Res 430:72–81. doi:10.1016/j.carres.2016.04.021
Sharma K, Goehe R, Beckta JM, Valerie K, Gewirtz DA (2014) Autophagy and radiosensitization in cancer. EXCLI J 13:178–191
Singh K, Sharma A, Mir MC et al (2014) Autophagic flux determines cell death and survival in response to Apo2L/TRAIL (dulanermin). Mol Cancer 13:70. doi:10.1186/1476-4598-13-70
Song X, Yin S, Huo Y et al (2015) Glycycoumarin ameliorates alcohol-induced hepatotoxicity via activation of Nrf2 and autophagy. Free Radic Biol Med 89:135–146. doi:10.1016/j.freeradbiomed.2015.07.006
Tang Q, Li G, Wei X et al (2013) Resveratrol-induced apoptosis is enhanced by inhibition of autophagy in esophageal squamous cell carcinoma. Cancer Lett 336(2):325–337. doi:10.1016/j.canlet.2013.03.023
Vucicevic L, Misirkic M, Janjetovic K et al (2011) Compound C induces protective autophagy in cancer cells through AMPK inhibition-independent blockade of Akt/mTOR pathway. Autophagy 7(1):40–50
Wang Y, Liang Y, Vanhoutte PM (2011) SIRT1 and AMPK in regulating mammalian senescence: a critical review and a working model. FEBS Lett 585(7):986–994. doi:10.1016/j.febslet.2010.11.047
Wang L, Gao C, Yao S, Xie B (2013) Blocking autophagic flux enhances matrine-induced apoptosis in human hepatoma cells. Int J Mol Sci 14(12):23212–23230. doi:10.3390/ijms141223212
Wei L, Huang N, Yang L et al (2009) [Sorafenib reverses multidrug resistance of hepatoma cells in vitro]. Nan fang yi ke da xue xue bao = J South Med Univ 29(5):1016–1019 (1023)
Wu Y, Li X, Zhu JX et al (2011) Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson’s disease. Neurosignals 19(3):163–174. doi:10.1159/000328516
Yang D, Li L, Liu H et al (2013) Induction of autophagy and senescence by knockdown of ROC1 E3 ubiquitin ligase to suppress the growth of liver cancer cells. Cell Death Differ 20(2):235–247. doi:10.1038/cdd.2012.113
Yip NK, Ho WS (2013) Berberine induces apoptosis via the mitochondrial pathway in liver cancer cells. Oncol Rep 30(3):1107–1112. doi:10.3892/or.2013.2543
Zhang J, Li L, Kim SH, Hagerman AE, Lu J (2009) Anti-cancer, anti-diabetic and other pharmacologic and biological activities of penta-galloyl-glucose. Pharm Res 26(9):2066–2080. doi:10.1007/s11095-009-9932-0
Zhang J, Nkhata K, Shaik AA et al (2011) Mouse prostate proteome changes induced by oral pentagalloylglucose treatment suggest targets for cancer chemoprevention. Curr Cancer Drug Targets 11(7):787–798
Zhang XJ, Chen S, Huang KX, Le WD (2013) Why should autophagic flux be assessed? Acta Pharmacol Sin 34(5):595–599. doi:10.1038/aps.2012.184
Zhao H, Darzynkiewicz Z (2014) Attenuation of replication stress-induced premature cellular senescence to assess anti-aging modalities. Curr Protoc Cytom 69:9.47.1–9.47.10. doi:10.1002/0471142956.cy0947s69
Zhao C, Yin S, Dong Y et al (2013) Autophagy-dependent EIF2AK3 activation compromises ursolic acid-induced apoptosis through upregulation of MCL1 in MCF-7 human breast cancer cells. Autophagy 9(2):196–207. doi:10.4161/auto.22805
Zhao T, Sun Q, del Rincon SV, Lovato A, Marques M, Witcher M (2014) Gallotannin imposes S phase arrest in breast cancer cells and suppresses the growth of triple-negative tumors in vivo. PLoS One 9(3):e92853. doi:10.1371/journal.pone.0092853
Zhou J, Hu SE, Tan SH et al (2012) Andrographolide sensitizes cisplatin-induced apoptosis via suppression of autophagosome-lysosome fusion in human cancer cells. Autophagy 8(3):338–349. doi:10.4161/auto.18721
Zhou ZW, Li XX, He ZX et al (2015) Induction of apoptosis and autophagy via sirtuin1- and PI3K/Akt/mTOR-mediated pathways by plumbagin in human prostate cancer cells. Drug Des Devel Ther 9:1511–1554. doi:10.2147/DDDT.S75976
Zu Y, Liu L, Lee MY et al (2010) SIRT1 promotes proliferation and prevents senescence through targeting LKB1 in primary porcine aortic endothelial cells. Circ Res 106(8):1384–1393. doi:10.1161/CIRCRESAHA.109.215483
Acknowledgements
This work was supported by the Korea Science and Engineering Foundation (KOSEF) grant funded by the Korea government (MEST) (2014R1A2A10052872).
Author information
Authors and Affiliations
Contributions
HY Kwon designed this project, performed most of the experiments, and drafted the manuscript; JH Kim contributed to animal study; SK. Srivastava and BL Kim advised us of the experiment design and helped proofreading; SH Kim supervised all experiments and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
204_2017_2021_MOESM1_ESM.jpg
Supplementary Figure 1. Gallotannin induced weak apoptosis in HCC cells in a time and concentration dependent fashion. a Cell cycle analysis of gallotannin treated HepG2 and SK-Hep1 cells. Cells were treated with gallotannin indicated concentrations and for different times. Then the cell cycle analysis was performed using FACs Calibur, and the cell cycle was analyzed using CellQuest Software. b SubG1 portion was increased dependent on time and concentration in gallotannin treated HepG2 and SK-Hep1 cells. The results of cell cycle analysis were represented as relative percentage ± SD. SubG1 portion of each treatment was represented (means ± SD of three independent experiments, **p < 0.01 and *** p < 0.001 vs. control by One-Way ANOVA). (JPEG 3334 kb)
Rights and permissions
About this article

{kind=link}
Cite this article
Kwon, H.Y., Kim, JH., Kim, B. et al. Regulation of SIRT1/AMPK axis is critically involved in gallotannin-induced senescence and impaired autophagy leading to cell death in hepatocellular carcinoma cells. Arch Toxicol 92, 241–257 (2018). https://doi.org/10.1007/s00204-017-2021-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-017-2021-y