
Abstract
We succeeded in homogeneously expressing and purifying l-asparaginase from Latilactobacillus sakei LK-145 (Ls-Asn1) and its mutated enzymes C196S, C264S, C290S, C196S/C264S, C196S/C290S, C264S/C290S, and C196S/C264S/C290S-Ls-Asn1. Enzymological studies using purified enzymes revealed that all cysteine residues of Ls-Asn1 were found to affect the catalytic activity of Ls-Asn1 to varying degrees. The mutation of Cys196 did not affect the specific activity, but the mutation of Cys264, even a single mutation, significantly decreased the specific activity. Furthermore, C264S/C290S- and C196S/C264S/C290S-Ls-Asn1 almost completely lost their activity, suggesting that C290 cooperates with C264 to influence the catalytic activity of Ls-Asn1. The detailed enzymatic properties of three single-mutated enzymes (C196S, C264S, and C290S-Ls-Asn1) were investigated for comparison with Ls-Asn1. We found that only C196S-Ls-Asn1 has almost the same enzymatic properties as that of Ls-Asn1 except for its increased stability for thermal, pH, and the metals NaCl, KCl, CaCl2, and FeCl2. We measured the growth inhibitory effect of Ls-Asn1 and C196S-Ls-Asn1 on Jurkat cells, a human T-cell acute lymphoblastic leukemia cell line, using l-asparaginase from Escherichia coli K-12 as a reference. Only C196S-Ls-Asn1 effectively and selectively inhibited the growth of Jurkat T-cell leukemia, which suggested that it exhibited antileukemic activity. Furthermore, based on alignment, phylogenetic tree analysis, and structural modeling, we also proposed that Ls-Asn1 is a so-called “Type IIb” novel type of asparaginase that is distinct from previously reported type I or type II asparaginases. Based on the above results, Ls-Asn1 is expected to be useful as a new leukemia therapeutic agent.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
l-asparaginase (EC 3.5.1.1) catalyzes the reversible production of l-aspartic acid and ammonia from l-asparagine and water stoichiometrically (Kumar and Verma 2012). l-asparaginase is located at the site where organic nitrogen and inorganic nitrogen are converted and plays an important biochemical role (Gaufichon et al. 2016). l-asparaginase is also biotechnologically important and used as a food additive for the degradation of l-asparagine, a source of toxic acrylamide that can be generated in food (Pedreschi et al. 2008). Furthermore, l-asparaginase is used as a therapeutic agent for cancers such as acute leukemia (Salzer et al. 2014). For this purpose, the Escherichia coli and Erwinia chrysanthemi enzymes are commercially available as Leunase® and Erwinase®, respectively. However, prolonged use of these enzyme medicines is known to cause side reactions such as allergies (Duval et al. 2002). Therefore, in the search for enzymes from new sources with low primary structural similarity to these enzymes, it is expected that the selected enzyme does not belong to the previously reported type I or type II asparaginase family and does not exhibit cross-immunity (Pokrovsky et al. 2016).
In 2017, we obtained the complete genome sequence of Latilactobacillus sakei LK-145, and found that two putative asparaginase homolog genes exist in the Latilactobacillus sakei LK-145 genome; accession no. AP017931 REGION: 1561151.1562083 (named Ls-asn1); AP017931 REGION: 178764.179717 (named Ls-asn2). Based on the homology search using the primary structure translated from the coding sequence of these two genes, we predicted that Ls-asn1 encodes l-asparaginase. In addition, we found that Ls-Asn1 (the gene product of Ls-asn1) has three characteristic cysteine residues in its primary structure. In this study, we expressed the Ls-asn1 gene in Escherichia coli and clarified the enzymatic properties of Ls-Asn1. We also evaluated the function of three characteristic cysteine residues in the primary structure of Ls-Asn1 by constructing 7 kinds of comprehensive mutated enzymes of Ls-Asn1 in which cysteine residues are replaced with serine residues and examined the possibility of applying Ls-Asn1 as a leukemia drug in Jurkat cells. Furthermore, based on alignment, phylogenetic tree analysis, and structural modeling, we also proposed that Ls-Asn1 is a so-called “Type IIb” novel type of asparaginase that is distinct from previously reported type I or type II asparaginases.
Materials and methods
Materials
Latilactobacillus sakei LK-145, which was isolated from Japanese a sake cellar, was kindly gifted by Kiku-Masamune Sake Brewing Co., Ltd., Hyogo, Japan. Escherichia coli Rosetta (DE3) and the pET21b vector were purchased from Novagen (Merck KGaA, Darmstadt, Germany). Escherichia coli NEB10β was purchased from New England BioLabs (Massachusetts, USA). l-Asparagine monohydrate (purity: > 99%), l-glutamine (purity: > 99%), d-asparagine (purity: > 99%), and d-glutamine (purity: > 99%) were purchased from Watanabe Chemical Ind. (Hiroshima, Japan). A protein assay mixture containing Coomassie Brilliant Blue (CBB) was purchased from Nacalai Tesque, Inc. (Tokyo, Japan). Ni–NTA agarose was purchased from QIAGEN (Hilden, Germany). Nessler’s solution was purchased from FUJIFILM Wako Pure Chemical Corp. (Osaka, Japan). The human T-cell leukemia cell line Jurkat (RCB0806) and a human cell line derived from the kidney of a fetus, HEK293 cells (RCB1637), were obtained from RIKEN BRC, Japan. Jurkat cells were cultured in advanced DMEM/F12 (Thermo Fisher Scientific, Waltham, MA) supplemented with 2% heat-inactivated fetal bovine serum (FBS), 100 U/mL penicillin and 100 μg/mL streptomycin (P/S), and HEK293 cells were cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum (FBS) and P/S at 37 °C and 5% CO2. Escherichia coli asparaginase B (Ec-AnsB; Accession No. NP_417432) was cloned from the Escherichia coli NEB10β genome, purified, and used as a control. All other reagents were of the best commercially available grade and were purchased from Invitrogen Co. (Carlsbad, CA, USA), Kanto Chemical Co., Inc. (Tokyo, Japan), Kishida Chemical Co., Ltd. (Tokyo, Japan), Nacalai Tesque, Inc. (Kyoto, Japan), Sigma–Aldrich Co. (St. Louis, MO, USA), Tokyo Chemical Ind. Co., Ltd. (Tokyo, Japan), FUJIFILM Wako Pure Chemical Corp. (Osaka, Japan), or Watanabe Chemical Ind., unless otherwise stated.
Expression of the l-asparaginase homologous gene from Latilactobacillus sakei LK-145
First, we examined the coding sequence of Ls-asn1 (accession no. AP017931 REGION: 1561151.1562083) and found another candidate start codon, ATG, which is 42 bases upstream of the start codon of Ls-asn1. Accordingly, we decided to express the gene (accession no. AP017931 REGION: 1561109.1562083) as Ls-asn1; hereafter, we used the word Ls-asn1 to describe the coding region of putative l-asparaginase (accession no. AP017931 REGION: 1561109.1562083) in this study.
L. sakei LK-145 was cultivated at 30 °C for 46 h in de Man Rogosa Sharpe (MRS) medium with shaking (130 rpm), after which its genomic DNA was extracted using the phenol/chloroform/isoamyl alcohol method (Miller et al. 1988). The Ls-asn1 gene was amplified by polymerase chain reaction (PCR) from the genomic DNA of L. sakei LK-145 using primers with the NdeI and XhoI restriction sites at the 5ʹ and 3ʹ termini of the gene, respectively: 5ʹ-AGATATACATATGAAAAAAATCCTCGTCTTGCAC-3ʹ and 5ʹ-GGTGGTGCTCGAGTGAGTCGGCGTTGCTGAC-3ʹ. The purified PCR product and a pET21 vector were double digested with the restriction enzymes NdeI and XhoI and ligated using T4 DNA ligase after dephosphorylation with alkaline phosphatase. After the DNA sequence of the pET21-Ls-asn1 vector was confirmed, competent Escherichia coli TOP 10 cells were transformed with the vector using the electroporation Gene Pulser Xcell™ (Bio-Rad Laboratories, Inc., California, USA) to amplify the vector. Then, the purified vector was transformed into Escherichia coli Rosetta (DE3) cells to generate pET21-Ls-asn1/E. coli Rosetta (DE3).
Purification of the l-asparaginase homolog from Latilactobacillus sakei LK-145 expressed in Escherichia coli rosetta (DE3)
The transformant obtained was pET21-Ls-asn1/E. coli Rosetta (DE3), which was cultivated in LB media (5 mL) in a Falcon tube® (15 mL) supplemented with 0.4% (w/v) glucose and 200 μg/mL carbenicillin at 30 °C for 16 h. The culture (1 mL) was transferred to a Sakaguchi flask (500 mL) containing the same medium above and cultivated at 30 °C (127 rpm) until the optical density at 600 nm reached approximately 0.6. After cultivation at 18 °C for 30 min, Ls-Asn1 was expressed by the addition of isopropyl β-d-1-thiogalactopyranoside (0.4 mM final concentration) and further cultivation at 18 °C for 20 h with shaking (130 rpm).
pET21-Ls-asn1/E. coli Rosetta (DE3) cells were collected by centrifugation at 7197×g for 5 min at 4 °C using a Centrifuge 5430R (Eppendorf, Tokyo, Japan) and suspended in 0.75% (w/v) saline (20 mL). The cell suspension was centrifuged again under the same conditions above, and the cells obtained were stored at − 80 °C after decantation. The cell pellets were resuspended in lysis buffer (10 mL) composed of 50 mM potassium phosphate buffer (KPB), pH 8.0, 300 mM NaCl (buffer A) and 10 mM imidazole. The cell suspension was transferred to a beaker (10 mL) and treated with a sonicator (UD-201, Tomy Seiko, Tokyo) in ice-cooled water to disrupt the cells (output, 6; duty cycle, 50%; 1 min × 3 times). After ultrasonication, the suspension was centrifuged at 7197×g for 15 min at 4 °C, and the supernatant was used as a celL-free extract. The celL-free extract was subsequently added to a column of Ni–NTA agarose (2 mL) (Qiagen, Hilden, Germany) that had been equilibrated with lysis buffer. After washing with wash buffer composed of buffer A plus 50 mM imidazole (15 mL), the adsorbed protein (Ls-Asn1) was eluted with elution buffer composed of buffer A plus 250 mM imidazole. The purified Ls-Asn1 was collected and dialyzed for 12 h against 1×phosphate-buffered saline (PBS) composed of 1 mM Na2HPO4, 0.2 mM KH2PO4, 13.7 mM NaCl, and 0.268 mM KCl. The mixture was diluted to 0.4 mg/mL with 1×PBS and stored at −80 °C without any cryoprotectant.
Standard assay conditions
l-asparaginase activity was assayed spectrophotometrically by measuring the amount of ammonia produced in a reaction mixture using Nessler’s reagent. The standard reaction mixture (total volume: 1.0 mL) contained 10 mM l-asparagine, 40 mM 3,3-dimethylglutaric acid/Tris(hydroxymethyl)amino methane/2-amino-2-methyl-1,3-propanediol (GTA) buffer (pH 7.5), and the enzyme mixture. The enzyme reaction was started by the addition of an enzyme solution containing 2 μg of purified enzyme as otherwise stated, and the mixture was shaken (500 rpm) at 37 °C for 0, 5, 10, 15, or 20 min. The enzyme reaction was terminated by the addition of 50% (v/v) trichloroacetic acid (TCA) (125 μL) and centrifuged at 16900 × g for 10 min at 4 °C.
The reaction mixture (50 μL) was subsequently mixed with Nessler’s reagent (50 μL) and deionized water (900 μL) in a microtube and incubated with shaking (500 rpm) at 20 °C after mixing using Vortex-Genie 2 (Kenis Ltd., Osaka, Japan). After 20 min, the absorption at 450 nm was measured using a spectrophotometer (UV-1800; Shimadzu Co. Ltd., Kyoto, Japan). One unit was defined as the amount of enzyme that produced 1 μmol of ammonia per min. The protein concentration was measured by the Bradford method (Bradford 1976) using a protein assay CBB solution with bovine serum albumin as a standard. The enzyme activity and protein concentration were measured repeatedly three times, and the average value and the standard deviation were calculated and are shown.
Basic enzymological properties
The substrate specificity of Ls-Asn1 was examined under standard assay conditions. The substrates used were l-asparagine, d-asparagine, l-glutamine, and d-glutamine. The effects of temperature and pH on Ls-Asn1 activity were examined by measuring the enzyme activity under various temperature and pH conditions. The reaction temperature was changed to 30, 40, or 50 °C. GTA buffer was used at pH 6, 7, 8, 9, and 10. The thermal and pH stabilities of Ls-Asn1 were examined by measuring the residual activity under standard assay conditions after heat or pH treatment. Heat treatment was carried out by incubating 100 μL of purified Ls-Asn1 (0.4 mg/mL) diluted tenfold with 1X PBS at 40 °C for 60 min (500 rpm). The pH treatment was carried out by mixing 100 μL of purified Ls-Asn1 (0.4 mg/mL) dissolved in 1 × PBS with 900 μL of 0.5 M GTA buffer (pH 7.0), and the mixture was kept in an ice box for 24 h. We measured the enzyme activity in the presence of various metal ions (final concentration: 1 mM). Ls-Asn1 (0.4 mg/mL, 100 μL) dissolved in 1 × PBS was mixed with 20 μL of 0.1 M NaCl, KCl, CaCl2, and FeCl2, and the mixture was subsequently kept in an ice bath for 60 min. The residual activity was subsequently measured under standard assay conditions.
Site-directed mutagenesis
The amino acid residues C196, C264, and C290 are characteristic of the primary structure of Ls-Asn1. To clarify the effects of these cysteine residues on enzyme activity, we constructed the mutated enzymes Ls-Asn1 C196S, C264S, C290S, C196S/C264S, C196S/C290S, C264S/C290S, and C196S/C264S/C290S-Ls-Asn1 by site-directed mutagenesis according to the methods of Takara Bio, Inc. (Kyoto, Japan). The primers used are listed in Table 1. In order to minimize the decrease in activity by the mutations, we chose firstly Ser as a substituent for a Cys residue. The O atom in the side chain of Ser which belongs to same Group 16 of the periodic table as the S atom in the side chain of Cys, expect to have similar chemical properties of S atom but less reactive than S atom. The activities of each mutated enzyme were measured under standard assay conditions. In addition, the basic enzymatic properties of the mutant enzyme were measured in the same way as those of Ls-Asn1.
Molecular mass and quaternary structure
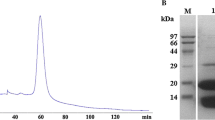
The molecular masses of Ls-Asn1 and its mutated enzymes were estimated by gel filtration according to the same method previously reported (Oikawa et al. 2022). Sodium dodecyl sulfate (SDS)-polyacrylamide electrophoresis was performed according to the methods of Laemmli (10%T) (Laemmli 1970).
Analysis of protein sequence alignment and phylogenetic tree analysis
Amino acid alignment analysis of Ls-Asn1 and related asparaginases and neighbor-joining phylogenetic tree analysis were performed using the ClustalW program and MAGA11 software (Tamura et al. 2021), respectively. For both analyses, the following sequences were used: E. coli AnsB (Accession No. NP_417432), E. coli AnsA (Accession No. NP_416281), Bacillus paralicheniformis (Accession No. WP_020451978), Aquabacterium sp. (Accession No. ANC70284), Pyrococcus horikoshii (Accession No. BAA29135), Vibrio cholerae (Accession No. WP_000101384), Pyrococcus furiosus (Accession No. AAL82171), Yersinia pestis (Accession No. WP_002211675), Campylobacter jejuni subsp. jejuni (Accession No. YP_002343501), Wolinella succinogenes (Accession No. CAE097909), Helicobacter pylori (Accession No. 2WLT_A), Dickeya chrysanthemi (Accession No. CAA31239), Pectobacterium atrosepticum (Accession No. AAP92666), Acinetobacter glutaminasificans (Accession No. P10172), Pseudomonas sp. (Accession No. 4PGA_A), Cavia porcellus (Accession No. 4R8K_A), Thermococcus kodakarensis (Accession No. BAD85845), Lacticaseibacillus casei (Accession No. BAN75184), Limosilactobacillus reuteri subsp. reuteri (Accession No. BAN75184), Anoxybacillus flavithermus (Accession No. ACJ34406).
Structural modeling and structural similarity analysis
A homodimer model of Ls-Asn1 was created using ColabFold:AlphaFold2 via MMseqs2 (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb, Mirdita et al. 2022) without a template. The structural similarity of the resultant model was compared to that of previously reported l-asparaginases ([PDBID: 7c8x], B. paralicheniformis; [PDBID: 6pab], E. coli AnsB; [PDBID: 6nxc], E. coli AnsA; [PDBID: 4r8l], C. porcellus; [PDBID: 1wls], P. horikoshii) using the Dali Server (Holm and Rosenstrom 2010).
Cell viability assay
Jurkat and HEK293 cells were seeded in 96-well plates at 5 × 103 cells/well and cultured at 37 °C. After overnight incubation, the cells were treated with Ls-Asn1, Ls-Asn1-C196S, and Ec-AnsB (0.02, 0.05, 0.2, 0.5, 2, 5, 10 U/mL) for 72 h, after which cell viability was assessed using the WST-8 reagent and the Cell Count Reagent SF (Nacalai Tesque, Kyoto, Japan) according to the manufacturer's protocol. Cell viability was expressed as a percentage relative to that of control cells not treated with Ls-Asn1, Ls-Asn1-C196S, or Ec-AnsB. Sigmoidal dose‒response curves were fitted using nonlinear regression analysis. The IC50 values for the enzymes were calculated using GraphPad Prism 6.0 (GraphPad Prism Software, Inc., San Diego, California).
Results
Expression and purification of Ls-Asn1 and its mutated enzymes
We succeeded in expressing and purifying Ls-Asn1 and its mutated enzymes C196S, C264S, C290S, C196S/C264S, C196S/C290S, C264S/C290S, and C196S/C264S/C290S-Ls-Asn1 homogeneously (Supplementary Fig. S1).
Comparison of the basic enzymatic properties of Ls-Asn1 with those of its mutated enzymes
The specific activities of the purified Ls-Asn1 and its mutated enzymes are summarized in Table 2. These results suggested that all cysteine residues of Ls-Asn1 were found to affect the catalytic activity of Ls-Asn1 to varying degrees and that the mutation of Cys196 did not affect the specific activity; however, the mutation of Cys264, even a single mutation, significantly decreased the specific activity. Cys264 was found to be an important amino acid residue for determining catalytic activity. Furthermore, C264S/C290S- and C196S/C264S/C290S-Ls-Asn1 almost completely lost their activity, suggesting that C290 cooperates with C264 to influence the catalytic activity of this enzyme. Further enzymatic properties were investigated for three single mutated enzymes (C196S, C264S, and C290S-Ls-Asn1) compared to the wild-type enzyme (Table 3). Ls-Asn1 and C196S-Ls-Asn1 exhibited maximum enzyme activity at 40 °C, but C264S and C290S-Ls-Asn1 exhibited maximum enzyme activity at 30 °C, and their specific activities dramatically decreased at 50 °C. Ls-Asn1 and C196S-Ls-Asn1 exhibited comparable enzymatic activity over a wide range of pH values (7 to 10), but C264S-Ls-Asn1 and C290S-Ls-Asn1 exhibited less activity at pH 6 to 7 than Ls-Asn1. After heat treatment at 35, 40, and 45 °C for 1 h, C196S-Ls-Asn1 showed higher stability than Ls-Asn1 (Table 3). In particular, C196S-Ls-Asn1 did not lose almost any activity at 35 and 40 degrees (Table 3). The half-life time of C196S-Ls-Asn1 at 45 °C is calculated to be 38.5 min based on the equation of y = 100e−0.018x (y: Relative activity (%); x: Heat treatment time at 45 °C; R2 = 0.90). After treatment at pH 7.0 for 24 h in an ice box, C196S-Ls-Asn1 showed higher stability than Ls-Asn1. In the case of Ls-Asn1, the enzyme activity decreased in the presence of NaCl, KCl, CaCl2, or FeCl2, but no such decrease in enzyme activity was observed for C196S-Ls-Asn1, and the activity increased markedly in the presence of CaCl2.
Substrate specificity of Ls-Asn1
Table 4 shows the substrate specificity of Ls-Asn1. Ls-Asn1 showed high specificity for l-Asn, and d-Asn, l-Gln, and d-Gln were not used as substrates (Table 4).
Quaternary structure of Ls-Asn1 and its mutated enzymes
As a result of gel filtration, Ls-Asn1 and C196S, C264S, C290S, C196S/C264S, C196S/C290S, C264S/C290S, and C196S/C264S/C290S-Ls-Asn1 were estimated to have molecular masses ranging from 74,000 to 80,000 (Table 3). Considering the calculated molecular mass of 35,514 Da, Ls-Asn1 and all other mutant enzymes were found to be homodimers. Based on the basic enzymatic properties, substrate specificity, and quaternary structures described above, we decided to carry out subsequent cell culture experiments on Ls-Asn1 and C196S-Ls-Asn1.
Analysis of protein sequence alignment and phylogenetic tree analysis
Amino acid alignment analysis (Fig. 1) showed that two catalytically important threonine residues (T12 and T86) are conserved in Ls-Asn1, similar to the other residues, and that the primary structure of Ls-Asn1 is quite similar to that of l-asparaginases from B. paralicheniformis (BlAsnase) and L. casei. Neighbor-joining phylogenetic tree analysis suggested that Ls-Asn1 is located on a novel branch that comprises BlAsnase and l-asparaginase from L. casei (Fig. 2).

Amino acid alignment Multiple alignments were created using ClustalW and BOXSHADE v3.31 C. Diamonds: catalytically important threonine residues (T12 and T86 in Ls-Asn1); circles: two putative motifs (P53, S54, P55, Y273, D274 and Y275 in Ls-Asn1); asterisks: three cysteine residues (C196, C264 and C290 in Ls-Asn1); squares: putative zinc binding sites (D97 and E264 in Ls-Asn1)

Neighbor-joining phylogenetic tree. The amino acids were aligned using the ClustalW program. Branches were labeled by bootstrap values from 10,000 replicates
Structural modeling and similarity
Based on the results of the quaternary structure analysis, a homodimer model of Ls-Asn1 was created ab initio using the ColabFold:AlphaFold2 tool in MMseqs2. Estimated mean pLDDT and pTM scores of the model were 95.5 and 0.947, respectively. Structural comparison of the resultant model with those of other l-asparaginases suggested that the overall structure of Ls-Asn1 is more similar to that of B. paralicheniformis l-asparaginase than to that of other welL-known type I and type II asparaginases (Fig. 3, Table 5). The amino acid architecture of the Ls-Asn1 active site was also more similar to that of B. paralicheniformis l-asparaginase (Fig. 4).

Structural comparison of l-asparaginases. Cartoons were drawn using PyMol 2.3.0 (Schrödinger LLC, New York, NY, USA). l-Asn, l-Asp, or citrate is shown in magenta and orange. A: Ls-Asn1 model (chains A (green) and B (cyan)) B: Bacillus paralicheniformis BlAsnase (chains A (green) and B (cyan), PDBID: 7c8x) C: Ec-AnsB (chains A (green) and B (cyan), PDBID: 6pab) D: Ec-AnsA (chains B (cyan) and D (green), PDBID: 6nxc) E: Cavia porcellus (chains A (green) and D (cyan), PDBID: 4r8l) F: Pyrococcus horikoshii (chains A (green) and B (cyan), PDBID: 1wls)

Structural comparison of active site residues. Stick images were drawn using PyMol 2.3.0 (Schrödinger LLC, New York, NY, USA). A: Ls-Asn1 model (chains A (green) and B (cyan))B: Bacillus paralicheniformis BlAsnase (chains A (green) and B (cyan), l-Asn (magenta) PDBID: 7c8x) C: Ec-AnsB (chains A (green) and B (cyan), l-Asp (magenta), PDBID: 6pab) D: Ec-AnsA (chains B (cyan) and D (green), Citrate (magenta) PDBID: 6nxc) E: Cavia porcellus (chains A (green) and D (cyan), l-Asp (magenta), PDBID: 4r8l) F: Pyrococcus horikoshii (chains A (green) and B (cyan), PDBID: 1wls)
Anti-leukemic activity of C196S-Ls-Asn1
To investigate the antileukemic activity of Ls-Asn1, C196S-Ls-Asn1, and Ec-AnsB, their cytotoxicities against Jurkat T-cell leukemia and immortalized nontumor HEK293 renal cell lines were measured using WST-8 reagents. Figure 5A shows that, compared with Ec-AnsB, Ls-Asn1 slightly reduced the viability of Jurkat cells at concentrations above 5 U/mL, but the effect was negligible. On the other hand, C196S-Ls-Asn1 (IC50 of 11.43 U/mL) was shown to decrease the viability of Jurkat cells in a concentration-dependent manner, similar to that of Ec-AnsB (IC50 of 9.69 U/mL). The increased cytotoxic activity of C196S-Ls-Asn1, compared to that of Ls-Asn1, is consistent with the stability of the enzyme in advanced DMEM/F12 used to culture Jurkat cells (Fig. 5). The viability of nontumor HEK293 cells was not affected by the concentration of any of the l-asparaginases used in this study (Fig. 5B). Thus, C196S-Ls-Asn1 effectively and selectively reduced the viability of Jurkat T-cell leukemia cells, suggesting that it exhibited antileukemic activity.

Effect of l-asparaginases on the viability of Jurkat cells and HEK293 cells Jurkat (A) or HEK293 (B) cells were cultured in the presence of C196S-Ls-Asn1 (orange, filled squares), Ls-Asn1 (gray, filled triangles), Ec-AsnB (blue, filled circles (0.02–10 U/mL) or PBS (black, open squares) as control for 72 h, after which cell viability was determined via the WST-8 assay. The data are presented as the mean ± SD of two independent experiments.
Discussion
We expressed and purified an asparaginase homolog (Ls-Asn1) found in the Latilactobacillus sakei LK-145 genome in Escherichia coli Rosetta (DE3) and characterized its basic enzymatic properties. This is the first report of asparaginase from Latilactobacillus sakei. In addition, we focused on three Cys residues characteristically present in Ls-Asn1, comprehensively produced single-, double-, and triple-mutated enzymes, and compared their enzymatic properties. We found significant differences in enzymatic activity between these mutated Ls-Asn1enzymes. The functions of these Cys residues are explained below based on the primary structure and molecular model of Ls-Asn1.
Although the three Cys residues are unlikely to be catalytic residues according to the present study, they still have several interesting features. The C196 residue of Ls-Asn1 was predicted to be located on the surface of the homodimeric structure (Fig. 3 and Fig. S2); therefore, the side chain thiol group of the residue might be sensitive to oxidative agents. A C196S single mutant showed no significant decrease in activity or stability, suggesting that the mutant is promising for various applications. Unlike the C196S mutation, the C264S and C290S mutations affect enzyme properties, including activity, stability, substrate specificity, and sensitivity to additives. Among the three Cys residues, only the C264 residue is partially conserved in Lactobacillaceae and type I enzymes from bacterial sources (Fig. 1). The C265 residue of type I l-asparaginase from A. flavithermus, which corresponds to C264 of Ls-Asn1, raises the possibility that this residue contributes to the allosteric regulation of the bacterial type I enzyme (Maqsood et al. 2020); however, Ls-Asn1 is not a member of the phylogenetic branch of type I enzymes. As shown in Fig. 3 and Supplemental Fig. S2, the C264 residue is located close to the active site, the side chain is positioned on the surface of the tunne l-like hole observed only in Ls-Asn1 and BlAsnase, and the loop containing the C290 residue lies nearby the other C264 residue-containing loop. In addition, calculated distance between Cβ atoms of C264 and C290 in the model structure was over 10 Å, suggesting that there is no disulfide bond formation between them. This structural feature may be a cause of changes in various enzyme properties when C264 and/or C290 are mutated.
The present phylogenetic analysis suggested the possibility that a novel branch diverged from type II enzymes in the l-asparaginase phylogenetic tree. The novel branch consists of Ls-Asn1, BlAsnase and L. casei l-asparaginase and appears to diverge earlier than a subbranch of l-asparaginase from Aquabacterium sp. previously reported as a novel type II enzyme (Sun et al. 2016). The structure of BlAsnase, which was identified as a type I enzyme in the original paper, is more similar to that of type II enzymes than to that of type I enzymes (Ran et al. 2020); moreover, Lubkowski and Wlodawer noted in their attentive review article (Lubkowski and Wlodawer 2021) that the available information about this enzyme was insufficient for proper comparison. The L. casei enzyme, whose primary structure is the closest to that of Ls-Asn1 among the reported enzymes, has been described as a type II enzyme (Aishwarya et al. 2019). Here, we note that these three l-asparaginases, Ls-Asn1, BlAsnase and the L. casei enzyme, share some unique features.
The quaternary structures of Ls-Asn1 (this study) and BlAsnase (Ran et al. 2020) are homodimers, similar to archaeal l-asparaginases (Guo et al. 2017; Yao et al. 2005). Structural modeling and comparison showed that Ls-Asn1 structure is predicted to be close to BlAsnase, which means that the structure of Ls-Asn1 is more similar to that of type II enzymes than type I enzymes (Fig. 3, Table 5), and that active site Tyr residue (Y273 of Ls-Asn1) is contributed by the adjacent protomer similar to BlAsnase (Y276), Ec-AnsA (Y283) and l-asparaginases from C. porcellus (Y308) and P. horikoshii (Y273) (Figs. 1 and 4). In addition to the active site Tyr residue corresponding to Tyr47 of Ec-AnsB, another Tyr residue in the active site (Y275 of Ls-Asn1 and Y278 of BlAsnase) is conserved only in the Ls-Asn1, BlAsnase and L. casei enzymes (YXY-motif) (Figs. 1 and 4). Considering the structural similarities, the reaction mechanism of Ls-Asn1 may be direct displacement (Schalk et al. 2016), as proposed by Ran et al. for BlAsnase (Ran et al. 2020).
Ls-Asn1 showed no activity toward l-Gln, similar to BlAsnase (Ran et al. 2020). A low affinity for l-Gln is one of the features of the bacterial type II l-asparaginase (Castro et al. 2021). Although further detailed kinetic analysis will be needed, the substrate specificity of Ls-Asn1 and BlAsnase might be similar to that of type II enzymes. Docking simulation analysis showed that l-Asn, l-Gln and l-Asp could bind in a similar manner with calculated binding affinities (kcal/mol) of -4.2, -3.9 and -3.6, respectively; however, the position of side chain amide group of l-Gln seems to differ from that of l-Asn (Supplemental Fig. S3). The substrate α-carboxylic acid is surrounded by the P53, S54 and P55 residues in the active site of Ls-Asn1. The corresponding residues are P55, S56 and M57 in BlAsnase and P54, S55 and P56 in the L. casei enzyme. Some changes in catalytic efficiency have been reported when the mutation was introduced into the corresponding region of P. furiosus archaeal l-asparaginase (Bansal et al. 2012), H. pylori type I l-asparaginase (Maggi et al. 2015) or E. coli type II l-asparaginase (AnsB) (Derst et al. 2000). The rigidity of the Pro residue may affect the enzymatic properties. The Pro residue in this region is observed only in the Ls-Asn1, BlAsnase and L. casei enzymes (Pro-motif) (Fig. 1 and 4).
The zinc binding site of type II enzymes has been shown to be composed of two amino acid residues, Asp (or Asn or His), and His (Borek et al. 2014). In Ls-Asn1, the corresponding Asp residue (D97) is conserved, while another His residue is not conserved like in type I enzymes (Fig. 1). The conservation pattern of this site in BlAsnase and the L. casei enzyme is consistent with that of Ls-Asn1 (Fig. 1).
Ls-Asn1 and BlAsnase seem to combine characteristics of both type I enzymes from bacterial, archaeal and eukaryotic sources and type II enzymes and have additional unique features that are not conserved among welL-characterized type I and type II enzymes. Basic Local Alignment Search Tool (BLAST) analysis revealed that putative l-asparaginase proteins that highly conserved both novel motifs (Pro-motif and YXY-motif) are widely distributed in bacteria and in some archaea and eukaryotes (Fig. S4). We propose to classify the Ls-Asn1, BlAsnase and L. casei enzymes into a novel group of l-asparaginases as type IIb enzymes.
In this study, structural model was created using AlphaFold2. Recently, it was suggested that structure model with very high (> 90) mean pLDDT score of pocket-associated residues can be applied for pocket detection (Akdel et al. 2022). Ls-Asn1 model presented here was with very high mean pLDDT (overall: 95.5, active site residues shown in Fig. 4A: 95.6), suggesting that at least the position and size of substrate binding pocket might be reliable. C196S-Ls-Asn1 model was also created and supplied to docking simulation analysis (Fig. S5). Mean pLDDT of the C196S model was also very high (overall: 94.2, active site residues shown in Fig. S5, B: 94.5), and the simulation analysis suggested that substrate binding manner (Fig. S5, C) and calculated binding affinity for l-Asn (−3.9 kcal/mol) are similar to those of Ls-Asn1. These predictions are good correlation with experimental results (Table 2,3 and 4, Fig. 5). However, experimental X-ray crystallographic structural analysis of Ls-Asn1 is needed for more detailed and credible analysis about classification of l-asparaginases and is our future task.
Figure 5 shows the growth inhibitory effect of the l-asparaginase mutant from L. sakei LK-145 on Jurkat cells, a human T-cell acute lymphoblastic leukemia cell line. Ls-Asn1 was inactivated in the cell culture medium and had minimal antitumor effects on Jurkat cells. However, C196S-Ls-Asn1, in which the Cys residue was replaced with a Ser residue, was stable and reduced Jurkat cell viability in a dose-dependent mannSer, comparable to the effect of clinically used E. coli-derived l-asparaginase. These results suggest that the introduction of mutation at the position of C196 in very effective to stabilize Ls-Asn1. We used advanced DMEM/F12 and DMEM as the cell culture medium for the experiments. Advanced DMEM/F12 medium is generally used in experiments on the inhibitory effect of asparaginase on leukemia cell growth and contain 0.05 mM of l-Asn as similar concentration of the human serum. As described one paragraph before, the binding affinity for l-Asn of both Ls-Asn1 and C196S-Ls-Asn1 is similar based on reliable in silico analysis. Accordingly, the affinity of l-Asn to Ls-Asn1 and C196S-Ls-Asn1 in the culture media is probably not so different one another and will not affect the enzyme activity. But after preparation of various mutated enzymes of Ls-Asn1 at a position of C196 in addition to C196S-Ls-Asn1, we are planning to measure Km value of them to compare each other to clarify the apparent binding affinity to l-Asn and will clarify the effect of l-Asn concentration on the enzyme activity in vitro. One of the reasons why C196S-Ls-Asn1 shows a higher growth inhibitory effect on Jurkat cells than Ls-Asn1 is thought to be that Ls-Asn1 showed high thermostability at 35 and 40 °C corresponding to human body temperature (Table 3).
The clinical effect of l-asparaginase is related to the depletion of serum l-asparagine, an amino acid necessary for leukemic cell growth (Dübbers et al. 2000; Hawkins et al. 2004). Normal cells can synthesize l-asparagine and are less affected by rapid l-asparagine depletion. C196S-Ls-Asn1 was not toxic to nontumor HEK293 cells even at high concentrations. These results suggest that C196S-Ls-Asn1 may exert selective cytotoxicity only against leukemia cells, which, when combined with its high substrate specificity, may make it an ideal candidate for cancer chemotherapy against leukemia.
We are currently studying the possibility of developing C196S-Ls-Asn1 and other C196X-mutated Ls-Asn1 as a therapeutic agent for cancers via various biological assay systems.
References
Aishwarya SS, Selvarajan E, Iyappan S, Rajnish KN (2019) Recombinant l-asparaginase II from Lactobacillus casei subsp. casei ATCC 393 and its anticancer activity. Ind J.microbiol 59:313–320
Akdel M, Pires DE, Pardo EP, Jänes J, Zalevsky AO, Mészáros B, Bryant P, Good LL, Laskowski RA, Pozzati G, Shenoy A, Zhu W, Kundrotas P, Serra VR, Rodrigues CH, Dunham AS, Burke D, Borkakoti N, Velankar S, Frost A, Basquin J, Lindorff-Larsen K, Bateman A, Kajava AV, Valencia A, Ovchinnikov S, Durairaj J, Ascher DB, Thornton JM, Davey NE, Stein A, Elofsson A, Croll TI, Beltrao P (2022) A structural biology community assessment of alphafold2 applications. Nat Struct Mol Biol 29(11):1056–1067
Bansal S, Srivastava A, Mukherjee G, Pandey R, Verma AK, Mishra P, Kundu B (2012) Hyperthermophilic asparaginase mutants with enhanced substrate affinity and antineoplastic activity: structural insights on their mechanism of action. FASEB J 26(3):1161–1171
Borek D, Kozak M, Pei J, Jaskolski M (2014) Crystal structure of active site mutant of antileukemic l-asparaginase reveals conserved zinc-binding site. FEBS J 281(18):4097–4111
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72(248):254
Castro D, Marques ASC, Almeida MR, de Paiva GB, Bento HB, Pedrolli DB, Freire MG, Tavares APM, Santos-Ebinuma VC (2021) l-asparaginase production review: bioprocess design and biochemical characteristics. Appl Microbiol Biotechnol 105:4515–4534
Derst C, Henseling J, Röhm KH (2000) Engineering the substrate specificity of Escherichia coli asparaginase II. selective reduction of glutaminase activity by amino acid replacements at position 248. Protein Sci 9(10):2009–2017
Dübbers A, Würthwein G, Müller HJ, Schulze-Westhoff P, Winkelhorst M, Kurzknabe E, Lanvers C, Pieters R, Kaspers GJ, Creutzig U, Ritter J, Boos J (2000) Asparagine synthetase activity in paediatric acute leukaemias: AML-M5 subtype shows lowest activity. Br J Haematol 109:427–429. https://doi.org/10.1046/j.1365-2141.2000.02015.x
Duval M, Suciu S, Ferster A, Rialland X, Nelken B, Lutz P, Benoit Y, Robert A, Manel AM, Vilmer E, Otten J, Phillipe N (2002) Comparison of Escherichia coli- asparaginase with Erwinia-asparaginase in the treatment of childhood lymphoid malignancies: results of a randomized european organization for research and treatment of cancer—children’s leukemia group phase 3 trials. Blood 99:2734–2739
Gaufichon L, Rothstein SJ, Suzuki A (2016) Asparagine Metabolic Pathways in Arabidopsis. Plant Cell Physiol 57:675–689. https://doi.org/10.1093/pcp/pcv184
Guo J, Coker AR, Wood SP, Cooper JB, Chohan SM, Rashid N, Akhtar M (2017) Structure and function of the thermostable l-asparaginase from Thermococcus kodakarensis. Acta Crystallogr Sect d Struc Biol 73(11):889–895
Hawkins DS, Park JR, Thomson BG, Holcenberg FJL, JS, Panosyan EH, Avramis VI, (2004) Asparaginase pharmacokinetics after intensive polyethylene glycoL-conjugated l-asparaginase therapy for children with relapsed acute lymphoblastic leukemia. Clin Cancer Res 10:5335–5341. https://doi.org/10.1158/1078-0432.CCR-04-0222
Holm L, Rosenstrom P (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res 38:W545-549
Kato S, Oikawa T (2017) Genome sequence of Lactobacillus sakei LK-145 isolated from a japanese sake cellar as a high producer of d-amino acids. Genome Announc 5:e00656-e717. https://doi.org/10.1128/genomeA.00656-17
Kumar K, Verma N (2012) The various sources and application of l-asparaginase. Asian J Biochem Pharm Res 3:197–205
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. https://doi.org/10.1038/227680a0
Lubkowski J, Wlodawer A (2021) Structural and biochemical properties of l-asparaginase. FEBS J 288(14):4183–4209
Maggi M, Chiarelli LR, Valentini G, Scotti C (2015) Engineering of Helicobacter pylori l-asparaginase: characterization of two functionally distinct groups of mutants. PLoS ONE 10(2):e0117025
Maqsood B, Basit A, Khurshid M, Bashir Q (2020) Characterization of a thermostable, allosteric l-asparaginase from Anoxybacillus flavithermus. Int J Biol Macromol 152:584–592
Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215
Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M (2022) ColabFold: making protein folding accessible to all. Nat Methods 19:679–682
Oikawa T, Okajima K, Yamanaka K, Kato S (2022) First enzymological characterization of selenocysteine β-lyase from a lactic acid bacterium, Leuconostoc mesenteroides. Amino Acids 54:787–798. https://doi.org/10.1007/s00726-022-03133-9
Pedreschi F, Kaack K, Granby K (2008) The effect of asparaginase on acrylamide formation in French fries. Food Chem 109:386–392
Pokrovsky VS, Kazanov MD, Dyakov IN, Pokrovskaya MV, Aleksandrova SS (2016) Comparative immunogenicity and structural analysis of epitopes of different bacterial l-asparaginases. BMC Cancer. https://doi.org/10.1186/s12885-016-2125-4
Ran T, Jiao L, Wang W, Chen J, Chi H, Lu Z, Zhang C, Xu D, Lu F (2020) Structures of l-asparaginase from Bacillus licheniformis reveal an essential residue for its substrate stereoselectivity. J Agric Food Chem 69(1):223–231
Salzer WL, Asselin BL, Plourde PV, Corn T, Hunger SP (2014) Development of asparaginase Erwinia chrysanthemi for the treatment of acute lymphoblastic leukemia. Ann N Y Acad Sci 1329:81–92
Schalk AM, Antansijevic A, Caffrey M, Lavie A (2016) Experimental data in support of a direct displacement mechanism for type I/II l-asparaginases. J Biol Chem 291(10):5088–5100
Sun Z, Qin R, Li D, Ji K, Wang T, Cui Z, Huang Y (2016) A novel bacterial type II l-asparaginase and evaluation of its enzymatic acrylamide reduction in French fries. Int J Biol Macromol 92:232–239
Tamura K, Stecher G, Kumar S (2021) MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol 38(7):3022–3027
Yao M, Yasutake Y, Morita H, Tanaka I (2005) Structure of the type i l-asparaginase from the hyperthermophilic archaeon pyrococcus horikoshii at 2 16 a resolution. Acta Crystallogr Sec D Biol Crystallogr 61(3):294–330
Acknowledgements
We thank Ms. Saho Kambe for the construction of pET21-Ls-asn1/Escherichia coli Rosetta (DE3). We thank Ms. Mai Sakata for her fundamental experiments on Ls-Asn1. We thank Ms. Miki Hatanaka for the construction of pET21-C196S, C264S, C290S, C196S/C264S, C196S/C290S, C264S/C290S, and C196S/C264S/C290S-Ls-asn1.
Funding
The authors have not disclosed any funding.
Author information
Authors and Affiliations
Contributions
T. O. designed and supervised the studies. S. K., Y. M., and T. O. wrote the manuscript. K. T. helped the preparation of manuscript partly. K. T., Y. M., and M. K. carried out the experiments. S. K. performed the primary structure, phylogenetic, and modeling analyses. S. K. carried out the molecular docking simulation. K. Y. advised the experiments part.
The authors have not disclosed any funding.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This manuscript does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by PANKAJ BHATT.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kato, S., Tamura, K., Masuda, Y. et al. A novel type IIb l-asparaginase from Latilactobacillus sakei LK-145: characterization and application. Arch Microbiol 206, 266 (2024). https://doi.org/10.1007/s00203-024-03979-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00203-024-03979-5