
Abstract
This article reports the results of quantitative intra- and intergeneric taxonomic relationships among Micrococcaceae strains and a novel endophytic bacterium (SG) isolated from a suspension culture of Arabidopsis thaliana (L.) Heynh in our laboratory. The known strain Rothia sp. ND6WE1A was used as a reference one for SG. Whole-genome sequencing and phylogenetic analysis were based on the 16S rRNA test. Quantitative analysis for the nucleotide identity (ANI) and calculation of evolutionary distances were based on the identified amino acids (AAI) test indicating the generic assignment of the reference strain within and between the identified monophyletic groups of Micrococcaceae. The amino acid data structure of Rothia sp. ND6WE1A was compared against the UniProt database (250 million records) of close lineage of Micrococcaceae, including other Rothia spp. These data presented unique and evolutionary amino acid alignments, eventually expected in the new SG isolate as well. The metagenomic entries of the respective genome and proteome, characterized at the genus and species levels, could be considered for evolutionary taxonomic reclassification of the isolated and the reference strain (SG + Rothia sp. ND6WE1A). Therefore, our results warrant further investigations on the isolated SG strain.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rothia is a genus of gram-positive, aerobic, coccoid or bacillary, nonmotile, non-spore-forming bacteria of the family Micrococcaceae, phylum Actinobacteria (Austin 2015). To date, 15 Rothia species have been identified. As part of the normal microflora of the gastrointestinal tract (especially the oral cavity and stomach) of humans and animals, it can also cause gastric atrophy and intestinal metaplasia and induce opportunistic infections of the upper respiratory tract in immunocompromised people (Nardone and Compare 2015; Odeberg et al. 2023). Various Rothia have been recovered from soil, water sources, benthos, rocks, atmosphere, and other sources (Austin 2015).
Endophytic Rothia strains of different species have been isolated from the rhizosphere and tissues of many plants, viz. Sphagnum magellanicum (Opelt et al. 2007), Dysophylla stellata (Xiong et al. 2013), Hedysarum perrauderianum (Torche et al. 2014), H. naudinianum (Torche et al. 2014), Musa acuminate (Sekhar and Thomas 2015), Oryza sativa (Evangelista et al. 2017), Seidlitzia rosmarinus (Shurigin et al. 2020), Camellia sinensis (Borah and Thakur 2020), Alnus glutinosa (Davis et al. 2020), A. incana (Mercurio et al. 2022), Zea mays (Elbahnasawy et al. 2021; Pisarska and Pietr 2015), Vaccinium myrtillus (Mažeikienė et al. 2021), Santalum album (Tuikhar et al. 2022), Miscanthus floridulus (Xiao et al. 2023), Arabidopsis thaliana (Sokolov et al. 2021), Vítis vinífera (Vaghari Souran et al. 2023), and Beta vulgaris (Petrović et al. 2024). Rothia endophytes are inhibitory to several pathogenic fungi, bacteria, parasitic nematodes, and insect larvae, and they can be used as biofertilizers (Asadu et al. 2020; Bano and Muqarab 2017; da Silva et al. 2018; Nuaima 2022). Some Actinobacteria, e.g. Rothia mucilaginosa, have been reported to have a nifH gene sequence representing their ability to fix atmospheric nitrogen, which is yet to be demonstrated (Gtari et al. 2012). Additionally, Rothia endophytica can be utilized as biofertilizer producing no adverse effect, has shown biocompatibility during cocultivation with crops by generating enough indole-3-acetic acid, tryptophan, and biomass, and demonstrates phosphate solubilizing ability (Asyakina et al. 2023; Sokolova et al. 2024).
Many Rothia genomes isolated from humans, animals, and the environment have biosynthetic gene clusters that supposedly produce antibiotic nonribosomal peptides, siderophores, and other secondary metabolites that modulate microbe–microbe (quorum sensing) and, potentially, microbe–host interactions (de Oliveira et al. 2022). Certain strains produce natural substances with antimicrobial activity, which are planned to be used to treat human and animal infections (Fatahi-Bafghi 2021). In particular, peptidoglycan hydrolases produced by Rothia dentocariosa can be used as highly specific therapeutic agents against nasal pathogens (Stubbendieck et al. 2023). Finally, a number of Rothia soil bacteria can degrade xenobiotics, specifically phthalate esters and aromatic hydrocarbons (Jia et al. 2022; Yastrebova and Plotnikova 2020).
In a suspension culture of Arabidopsis thaliana (L.) Heynh (All-Russian Collection of Higher Plant Cell Cultures, Timiryazev Institute of Plant Physiology of the Russian Academy of Sciences, Moscow, Russia), a bacterial microflora was found that did not inhibit plant growth (Sokolov et al. 2021). A number of microbiological characteristics of this strain are presented in Methods. DNA sequencing of the isolate’s 16S rRNA gene sequence (GenBank accession number OQ702765.1) was done by the Research and Production Company SYNTOL (2023). The results obtained by the 16S rRNA test (Sokolov et al. 2021; Shchyogolev et al. 2023) showed that this isolate (hereafter Isolate SG) is taxonomically close to members of the genus Rothia.
Analysis of the systematic position of Isolate SG (Sokolov et al. 2021) and phylogenetic studies of Rothia members in the 16S rRNA-based test (de Oliveira et al. 2022; Elbahnasawy et al. 2021; Fan et al. 2002; Fatahi-Bafghi 2021; Ko et al. 2009; Stubbendieck et al. 2023; Tuikhar et al. 2022; Xiong et al. 2013; Yastrebova and Plotnikova 2020) have shown their taxonomic proximity to other Micrococcaceae, including Kocuria, Arthrobacter, Micrococcus, and other genera.
This article reports the results for the quantitative intra- and intergeneric taxonomic relationships between the Micrococcaceae strains closely related to Isolate SG (Sokolov et al. 2021). The historically given assignments (names) of the strains were validated by using (1) the known results from whole-genome sequencing of the strains’ DNA and (2) a set of five whole-genome phylogenetic tests.
Methods
Microbiological characteristics of Isolate SG
Isolate SG was recovered from a suspension culture of Ar. thaliana (L.) Heynh (Sokolov et al. 2021) and was deposited in the Collection of Rhizosphere Microorganisms (Collection of Rhizosphere Microorganisms 2023), Institute of Biochemistry and Physiology of Plants and Microorganisms, Russian Academy of Sciences (IBPPM RAS) under accession number IBPPM 684. Bacteria were grown on tryptic soy broth or on tryptic soy agar (Becton Dickinson, NJ, USA).
Morphologically, Isolate SG is a gram-positive, facultatively anaerobic, coccoid bacterium that is non-acid-resistant and is shaped closely to a sphere (diameter, about 1 µm). The optimal growth temperature is 30–37 °C. When growing in liquid tryptic soy broth, the cells spread by gliding motility. Incubation on tryptic soy agar at 30 °C gives rise to creamy, rounded colonies with an even edge and a smooth surface. The cells utilize sucrose and lactose as their sole sources of carbon and energy; they are sensitive to rifamycin, tetracycline, and cefoxitin and are resistant to laevomycetin (chloramphenicol), penicillin, and amoxycillin.
16S rRNA test
Isolate SG was taxonomically identified by its 16S rRNA sequence. The sequence was obtained by the Research and Production Company SYNTOL (SYNTOL 2023) by polymerase chain reaction with a set of universal primers, by using a cell suspension of Isolate SG provided by us. Bacteria were isolated from a 10-day-old suspension culture of Ar. thaliana (L.) Heynh by fractional centrifugation. The first centrifugation of the Arabidopsis cell suspension was done with a Jouan BR4i centrifuge (Jouan, France) at 300 g for 30 min. The supernatant liquid was passed through a fine-pore paper filter to remove plant cell debris and was centrifuged again with a 5810R centrifuge (Eppendorf, USA) at 15,000g for 30 min. The bacterial cell sediment was resuspended in phosphate-buffered saline. The obtained 16S rRNA gene sequence of Isolate SG (GenBank accession number OQ702765) covers V2–V8 hypervariable regions and can be used for genus and species identification of the strain (Shchyogolev et al. 2023).
Sequence alignment of the 16S rRNA test
The results generated by the 16S rRNA-based tool (Search EzBioCloud Database 2023; Table 1) with the 16S rRNA gene sequence of Isolate SG were used as the initial sets of strains, 16S rRNA gene sequences, and genomes. A 16S rRNA phylogenetic tree was constructed with the “Advanced” option of the NGPhylogeny web service (Lemoine et al. 2019; NGPhylogeny.fr 2023) and with the FastME 2.0 program (FastME 2.02023). This method is distance based (Lefort et al. 2015) and was selected to facilitate comparison of its results with those of the ANI and AAI tests. The 16S rRNA genes of Isolate SG and of the strains listed in Table 1 were subjected to multiple sequence alignment (MSA). The MAFFT MSA tool used is part of the NGPhylogeny toolkit, can be combined with FastME, PhyML, and other tools, and is commonly used in phylogenetic studies.
Evolutionary distance matrices of average nucleotide and amino acid identities (ANI and AAI)
The average nucleotide identity (ANI) and average amino acid identity (AAI) matrices (ANI/AAI-Matrix 2023; Goris et al. 2007; Jain et al. 2018; Rodriguez-R and Konstantinidis 2014) and their corresponding evolutionary distance matrices were used to obtain quantitative data on the belonging of strains to a particular taxonomic category on the basis of whole-genome sequencing data. The matrices were generated by linear transformation of the ANI/AAI values. References to the genomes of 20 strains marked with asterisks in Table 1 are given in the output data of the program (Search EzBioCloud Database 2023). Additionally, when this study was in progress, only six more references to the genomes of strains of the species listed in Table 1 and marked with a double asterisk could be found on the website (Genome Information by Organism 2023). Thus, it became possible to take into consideration 26 whole genomes [references for 26 of the 33 strains in Table 1 (column 5)] across the range of I values from Table 1.
Tests based on ANI and AAI form the basis of the Microbial Genomes Atlas (MiGA) genomic and metagenomic data management and processing system (Rodriguez-R et al. 2020) for the phylogenetic classification and cataloging of microbial genomes and analysis of their gene content. In this study, the TypeMat version (New query datasets 2023) was used, which determines the taxonomic position, novelty rank, and content of the query genome and finds the closest genomes of the type strains of officially named species from the TypeMat specialized database (Rodriguez-R et al. 2020; 20,809 reference genomes as of summer 2023).
In the output generated by the programs (ANI/AAI-Matrix 2023), the phylogenetic trees built from evolutionary distance matrices by the BioNJ method (and some other methods) do not include data to characterize branch significance. In MSA-using taxonomic studies, this function is fulfilled by bootstrap analysis (Lemoine et al. 2018). This gap was addressed by applying the rate of elementary quartets (RΕQ) program (REQ 2023), which is designed to estimate branch significance for phylogenetic trees based on distance matrices. The closer to 100 is the quantitative value in the RΕQ output (Guénoche and Garreta 2000), the more fully the corresponding branch is supported by the pairwise evolutionary distances.
The systematic position of Isolate SG at the whole-genome level with respect to the entries from the UniProt protein structure database (UniProtKB 2023) (250 million records) was evaluated with the AAI-profiler program (AAI-profiler 2023; Medlar et al. 2018).
GTDB-based taxonomic classification
The taxonomic position of Isolate SG was also determined on the basis of the phylogenetic system described by Parks et al. (2017, 2018, 2020). This system is aimed at constructing a Genome Taxonomy Database (GTDB; Genome Taxonomy Database 2023) by using sets of 120 (bacteria) or 122 (archaea) concatenated amino acid sequences of ubiquitous single-copy proteins as phylogenetic markers. The online version of the GTDB-Tk taxonomic classification toolkit (Chaumeil et al. 2022; GTDB-Tk Classify-v1.6.0 2023) was used.
Phylogenetic trees
Phylogenetic structures with relatively small numbers of operational taxonomic units (OTUs) (tens and hundreds) were visualized and analyzed with the MEGA11 program (MEGA 2023). For trees with hundreds of thousands of OTUs, the Archaeopteryx program (Archaeopteryx 2023) was used. Table S1 (Supplementary Material) summarizes the phylogenetic tests used, with quantitative criteria for taxon demarcation.
Results and discussion
16S rRNA-based test
Table 1 summarizes the data obtained by the 16S rRNA-based tool (Search EzBioCloud Database 2023) for 33 type (T) strains of the genera Rothia, Kocuria, and Arthrobacter in the “Valid names only” category. On the basis of pairwise alignment of the 16S rRNA gene sequences, these strains were identified as related to Isolate SG, with values of pairwise genetic sequence identity I > 95%. Following the commonly accepted convention and as stated on the web page (Understanding Results 2023), I values of 95–98.6% correspond to strains belonging to the same genus and those of 92–95% correspond to strains belonging to the same family. According to Yarza et al. (2014), the taxonomic threshold for genera is I > 94.5% and that for families is I > 86.5%. For species, the condition I > 98.6% is now accepted (Kim et al. 2014; Stackebrandt and Ebers 2006), in contrast to the previously used I > 97% (Tindall et al. 2010).
The I value (Table 1, line 2, column 6, red color) indicates that Isolate SG and R. amarae strain JCM 11375 belong to the same species. The > 95% pairwise I values, obtained for Isolate SG with all strains (Table 1, column 6), are the basis for assigning Isolate SG to the same genus with each of the indicated 33 type strains (16S rRNA-based test). According to the traditional taxonomical classification trends, these strains have been assigned by their authors as members of three genera: Rothia, Kocuria, and Arthrobacter. These conventional illustrations of conducting traditional prokaryotic classifications essentially need further revision on the basis of new emerging methods. These clarifications must be identified on the basis of phylogenetic studies of strains by using whole genome data (Genome Taxonomy Database 2023; Parks et al. 2017, 2018, 2020).
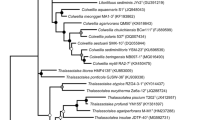
Figure 1 shows the phylogram of 16S rRNA gene sequences obtained with FastME 2.0 (FastME 2.02023) for the strains listed in Table 1. The numbers near the nodes indicate statistical support for the branches (absolute units) in 1000 bootstrapping cycles (Lemoine et al. 2018). The red dots mark the nodes of four clusters (monophyletic groups a–d) comprising sets of 5 to 12 Rothia, Kocuria, and Arthrobacter strains. As it follows from the results of determination of the percent identity matrix (Fig. S1, Supplementary Material), the pairwise values of I between strains within these monophyletic groups mostly satisfy the condition I > 95%. This indicates that by the criteria used (Kim et al. 2014; Stackebrandt and Ebers 2006; Understanding Results 2023; Yarza et al. 2014), these strains belong to the same genus according to 16S rRNA technology. Meanwhile, between clusters, I values generally satisfy the 87–92% < I < 95% condition, which demarcates genera within a family. However, both within and outside the clusters, there are a notable number of deviations of I values from these quantitative criteria of taxon demarcation. This fact illustrates their conventionality as averaged characteristics within fairly broad distributions of the corresponding statistical data with 16S rRNA sequences (Luo et al. 2014).

Phylogram (FastME) of the 16S rRNA gene sequences of the Rothia, Kocuria, and Arthrobacter type strains related to Isolate SG (Table 1). The branch length reflects the FastME distance. The numbers near the nodes indicate the bootstrapping results
Whole-genome GTDB, ANI, and AAI tests
More unambiguous taxonomic estimates can be expected from the use of the ANI and AAI matrices on the basis of whole-genome DNA sequencing of the strains (ANI/AAI-Matrix 2023; Goris et al. 2007; Jain et al. 2018; Rodriguez-R and Konstantinidis 2014). In Table 1, GCA_007666515.1 was initially specified as the genome corresponding to R. amarae strain JCM 11375T, which is the most closely related to Isolate SG in the 16S rRNA test. However, the GCA_007666515.1 genome is described in the NCBI database record as an “anomalous assembly” owing to the “unverified source organism.” We examined it by using the GTDB-Tk software package (GTDB-Tk Classify-v1.6.0 2023) to determine its most probable taxonomic classification and to clarify the species identity of the strain under study on the basis of ANI. The resulting taxonomic classification of this genome was as follows: domain, Bacteria; phylum, Actinobacteriota; class, Actinomycetia; order, Actinomycetales; family, Micrococcaceae; genus, Rothia; and species, Rothia sp001683935 (reference genome ID), when assigning to it the taxonomy of the reference genome GCF_001683935.1 of Rothia sp. ND6WE1A. The resulting pairwise value of ANI = 98.87% and the value of AF = 96% (the percentage of orthologous regions) indicate high intraspecies relatedness between R. amarae JCM 11375T (GCA_007666515.1) and Rothia sp. ND6WE1A (GCF_001683935.1). The program output contains the item “note,” which states that “topological placement and ANI have congruent species assignments.”
In view of the results given just above, the GCF_001683935.1 genome [the reference genome for GCA_007666515.1 in the GTDB (Genome Taxonomy Database 2023)] was accepted in our laboratory as a verified reference genome for our new isolate SG. Hence, the GTDB test was the subsequent whole-genome test method to determine the differences, in contrast to GCA_007666515.1 (the reference genome for SG in the 16S rRNA test).
Like the aforementioned 16S rRNA-, ANI-, and AAI-based tests, the GTDB technology remains within the paradigm of vertically inherited genotypic traits of prokaryotes, because it uses the markers from the core part of the pangenome without consideration of the effects of horizontal gene transfer (HGT) in its accessory and strain-specific parts (Koonin 2012). This probably explains, in particular, the need to verify prokaryote classification and nomenclature with the corresponding classification changes, which turned out relevant for about 60% of the GTDB entries (Genome Taxonomy Database 2023) analyzed in Parks et al. (2018).
Figure 2 shows the phylogram of the ANI of the 26 strains being considered. The phylogram is presented in the output data of the software package (ANI/AAI-Matrix 2023) and was constructed by the BioNJ method by using a matrix of ANI values. The red dots mark the nodes of five monophyletic groups (a–e) comprising sets of three to seven Rothia, Kocuria, and Arthrobacter strains. The results reflected in the matrix of pairwise ANI values (Fig. S2, Supplementary Material) show that there is no significant nucleotide identity between the genome of Rothia sp. ND6WE1A (the reference strain for Isolate SG) and the genomes of a number of strains. This explains the greatest evolutionary distance for Rothia sp. ND6WE1A among all the 26 strains taken into consideration (Fig. 2).

There are ambiguous taxonomic % values (Fig S1–S2, Supplementary Material) in the 16S rRNA test (based on sequence similarity) and ANI matrix pairwise values (based on the evolutionary distance) at the genus and species levels. ANI values ≤ 80% were considered within and between the monophyletic groups in Fig. S2. This is associated with the limited sensitivity of the ANI-based test at ANI values ≤ 80% (Rodriguez-R and Konstantinidis 2014). For more reliable quantitative demarcation of strains at the family level, the developers of the resource (ANI/AAI-Matrix 2023) recommend the use of the AAI-based test.
It is recommended that the ANI/AAI-Matrix software package (ANI/AAI-Matrix 2023) be used to generate a matrix of AAI values and the corresponding phylogenetic constructs by using the whole proteomes of the same 26 strains of Rothia, Kocuria, and Arthrobacter. The red dots (Fig. 3) indicate 5 nodes of monophyletic groups a–e, comprising 2–13 strains in the phylogram. The pairwise AAI matrix values showed a distribution of strains between the five groups, marked with dashed rectangles (Fig. 4), with intrageneric (AAI ≥ 65%, red) and intraspecies (AAI > 90%, yellow) AAI values (Understanding Results 2023). For demarcating genera at the family level, the condition 45% < AAI < 65% (colorless cells) is given on the web page (Understanding Results 2023). These inequalities are based on averaged characteristics and are generally consistent with the statistical analysis results given in Luo et al. (2014). Note the higher branch support in the AAI-based test (Fig. 3), as compared with that in the ANI-based (Fig. 2) and 16S rRNA-based (Fig. 1) tests.


AAI value matrix obtained from the distance matrix presented in the output data of the software package (ANI/AAI-Matrix 2023). The dashed rectangles mark the fragments corresponding to monophyletic groups a–e, whose nodes are marked with red dots in Fig. 3. The cells with AAI ≥ 65% (red, same genus) and AAI > 90% (yellow, same species) are highlighted in color
In the AAI-based test, the strains collected within separate monophyletic groups a–e in Figs. 3 and 4 should be considered belonging to the same genus (AAI ≥ 65%). By the AAI < 65% criterion, however, the evolutionary distances between these groups correspond to the intergeneric level within the family Micrococcaceae. Consequently, the genus affiliation of the strains in monophyletic groups a, b, and c, originally assigned by their authors to the genus Rothia, and d and e, originally assigned to the genus Kocuria, requires clarification (with possible renaming). Rothia sp. ND6WE1A, the reference strain for Isolate SG at the whole-genome level, and R. dentocariosa ATCC 17931T, the type species for the genus Rothia, are part of groups a and b (Figs. 3, 4), separated by intergeneric AAI values. In the AAI-based test, Art. crystallopoietes strain DSM 20117T and partly R. mucilaginosa ATCC 25296T are found outside the genera Rothia and Kocuria. It is noteworthy that in the 16S rRNA-based test (Table 1, column 5), these strains show I > 95% (an intrageneric level of sequence identity to Isolate SG) (see also Shchyogolev et al. 2023). This fact illustrates limitations of the 16S rRNA test in the taxonomic identification of bacterial isolates, which have been repeatedly considered in the past two decades. For example, Konstantinidis and Tiedje (2007) pointed out that the frequent result of the high sequence conservation of prokaryotic 16S rRNA genes is that strains differing in their phenotype, ecology, or complete genome may have very similar 16S rRNA gene sequences.
The AAI-based test results (Figs. 3 and 4) indicate that Rothia sp. ND6WE1A, the reference strain for Isolate SG at the level of the genomes studied, belongs to the genus Rothia and forms a monophyletic group with R. aerolata CCM 8669 (AAI = 71%), R. terrae KJZ-14 (AAI = 67%), and R. nasimurium E1706032 (AAI = 67%) in cluster a (Figs. 3 and 4). The sources of the strains are highly diverse in type and geography. Rothia sp. ND6WE1A is a member of a microbial consortium at the cathode of a solar microbial fuel cell originally enriched in seawater, New Jersey, USA. R. aerolata CCM 8669 is a type strain from the microbial culture collection in Beijing, China. R. terrae KJZ-14 and R. amarae KJZ-9 were isolated from soil dirt in Minhang District of Shanghai, China, and R. nasimurium E1706032 was isolated from a bird brain at a duck farm in Jining, Shandong Province, China. The adaptation of these strains to life in such contrasting ecological niches is reflected in their species diversity (AAI < 90%) within the genus Rothia, to which they were originally assigned by their authors.
These observations on Rothia sp. ND6WE1A are consistent with the results from the use of the MiGA webserver (New query datasets 2023; Rodriguez-R et al. 2020). These results indicate that this genome most probably belongs to the order Micrococcales (p = 0.0024) and probably belongs to the genus Rothia (p = 0.31). However, the “Taxonomic novelty” section states that it most probably belongs to a species not included in the TypMat database (New query datasets 2023; p = 0.0021), which comprises genomes of type strains (including Rothia dentocariosa ATCC 17931T), the highest taxonomic rank with p ≤ 0.01. The data presented in MyTaxa Scan testify to the detection in the genome of Rothia sp. ND6WE1A of regions with an unusual taxonomic distribution. These regions are shown and enumerated in the MyTaxa Scan diagram (Supplementary file MyTaxa_Scan_diagram_Rothia_sp_ND6WE1A.pdf). According to Luo et al. (2014) and Rodriguez-R et al. (2018, 2020), they can be interpreted as the result of HGT, because the estimates presented in the “Quality (essential genes)” section show zero contamination of the genome core. These regions are highly probably in the accessory and strain-specific parts of the pangenome (Koonin 2012; Tettelin and Medini 2020) and are not accounted for in the other tests we used.
Rothia sp. ND6WE1A, the reference strain for Isolate SG, is part of the GTDB R06-RS202 reference tree, included in the file bac120_r202.tree, with 254,091 OTUs (GTDB Data 2023). Figure 5 shows a Micrococcaceae subtree fragment that is a monophyletic group comprising strains assigned to the genus Rothia in GTDB R06-RS202. This fragment was extracted from the file bac120_r202.tree with Archaeopteryx (Archaeopteryx 2023). In the phylogenetic tree of Fig. 5, including 22 OTUs, the genome designations in the file bac120_r202.tree (GTDB Data 2023) [access codes to the NCBI genome assemblies (Genome Information by Organism 2023)] were replaced by the strain names given in the corresponding entries in the NCBI database. The red dots denote cluster nodes—three isolated monophyletic groups a–c within the g_Rothia fragment of the file bac120_r202.tree (GTDB Data 2023).

Fragment of the reference phylogenetic tree bac120_r202.tree (GTDB Data 2023) for the monophyletic group assigned to the genus Rothia in GTDB R06-RS202. The branch length reflects the similarity between the sets of 120 concatenated amino acid sequences of ubiquitous single-copy proteins. The numbers near the nodes indicate the bootstrapping results
As an example of the abovementioned need for detailing of the taxonomic affiliations detected by GTDB technology (Parks et al. 2018), one can note that the entries attributed by their authors to the genera Curtobacterium and Kocuria are found in the monophyletic group c of the g_Rothia fragment of the file bac120_r202.tree (GTDB Data 2023) (Fig. 5). A relevant positive finding here is the clarification of the systematic position of the metagenomic entries Micrococcaceae bacterium UBA7136 (rat intestinal metagenome) and M. bacterium UBA5788 (urban ground metagenome), having the status “unclassified Micrococcaceae” and found as part of one cluster in the monophyletic group a of the g_Rothia fragment (Fig. 5).
For estimating the validity of the generic assignment of the strains and metagenomic entries belonging to monophyletic groups a–c of the Fig. 5 phylogram, whose nodes are marked with red dots, the AAI test (ANI/AAI-Matrix 2023) was used as the most reliable means currently available to distinguish between strains within the family (Goris et al. 2007; Rodriguez-R and Konstantinidis 2014; Rodriguez-R et al. 2018). Among the 22 strains in Fig. 5, whose genomes are used in GTDB R06-RS202 (GTDB Data 2023), the proteomes of M. bacterium UBA7136, M. bacterium UBA5788, and Rothia sp. MGYG-HGUT-01258 were not found in the NCBI database (Genome Information by Organism 2023). The results obtained for the remaining 19 strains are shown in Figs. 6 and 7.

Phylogram obtained by the BioNJ method by using the distance matrix in the AAI-based test with the proteomes of the strains from the monophyletic group assigned to the genus Rothia in GTDB R06-RS202 (Fig. 5). The branch length reflects the AAI distance. The numbers near the nodes are branch significance estimates made with REQ (2023)

AAI value matrix obtained from the distance matrix presented in the output data of the software package (ANI/AAI-Matrix 2023). The dashed rectangles mark the fragments corresponding to monophyletic groups a–c, whose nodes are marked with red dots in Fig. 6. The cells with AAI ≥ 65% (red, same genus) and AAI ≥ 90% (yellow, same species) are highlighted in color
The red dots in Fig. 6 mark the nodes of three clusters corresponding to those in Fig. 5, with the same designations (a–c). The species (strain-level) and genus (cluster-level) demarcation of the OTUs in Figs. 5 and 6 is quantitatively ensured by their comparison with the matrix of AAI values in Fig. 7, in which the areas corresponding to clusters a–c in Figs. 5 and 6 are marked with dashed lines. The coloring shows cells satisfying the AAI ≥ 90% (yellow, same species) and AAI ≥ 65% (red, same genus) conditions (Luo et al. 2014; Understanding Results 2023). The colorless cells correspond to intergeneric pairwise values of 45% < AAI < 65%, joining clusters a–c together (Figs. 5 and 6) within the same family (Luo et al. 2014; Understanding Results 2023). The belonging to the same species in the AAI-based test (Fig. 7, yellow cells) is confirmed for the strains R. nasimurium irhom31/R. nasimurium E1706032; Rothia sp. HMSC078H08, with the pair R. mucilaginosa ATCC 25296T/R. mucilaginosa NUM-Rm6536; Kocuria sp. TGY1127_2/Kocuria sp. 36.
The most substantial result of using the AAI-based test in the analysis of the phylograms in Figs. 5 and 6 for the strains assigned to Rothia in GTDB R06-RS202 (GTDB Data 2023) is the < 65% intergeneric level of AAI values (Luo et al. 2014; Understanding Results 2023) for clusters a, b, and c (Fig. 7). In accordance with the conditions (Luo et al. 2014; Understanding Results 2023), these clusters should be assigned to different genera within the same family (AAI > 45%).
AAI-profiler
Figure 8 shows the AAI distribution diagram for the proteome of the reference strain Rothia sp. ND6WE1A, used as a query in the AAI-profiler test (AAI-profiler 2023; Medlar et al. 2018). On the horizontal axis are plotted AAI values between the query and the species members in the UniProt database (UniProtKB 2023). The vertical axis shows the matched fraction (MF, coverage), which is the fraction of query proteins that have a match in the species. MF values are averaged characteristics over a large dataset in the UniProt database, including proteins from partially sequenced genomes. The icons in the diagram correspond to the species that have scored the highest marks with allowance for AAI and MF, i.e., the sum of the sequence identity values for all query proteins with established matches. Related species, grouped and colored on the basis of genus, form a characteristic “cloud” in the diagram, with AAI values reflecting the evolutionary proximity of the UniProt strains to the query strain. The horizontal axis has icons for the species for which DNA sequencing results have been obtained for individual proteins only.

AAI distribution diagram for Rothia sp. ND6WE1A, as found in the AAI-profiler output data. The vertical dashed lines in Fig. 8 correspond to the AAI cutoff values for grouping strains into genera (AAI > 0.65) and species (AAI > 0.9). The icons are colored according to genus (bacteria) or order (eukaryotes). Eukaryotic species are marked with rhombuses; bacteria, with circles; archaea, with crosses; and everything else (viruses, metagenomes, unclassified samples), with squares
The vertical dashed lines in Fig. 8 correspond to the AAI threshold values for grouping strains into genera (AAI > 0.65) and species (AAI > 0.9) according to the conditions specified on the web page (Understanding Results 2023). The strain most closely related to Rothia sp. ND6WE1A is R. amarae KJZ-9, with AAI = 98.9%, ANI = 98.6%, and MF = 0.946. Thus, all three genomes [GCA_007666515.1, R. amarae (mucilaginosa) DE0531 (originally listed in Table 1 as corresponding to R. amarae JCM 11375T but excluded from our consideration; see above); GCA_001683935.1, Rothia sp. ND6WE1A; and GCA_014705945.2, R. amarae KJZ-9] represent the same species with high interstrain evolutionary relatedness, characterized by ANI and AAI values of about 99%.
Another strain closely related to Rothia sp. ND6WE1A [AAI = 94%, which exceeds the threshold value for grouping strains at the species level (AAI > 0.9)] is Arthrobacter sp. SMCC G919, whose icon is located on the horizontal axis of the diagram (Fig. 8). Then come members of Thermocrinis, Planobispora, Mycobacterium, Glutamicibacter, Micrococcus, and other genera, which too are located on the horizontal axis in descending order of AAI values ≈ 0.9. The appearance in this diagram, grouping mostly Micrococcaceae members, of “noncongruent” species with a small (almost zero) MF, may be associated with HGT (Medlar et al. 2018; Treangen and Rocha 2011).
On the other hand, Arthrobacter occupies a certain position in the main part of the diagram with an MF in the range 0.03–0.25 and AAI = 62–67% in the region of 60–80%, which groups strains at the genus level (Luo et al. 2014). Members of Rothia and Kocuria also fall into the same area in the diagram (Fig. 8), with MFs ranging from 0.28 to 0.95. Such “incorporation” of Kocuria and Arthrobacter members (and other bacterial species/strains marked with pink, green, and light green dots in Fig. 8) into the main cluster of Rothia, comprising species related to the query strain Rothia sp. ND6WE1A, should be considered a signal for eventual taxonomic reclassification of these members, which was also shown above with the pairwise whole-genome tests based on ANI and AAI. According to Medlar et al. (2018), at high MFs, such overlapping is due to misclassified or mislabeled samples, whereas at lower MFs, it may indicate contamination or, possibly, HGT.
As found with AAI-profiler, the strain most closely related to Rothia sp. ND6WE1A (after R. amarae KJZ-9) is R. nasimurium PT-32 (Fig. 8; AAI = 71.7%, MF = 0.69). These data show a noticeable gap between Rothia sp. ND6WE1A (and its closely related R. amarae KJZ-9) and the main group of species in Fig. 8 (MF, 25% and greater), which indicates that its genome differs strongly from those of the related species, found with AAI-profiler in the UniProt database. This observation is in harmony with the results of phylogenetic studies using ANI (Fig. 2), which showed a considerable evolutionary deviation of Rothia sp. ND6WE1A from the 26 Rothia, Kocuria, and Arthrobacter strains considered.
According to the results in Figs. 3, 4, 5, 6 and 7, Rothia sp. ND6WE1A, which is the reference strain for Isolate SG at the reference genome level, and R. dentocariosa ATCC17931T, which represents the type species of Rothia (Austin 2015), may be members of different monophyletic groups separated by intergeneric AAI values < 65%. Thus, when the strains belonging to such groups are appropriately verified (Figs. 3, 4, 5, 6, 7), priority in the genus name Rothia should be given to the cluster with a member of the type species R. dentocariosa. It follows that the strains comprising cluster a in Figs. 3, 4, 5, 6 and 7 (including Rothia sp. ND6WE1A) could be assigned an appropriate genus name after additional genotypic and phenotypic data are obtained (Oren and Garrity 2014). This is consistent with the results of the MiGA-based test for Rothia sp. ND6WE1A, which showed its statistically valid taxonomic novelty in relation to the TypeMat reference database, containing type whole-genome material. Additional physiological–biochemical and genetic studies are needed to resolve this issue with respect to Isolate SG.
In the AAI-profiler test (Fig. 8), Rothia sp. ND6WE1A was the same species as R. amarae KJZ-9, with ANI and AAI values of about 99%. That they live in largely different ecological niches (see above) is very probably ensured by the phenotypic traits controlled by the genes in the accessory and strain-specific parts of the pangenome (Koonin 2012; Tettelin and Medini 2020). These genes, introduced most commonly by HGT, and their products are not accounted for in the tests based on 16S rRNA, ANI, AAI, GTDB-Tk, and MiGA (outside the “MyTaxa Scan” section), which handle “housekeeping” genes.
Conclusions
The obtained results show that the introduction into consideration of the ANI and AF, which ensure quantitative identification of OTUs at the species level for the GTDB bacterial and archaeal genomes, solves the problem of constructing a taxonomic structure from domain to species only in part. This is supported by the contradictions that we have identified between the obtained results and the genus names of the strains (metagenomic entries) in GTDB R06-RS202. We have shown that as the first step to eliminate them, one can use AAI-based tests. However, additional genotypic and phenotypic characteristics need to be considered before an appropriate reclassification of the strains/metagenomic entries can be made.
Thus, the presented results should be considered as identifying the problem and providing a signal for the subsequent reclassification of the strains in question. Such reclassification could be done by the authors of the strains (or any other interested researchers), by using appropriate additional genotypic and phenotypic criteria within a polyphasic approach.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
AAI-profiler: fast proteome-wide search reveals taxonomic outliers. http://ekhidna2.biocenter.helsinki.fi/AAI. Accessed 18 Sept 2023
ANI/AAI-Matrix. Genome-based distance matrix calculator. http://enve-omics.ce.gatech.edu/g-matrix. Accessed 18 Sept 2023
Archaeopteryx. https://sites.google.com/site/cmzmasek/christian-zmasek/software/archaeopteryx?authuser=0. Accessed 18 Sept 2023.
Asadu CO, Ike IS, Onu CE, Egbuna SO, Onoh M, Mbah GO, Eze CN (2020) Investigation of the influence of biofertilizer synthesized using microbial inoculums on the growth performance of two agricultural crops. Biotechnol Rep 27:e00493. https://doi.org/10.1016/j.btre.2020.e00493
Asyakina LK, Vorob’eva EE, Proskuryakova LA, Zharko MY (2023) Evaluating extremophilic microorganisms in industrial regions. Foods Raw Mater 11:162–171. https://doi.org/10.21603/2308-4057-2023-1-556
Austin B (2015) Rothia. In: Whitman WB (ed) Bergey’s manual of systematics of archaea and bacteria. Wiley, Hoboken, pp 1–13. https://doi.org/10.1002/9781118960608.gbm00124
Bano A, Muqarab R (2017) Plant defense induced by PGPR against Spodoptera litura in tomato (Solanum lycopersicum L.). Plant Biol 19:406–412. https://doi.org/10.1111/plb.12535
Borah A, Thakur D (2020) Phylogenetic and functional characterization of culturable endophytic actinobacteria associated with Camellia spp. for growth promotion in commercial tea cultivars. Front Microbiol 11:318. https://doi.org/10.3389/fmicb.2020.00318
Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH (2022) GTDB-Tk v2: memory friendly classification with the genome taxonomy database. Bioinformatics 38:5315–5316. https://doi.org/10.1093/bioinformatics/btac672
Collection of Rhizosphere Microorganisms. https://collection.ibppm.ru. Accessed 18 Sept 2023
da Silva JCP, de Medeiros FHV, Campos VP (2018) Building soil suppressiveness against plant-parasitic nematodes. Biocontrol Sci Technol 28:423–445. https://doi.org/10.1080/09583157.2018.1460316
Davis I, Sevigny J, Kleiner V, Mercurio K, Pesce C, Swanson E, Thomas WK, Tisa LS (2020) Draft genome sequences of 10 bacterial strains isolated from root nodules of Alnus trees in New Hampshire. Microbiol Resour Announc 9:e01440-e1519. https://doi.org/10.1128/MRA.01440-19
de Oliveira IMF, Ng DYK, van Baarlen P, Stegger M, Andersen PS, Wells JM (2022) Comparative genomics of Rothia species reveals diversity in novel biosynthetic gene clusters and ecological adaptation to different eukaryotic hosts and host niches. Microb Genom 8:mgmen000854. https://doi.org/10.1099/mgen.0.000854
Elbahnasawy MA, Shehabeldine AM, Khattab AM, Amin BH, Hashem AH (2021) Green biosynthesis of silver nanoparticles using novel endophytic Rothia endophytica: characterization and anticandidal activity. J Drug Deliv Sci Technol 62:102401. https://doi.org/10.1016/j.jddst.2021.102401
Evangelista EV, Garcia FC, Cruz JA (2017) Isolation, characterization and identification of plant growth-promoting rhizobacteria. Int J Agric Technol 13:715–727
Fan Y, Jin Z, Tong J, Li W, Pasciak M, Gamian A, Liu Z, Huang Y (2002) Rothia amarae sp. nov., from sludge of a foul water sewer. Int J Syst Evol Microbiol 52:2257–2260. https://doi.org/10.1099/00207713-52-6-2257
FastME 2.0: a comprehensive, accurate, and fast distance-based phylogeny inference program. http://www.atgc-montpellier.fr/fastme. Accessed 18 Sept 2023.
Fatahi-Bafghi M (2021) Characterization of the Rothia spp. and their role in human clinical infections. Infect Genet Evol 93:104877. https://doi.org/10.1016/j.meegid.2021.104877
Genome Information by Organism. https://www.ncbi.nlm.nih.gov/genome/browse/#!/overview. Accessed 18 Sept 2023
Genome taxonomy database. https://gtdb.ecogenomic.org. Accessed 18 Sept 2023.
Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM (2007) DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol 57:81–91. https://doi.org/10.1099/ijs.0.64483-0
Gtari M, Ghodhbane-Gtari F, Nouioui I, Beauchemin N, Tisa LS (2012) Phylogenetic perspectives of nitrogen-fixing actinobacteria. Arch Microbiol 194:3–11. https://doi.org/10.1007/s00203-011-0733-6
GTDB Data. Index of /releases/release202/202.0/. https://data.gtdb.ecogenomic.org/releases/release202/202.0/. Accessed 18 Sept 2023
GTDB-Tk Classify-v1.6.0. https://kbase.us/applist/apps/kb_gtdbtk/run_kb_gtdbtk. Accessed 18 Sept 2023.
Guénoche A, Garreta H (2000) Can we have confidence in a tree representation? In: Gascuel O, Sagot MF (eds) Computational biology. JOBIM 2000. Lecture Notes in Computer Science, vol 2066. Springer. Berlin, Heidelberg, pp 45–56. https://doi.org/10.1007/3-540-45727-5_5
Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S (2018) High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun 9:5114. https://doi.org/10.1038/s41467-018-07641-9
Jia Y, Li X, Xu F, Liu Z, Fu Y, Xu X, Yang J, Zhang S, Shen C (2022) Single-cell-level microfluidics assisted with resuscitation-promoting factor technology (SMART) to isolate novel biphenyl-degrading bacteria from typical soils in eastern China. Environ Pollut 311:119864. https://doi.org/10.1016/j.envpol.2022.119864
Kim M, Oh HS, Park SC, Chun J (2014) Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol 64:346–351. https://doi.org/10.1099/ijs.0.059774-0
Ko KS, Lee MY, Park YK, Peck KR, Song J-H (2009) Molecular identification of clinical Rothia isolates from human patients: proposal of a novel Rothia species, Rothia arfidiae sp. nov. J Bacteriol Virol 39:159–164. https://doi.org/10.4167/jbv.2009.39.3.159
Konstantinidis KT, Tiedje JM (2007) Prokaryotic taxonomy and phylogeny in the genomic era: advancements and challenges ahead. Curr Opin Microbiol 10:504–509. https://doi.org/10.1016/j.mib.2007.08.006
Koonin EV (2012) The logic of chance: the nature and origin of biological evolution. Pearson Education, Upper Saddle River
Lefort V, Desper R, Gascuel O (2015) FastME 2.0: a comprehensive, accurate, and fast distance-based phylogeny inference program. Mol Biol Evol 32:2798–2800. https://doi.org/10.1093/molbev/msv150
Lemoine F, Entfellner J-BD, Wilkinson E, Correia D, Felipe MD, De Oliveira T, Gascuel O (2018) Renewing Felsenstein’s phylogenetic bootstrap in the era of big data. Nature 556:452–456. https://doi.org/10.1038/s41586-018-0043-0
Lemoine F, Correia D, Lefort V, Doppelt-Azeroual O, Mareuil F, Cohen-Boulakia S, Gascuel O (2019) NGPhylogeny.fr: new generation phylogenetic services for non-specialists. Nucleic Acids Res 47:W260–W265. https://doi.org/10.1093/nar/gkz303
Luo C, Rodriguez-R LM, Konstantinidis KT (2014) MyTaxa: an advanced taxonomic classifier for genomic and metagenomic sequences. Nucleic Acids Res 42:e73. https://doi.org/10.1093/nar/gku169
Mažeikienė I, Frercks B, Burokienė D, Mačionienė I, Šalaševičiene A (2021) Endophytic community composition and genetic-enzymatic features of cultivable bacteria in Vaccinium myrtillus L. in forests of the Baltic-Nordic region. Forests 12:1647. https://doi.org/10.3390/f12121647
Medlar AJ, Törönen P, Holm L (2018) AAI-profiler: fast proteome-wide exploratory analysis reveals taxonomic identity, misclassification and contamination. Nucleic Acids Res 46:W479–W485. https://doi.org/10.1093/nar/gky359
MEGA. Molecular Evolutionary Genetics Analysis. https://www.megasoftware.net. Accessed 18 September 2023.
Mercurio K, Sevigny J, Pesce C, Thomas WK, Tisa LS (2022) Draft genome sequences for bacteria associated with root nodules of Alnus incana in New England. Microbiol Resour Announc 11:e0091422. https://doi.org/10.1128/mra.00914-22
Nardone G, Compare D (2015) The human gastric microbiota: is it time to rethink the pathogenesis of stomach diseases? United Eur Gastroenterol J 3:255–260. https://doi.org/10.1177/2050640614566846
New query datasets. https://disc-genomics.uibk.ac.at/miga/query_datasets/new?project_id=4. Accessed 18 Sept 2023
NGPhylogeny.fr. https://ngphylogeny.fr. Accessed 18 Sept 2023
Nuaima RH (2022) The difference in the bacterial attachment among Pratylenchus neglectus populations and its effect on the nematode infection. Microorganisms 10:1524. https://doi.org/10.3390/microorganisms10081524
Odeberg G, Bläckberg A, Sunnerhagen T (2023) Infection or contamination with Rothia, Kocuria, Arthrobacter and Pseudoglutamicibacter—a retrospective observational study of non-Micrococcus Micrococcaceae in the clinic. J Clin Microbiol 61:e0148422. https://doi.org/10.1128/jcm.01484-22
Opelt K, Berg C, Berg G (2007) The bryophyte genus Sphagnum is a reservoir for powerful and extraordinary antagonists and potentially facultative human pathogens. FEMS Microbiol Ecol 61:38–53. https://doi.org/10.1111/j.1574-6941.2007.00323.x
Oren A, Garrity GM (2014) Then and now: a systematic review of the systematics of prokaryotes in the last 80 years. Antonie Van Leeuwenhoek 106:43–56. https://doi.org/10.1007/s10482-013-0084-1
Parks DH, Rinke C, Chuvochina M, Chaumeil P-A, Woodcroft BJ, Evans PN, Hugenholtz P, Tyson GW (2017) Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat Microbiol 2:1533–1542. https://doi.org/10.1038/s41564-017-0012-7
Parks DH, Chuvochina M, Waite DW, Rinke C, Skarshewski A, Chaumeil P-A, Hugenholtz P (2018) A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol 36:996–1004. https://doi.org/10.1038/nbt.4229
Parks DH, Chuvochina M, Chaumeil P-A, Rinke C, Mussig AJ, Hugenholtz P (2020) A complete domain-to-species taxonomy for Bacteria and Archaea. Nat Biotechnol 38:1079–1086. https://doi.org/10.1038/s41587-020-0501-8
Petrović M, Janakiev T, Grbić ML, Unković N, Stević T, Vukićević S, Dimkić S (2024) Insights into endophytic and rhizospheric bacteria of five sugar beet hybrids in terms of their diversity, plant-growth promoting, and biocontrol properties. Microb Ecol 87:19. https://doi.org/10.1007/s00248-023-02329-0
Pisarska K, Pietr SJ (2015) Biodiversity of dominant cultivable endophytic bacteria inhabiting tissues of six different cultivars of maize (Zea mays L. ssp. mays) cropped under field conditions. Pol J Microbiol 64:163–170. https://doi.org/10.33073/pjm-2015-024
REQ. https://gitlab.pasteur.fr/GIPhy/REQ. Accessed 18 Sept 2023
Rodriguez-R LM, Konstantinidis KT (2014) Bypassing cultivation to identify bacterial species. Microbe 9:111–118. https://doi.org/10.1128/MICROBE.9.111.1
Rodriguez-R LM, Gunturu S, Harvey WT, Rossello-Mora R, Tiedje JM, Cole JR, Konstantinidis KT (2018) The Microbial Genomes Atlas (MiGA) webserver: taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Res 46:W282–W288. https://doi.org/10.1093/nar/gky467
Rodriguez-R LM, Harvey WT, Rosselló-Mora R, Tiedje JM, Cole JR, Konstantinidis KT (2020) Classifying prokaryotic genomes using the Microbial Genomes Atlas (MiGA) webserver. In: Whitman WB (ed) Bergey’s manual of systematics of archaea and bacteria. Wiley, Hoboken, pp 1–11. https://doi.org/10.1002/9781118960608.bm00042
Search EzBioCloud Database. https://www.ezbiocloud.net. Accessed 18 Sept 2023
Sekhar AC, Thomas P (2015) Isolation and identification of shoot-tip associated endophytic bacteria from banana cv. Grand Naine and testing for antagonistic activity against Fusarium oxysporum f. sp. cubense. Am J Plant Sci 6:943–954. https://doi.org/10.4236/ajps.2015.67101
Shchyogolev SY, Dykman LA, Sokolov AO, Sokolov OI (2023) Matora LY (2023) Elimination of contamination in the published 16S rRNA gene sequence of Rothia amarae type strain J18T during phylogenetic studies of a bacterial isolate from a suspension cell culture of Arabidopsis thaliana (Heynh.). BioRxiv 6:273. https://doi.org/10.1101/2023.11.08.566172
Shurigin V, Egamberdieva D, Li L, Davranov K, Panosyan H, Birkeland N-K, Wirth S, Bellingrath-Kimura SD (2020) Endophytic bacteria associated with halophyte Seidlitzia rosmarinus Ehrenb. ex Boiss. from saline soil of Uzbekistan and their plant beneficial traits. J Arid Land 12:730–740. https://doi.org/10.1007/s40333-020-0019-4
Sokolov AO, Dykman LA, Galitskaya AA, Sokolov OI (2021) Identification and characterization of Rothia amarae sp. nov. in a suspension culture of Arabidopsis thaliana (Heynh.) cells. Biol Life Sci Forum 4:92. https://doi.org/10.3390/IECPS2020-08753
Sokolova EA, Mishukova OV, Hlistun IV, Tromenschleger IN, Tikunov AY, Manakhov AD, Rogaev EI, Savenkov OA, Buyanova MD, Ivanov IV, Smirnova NV, Voronina EN (2024) The effectiveness of co-inoculation by consortia of microorganisms depends on the type of plant and the soil microbiome. Plants 13:116. https://doi.org/10.3390/plants13010116
Stackebrandt E, Ebers J (2006) Taxonomic parameters revisited: tarnished gold standards. Microbiol Today 33:152–155
Stubbendieck RM, Dissanayake E, Burnham PM, Zelasko SE, Temkin MI, Wisdorf SS, Vrtis RF, Gern JE, Currie CR (2023) Rothia from the human nose inhibit Moraxella catarrhalis colonization with a secreted peptidoglycan endopeptidase. mBio 14:e0046423. https://doi.org/10.1128/mbio.00464-23
SYNTOL. http://www.syntol.ru. Accessed 18 Sept 2023
Tettelin H, Medini D (eds) (2020) The Pangenome. In: Diversity, dynamics and evolution of genomes. Springer, Cham
Tindall BJ, Rosselló-Móra R, Busse HJ, Ludwig W, Kämpfer P (2010) Notes on the characterization of prokaryote strains for taxonomic purposes. Int J Syst Evol Microbiol 60:249–266. https://doi.org/10.1099/ijs.0.016949-0
Torche A, Benhizia H, Rosselli R, Romoli O, Zanardo M, Baldan E, Alberghini S, Tondello A, Baldan B, Benguedouar A, Squartini A, Benhizia Y (2014) Characterization of bacteria associated with nodules of two endemic legumes of Algeria, Hedysarum naudinianum and H. perrauderianum. Ann Microbiol 64:1065–1071. https://doi.org/10.1007/s13213-013-0745-3
Treangen TJ, Rocha EPC (2011) Horizontal transfer, not duplication, drives the expansion of protein families in prokaryotes. PLoS Genet 7:e1001284. https://doi.org/10.1371/journal.pgen.1001284
Tuikhar N, Kirdat K, Nair G, Padma S, Thorat V, Swetha P, Sathe S, Sundararaj R, Yadav A (2022) Rothia santali sp. nov. endophytic bacteria isolated from sandalwood (Santalum album L.) seedling. Arch Microbiol 204:609. https://doi.org/10.1007/s00203-022-03237-6
Understanding Results. https://help.microbial-genomes.org/understanding-results#distance. Accessed 18 Sept 2023
UniProtKB. Find your protein. https://www.uniprot.org. Accessed 18 Sept 2023
Vaghari Souran SE, Shekariesfahlan A, Ashrafi F, Naeimi S, Ghasemi A (2023) Isolation and identification of grapevine endophytic bacteria with antagonistic potential against Fomitiporia mediterranea, a pathogen involved in grapevine trunk disease. J Plant Dis Prot 130:1371–1384. https://doi.org/10.1007/s41348-023-00788-8
Xiao Y, Chen L, Teng K, Ma J, Xiang S, Jiang L, Liu G, Yang B, Fang J (2023) Potential roles of the rhizospheric bacterial community in assisting Miscanthus floridulus in remediating multi-metal(loid)s contaminated soils. Environ Res 227:115749. https://doi.org/10.1016/j.envres.2023.115749
Xiong Z-J, Zhang J-L, Zhang D-F, Zhou Z-L, Liu M-J, Zhu W-Y, Zhao L-X, Xu L-H, Li W-J (2013) Rothia endophytica sp. nov., an actinobacterium isolated from Dysophylla stellata (Lour.) Benth. Int J Syst Evol Microbiol 63:3964–3969. https://doi.org/10.1099/ijs.0.052522-0
Yarza P, Yilmaz P, Pruesse E, Glöckner FO, Ludwig W, Schleifer KH, Whitman WB, Euzéby J, Amann R, Rosselló-Móra R (2014) Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol 12:635–645. https://doi.org/10.1038/nrmicro3330
Yastrebova OV, Plotnikova EG (2020) Phylogenetic diversity of bacteria of the family Micrococcaceae isolated from biotopes with different anthropogenic impact. Bull Perm Univ Biol 4:321–333. https://doi.org/10.17072/1994-9952-2020-4-321-333
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design, including data collection and analysis. The first draft of the manuscript was written by S.Yu. Shchyogolev and L.A. Dykman. All authors commented on initial versions of the manuscript and all authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interests
The authors declare no conflict of interests.
Additional information
Communicated by Yusuf Akhter.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shchyogolev, S.Y., Dykman, L.A., Sokolov, A.O. et al. Quantitative intra- and intergeneric taxonomic relationships among Micrococcaceae strains reveal contradictions in the historical assignments of the strains and indicate the need for species reclassification. Arch Microbiol 206, 165 (2024). https://doi.org/10.1007/s00203-024-03896-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00203-024-03896-7