
Abstract
An isolate, designated MBLB2552T, was isolated from the gut of the honey bees (Apis mellifera L.) and identified as a member of the genus Paenibacillus based on the sequences of the 16S rRNA gene sequences. The most closely related species to strain MBLB2552T were Paenibacillus timonensis 2301032 T, Paenibacillus barengoltzii NBRC 101215 T, and Paenibacillus macerans IAM 12467 T, with similarity values of 98.1, 97.21 and 97.0%, respectively, based on 16S rRNA gene sequences. The genome size and G + C content of MBLB2552T were 5.2 Mb and 52.4%, respectively. The Ortho average nucleotide identity (OrthoANI) and in silico DNA–DNA hybridization (isDDH) values between strain MBLB2552T and the type strains of the closest species were below the species delineation threshold. Comparative genomic analysis showed that most core POGs of strain MBLB2552T and other related taxa were related to translation, ribosomal structure and biogenesis (J) and carbohydrate metabolism in the COG category and KEGG pathways, respectively. Strain MBLB2552T was Gram stain-positive, spore-forming, rod-shaped, facultative anaerobic, motile, and grew at 20‒45 °C in 0‒2% (w/v) NaCl at pH 6.0‒9.0. The major polar lipids identified were diphosphatidylglycerol, phosphatidylglycerol, phosphatidylcholine, phosphatidylserine, unidentified polar lipids, and an unidentified glycolipid. We propose that strain MBLB2552T represents the type strain of the genus Paenibacillus and its name Paenibacillus mellifer sp. nov. is proposed. The type of strain was MBLB2552T (= JCM 35371 T = KCTC 43386 T).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Honey bees (Apis mellifera L.) are the most important insects, both economically and ecologically, to humans, as they are critical pollinating insects globally and honey producers (Batra 1995; Dedej and Delaplane 2003). Therefore, the health of honey bees has become a significant concern in many countries (Goulson et al. 2015). Culture-dependent gut microbial communities in A. mellifera (Gilliam et al. 1997; Inglis et al. 1998; Saccà and Lodesani 2020) are complex and not fully understood or even known (Elzeini et al. 2021). Microflora found in the gut are members of: yeasts, Gram-positive bacteria, Gram-negative or gram variable bacteria (Mohr and Tebbe 2006; Elzeini et al. 2021).
The genus Paenibacillus was proposed in 1993 after Ash et al. (1993) conducted a broad comparative analysis of the 16S rRNA gene sequences of the genus Bacillus. The genus Paenibacillus has increased to 277 validly named species and 5 subspecies of P. polymyxa as the type species (http://www.bacterio.net/paenibacillus.html). Members of this genus can be widely found in various ecological environments and harbor strains of industrial and agricultural importance relevant to humans, animals, and plants (Grady et al. 2016).
During the isolation and characterization of the gut bacterial community in Apis mellifera, 81 isolates were obtained. Among them, strain MBLB2552T belonging to the genus Paenibacillus was identified as a candidate novel strains by comparative 16S rRNA gene sequence analysis. In this study, we report a polyphasic taxonomic description of strain MBLB2552T isolated from the honey bee gut.
Materials and methods
Isolation of the bacterial strain and culture condition
Healthy nurse honey bees (A. mellifera L.) were collected from Incheon National University, Incheon, Republic of Korea in the summer (June 2021) for bacterial isolation. The bee surface was sterilized with 100% ethanol and dissected the whole intestinal tract (esophagus to rectum). The whole intestinal tract of the bees was suspended and diluted in PBS. The suspensions were spread onto tryptic soy agar (TSA, Becton, Dickinson and Company, USA) medium and then incubated at 30 °C for 1 week. Cycloheximide at a concentration of 0.01% (v/v) was added to the media to prevent fungi growth. Colonies with distinct morphological characteristics based on color, shape, size, and rough or smooth surfaces were selected and subsequently re-streaked at least three times on the same type of fresh medium to obtain pure colonies.
Identification, sequence similarity analysis and genomic analysis
After obtaining pure single colonies, genomic DNA from each colony was extracted using the HiYield™ Genomic DNA Mini Kit (RBC, Taiwan) according to the manufacturer’s protocol. Universal bacterial primer pairs 27F (5'-AGAGTTTGATCMTGGCTCAG-3') and 1492R (5'-TACGGYTACCTTGTTACGACTT-3') were used to amplify the 16S rRNA gene for bacterial identification (Lane 1991). The PCR products of the amplified 16S rRNA genes were sequenced by Macrogen Co., Ltd and assembled as previously described by Roh et al. (2008) using the SeqMan program (DNA star). The obtained 16S rRNA gene sequence was analyzed using the BLAST program provided by GenBank (https://www.ncbi.nlm.nih.gov/) and EzBioCloud server (http://www.ezbiocloud.net/) (Yoon et al. 2017) for phylogenetic analysis. A phylogenetic tree was constructed using MEGA 7 (Kumar et al. 2016) after a gap deletion and multiple alignments of data via the CLUSTAL W program (Thompson et al. 1994). Evolutionary distances were calculated by the Kimura two-parameter model (Kimura 1983). A phylogenetic tree was built using the neighbor-joining (NJ) (Saitou and Nei 1987), maximum-likelihood (ML) (Felsenstein 1981), and maximum-parsimony (MP) (Kluge and Farris 1969) algorithms (each employed 1000 replicates) with the MEGA7 program.
The genome of strain MBLB2552T was sequenced and assembled using the Illumina SPAdes v. 3.10.1 next-generation sequencing platform and SOAP denovo v. 3.10.1 de novo assembles. The genome sequence authenticity and the absence of contamination by strain MBLB2552T were confirmed by the proposed minimal standards for using prokaryotes genome data (Chun et al. 2018). Authenticity was checked by comparing the 16S rRNA gene sequences obtained, one was the conventional Sanger sequencing method as previously mentioned, and the other was whole-genome sequencing. Contamination in the strain MBLB2552T genome was evaluated using the ContEst16S (https://www.ezbiocloud.net/tools/contest16s) (Lee et al. 2017).
Genome annotation, phylogenetic analysis
Clusters of Orthologous Groups (COG) category analysis was conducted using EggNOG v5.0 for homologous gene predicted search (Tatusov et al. 2000; Huerta-Cepas et al. 2015). To perform comparative genomic analysis, the NCBI genome database (http://www.ncbi.nlm.nih.gov/genome/) was referred to obtain closely related species of the genus Paenibacillus with strain MBLB2552T. The OrthoANI and isDDH values among the strain MBLB2552T and other species of the genus Paenibacillus were measured using OAT software version 0.93.1 (Lee et al. 2015) and Genome-to-Genome Distance Calculator program (GGDC 2.1; http://ggdc.dsmz.de/distcalc2.php) (Meier-Kolthoff et al. 2013) with the recommended formula 2.2 based on DNA–DNA hybridization, respectively.
Comparative genomic analysis
Pan-genome analysis was performed using the Bacterial Pan Genome Analysis (BPGA) software (Chaudhari et al. 2016). The genomes of strain MBLB2552T and related genus Paenibacillus strains were categorized as core (conserved for all strains), accessory (more than two species shared but not core), and unique (only strain-specific) genes. Pan-genome function and pathway analyses were assigned based on the COG database https://www.ncbi.nlm.nih.gov/research/cog/) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (https://www.genome.jp/kegg/) (Kanehisa and Goto 2000), respectively. COG and KEGG pan-genome orthologous groups (POGs) were clustered using the USEARCH algorithm with a 50% sequence identity cut-off value. For sequence alignment of the concatenated core POG sequences, the MAFFT v. 7.427 algorithms was used (Katoh et al. 2002) and a phylogenetic tree based on core POG sequences was constructed using the UPGMA method with 1000 bootstrap replications (Sneath and Sokal 1973). To construct a balanced minimum evolution tree with branch support via FASTME 2.1.4 including SPR postprocessing, intergenomic distances were calculated by Type Strain Genome Server (TYGS) (http://tygs.dsmz.de/) (Lefort et al. 2015; Meier-Kolthoff and Göker 2019).
Phenotype and biochemical characteristics
The morphology of strain MBLB2552T was observed by light microscopy (model CX 23; Olympus) and transmission electron microscopy (JEM-101; JEOL). Gram staining was assessed with a BD Gram stain kit according to the manufacturer’s protocols. Anaerobic growth was performed using a GasPak™ EZ anaerobe gas generating pouch system with an indicator (BD) on TSA at 30 °C for 3 weeks. To determine the condition for temperature growth, strain MBLB2552T was incubated in TSB at 4, 10, 15, 20, 25, 30, 35, 37, 40, 45, 50 and 55 °C for 1 week. To measure the growth range of different NaCl concentration, TSB as the basal medium was modified that NaCl in the medium was changed to 0, 0.5% (w/v) and 1.0‒5.0% (w/v) at intervals of 0.5%. The growth range in different pH was tested in TSB using the following buffer systems: pH 5.0 and 6.0 with 10 mM 2-(N-morpholino) ethanesulfonic acid; pH 7.0–9.0 with 10 mM Bis–Tris propane; pH 10.0 and 11.0 with 10 mM 3-(cyclohexylamino)-1-propanesulfonic acid, which was incubated at 30 °C. The growth of each medium was measured based on the optical density every 24 h at 600 nm using a UV-1280 spectrophotometer (Shimadzu, Japan). Catalase and oxidase activity were checked by transferring fresh colonies from TSA to a glass slide and adding 3% H2O2 and 1% tetramethyl-p-phenylenediamine solution, respectively. Hydrolysis of starch, casein, Tweens 20 and 80, and gelatin was performed as described by Benson (2002). H2S production was detected by cultivation in TSB broth containing sodium thiosulfate (5 g l−1) with lead acetate paper. Other biochemical characterization of strain MBLB2552T was experimented using the API 20NE and API 50CHE (bioMérieux, France) strips according to the manufacturer protocols. The susceptibility of the strain MBLB2552T was checked by the disc diffusion plate method as described by Bauer et al. (1996). Discs were contained with the following antibiotics (µg ml−1 disc unless indicated): ampicillin (10), cephalothin (30), ciprofloxacin (10), erythromycin (25), gentamicin (30), kanamycin (30), lincomycin (15), neomycin (30), norfloxacin (20), novobiocin (10), penicillin G (20 IU), streptomycin (50), and tetracycline (30). The strain was incubated at 30 °C for one week.
Chemotaxonomic characteristics
Cells of strain MBLB2552T for chemotaxonomic characterization were prepared by cultivation in TSB at 30 °C for 2 days. Cellular fatty acid and polar lipid profiles of strain MBLB2552T were analyzed according to the methods described by Miller (1982) and Minnikin et al. (1984). Fatty acid profiles were obtained using the Sherlock MIS Software version 6.2 based on the TSBA6 database (Sasser 1990). Polar lipids of strain MBLB2552T were analyzed by two-dimensional TLC on silica gel 60 F254 (10 × 10 cm; Merck) by spraying with the following staining reagents: sulfuric acid–ethanol (1:2, v/v) for total polar lipids, zinzade regent (5% of ethanolic molybdatophosphoric acid), and ninhydrin for aminolipids (Minnikin et al. 1984; Komagata and Suzuki 1987). Freeze-dry cells of the strain MBLB2552.T were used to investigate isoprenoid quinone according to Collins and Jones method (1981) and identified using an HPLC system (YL9100; Younglin). The diamino acid type in the cell wall analysis was determined according to the protocols reported by Staneck and Roberts (1974)
Results and discussion
Whole-genome sequencing and phylogenetic analysis
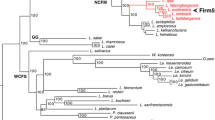
The full-length 16S rRNA gene sequence (1483 bp) was obtained by conventional Sanger sequencing, as described above for the phylogenetic analysis which revealed that the isolated strain MBLB2552T belongs to the genus Paenibacillus. Similarities in the 16S rRNA gene sequences between strains MBLB2552T and P. timonensis 2301032 T, P. barengoltzii NBRC 101215 T, P. macerans IAM 12467 T, P. phoenicis 3PO2SAT, P. faecis CIP 101062 T were 98.17, 97.21, 97.0, 96.96, and 96.96%, respectively. Similarities with another valid published species of the genus Paenibacillus were less than 96.8%. In the phylogenetic tree, strain MBLB2552T was grouped with P. timonensis 2301032 T and the topologies of the phylogenetic trees built using NJ and MP algorithms supported the finding that strain MBLB2552T formed a stable cluster with P. timonensis 2301032 T (Fig. 1). The genome sequences of P. timonensis 2301032 T (GCA009725205), P. barengoltzii NBRC 101215 T (GCA004000745), P. macerans IAM 12467 T (GCA004000965), P. faecis CIP 101062 T (GCA008084145), P. physcomitrellae XBT (GCA014640575), and P. polymyxa G02T (GCA900454525) were chosen for a more detailed comparison of genomic properties.

Maximum-likelihood (ML) phylogenetic tree, based on the 16S rRNA gene sequences, showing the position and relationship between strain MBLB2552T and related taxa of the genus Paenibacillus. Numbers at nodes indicate bootstrap values (> 70%) calculated based on the ML/ neighbor-joining (NJ) / maximum-parsimony (MP) algorithms for the branch point based on 1000 replications. Closed circles indicate that the corresponding nodes are also recovered by the NJ and MP. Open circles indicate that the corresponding nodes are also recovered by the NJ or MP. Bacillus subtilis DSM10T served as an outgroup. Bar, 0.01 substitutions per nucleotide position
The genome size was 5,176,139 bp, and the G + C content was 52.4 mol %. Of the 4800 predicted genes, 4655 were coding genes and 62 were pseudogenes (Table 1). The whole-genome sequence of strain MBLB2552T was deposited into DDBJ/EMBL/GenBank under the accession number JALPRK000000000. The OrthoANI values between strain MBLB2552T and other strains in the genus Paenibacillus were 89.78% for P. timonensis 2301032 T, 82.34% for P. barengoltzii NBRC 101215 T, and 76.62% for P. macerans IAM 12467 T (Supplementary Fig. S1). The isDDH values between strain MBLB2552T, and P. timonensis 2301032 T, P. polymyxa G02T, P. barengoltzii NBRC 101215 T, P. macerans NBRC 15307 T, P. physcomitrellae CGMCC 1.15044 T, and P. faecis DSM 23593 T were 39%, 25.3%, 25.0%, 20.9%, 19.9%, and 19.4%, respectively. The values of strain MBLB2552T were below the thresholds for generally recognized species descriptions (95–96% and 70%, respectively) (Lee et al. 2015; Meier-Kothoff et al. 2013).
Based on COG database analysis, the predicted functional genes of the strain MBLB2552T mainly belonged to Carbohydrate transport and metabolism (G:487 orthologs), transcription (K:464 orthologs), and carbohydrate transport and metabolism (G:353 orthologs), excluding an unknown functional category (S:946 orthologs) (Supplementary Table S1). According to the KEGG database, most annotated genes of strain MBLB2552T were predicted to be involved in environmental information processing (453), protein families: signaling and cellular processes (320), and protein families: genetic information processing (312) (Supplementary Fig. S2).
Based on the pan-genomic analysis, strain MBLB2552T and other genus Paenibacillus species had 13,958 POGs, 1448 core POGs, 3831 accessory POGs, 8122 unique POGs, and 557 POGs that were exclusively absent (Fig. 2, Supplementary Table S2). Most of the core POGs were related to translation, ribosomal structure, and biogenesis (J), distributed by the COG database and related to carbohydrate metabolism, distributed by the KEGG database. According to the KEGG database, the number of unique POGs in strain MBLB2552T was 232, of which 130 were annotated (Supplementary Table S3). The most annotated unique POGs of strain MBLB2552T were predicted to be ABC transporter pathways (Supplementary Table S3). Based on the COG database, the number of unique POGs of strain MBLB2552T was 234, and unique POGs were related to carbohydrate transport and metabolism (G, 35, 14.96%), transcription (K, 29, 12.40%), and signal transduction mechanisms (T, 24, 10.30%). (Supplementary Fig. S3). The concatenated core POG-based phylogenomic tree showed that MBLB2552T was clustered with the other six Paenibacillus strains (Supplementary Fig. S4). To provide more distinctive evidence of the strain within the genus Paenibacillus, the genome-based phylogenetic tree using TYGS also showed similar aspects, confirming the relatedness between strain MBLB2552T and other genus Paenibacillus species (Fig. 3). Identification of species and subspecies clustering showed that there were 7 species and subspecies, including strain MBLB2552T. In fact, the current genomic analysis of strain MBLB2552T revealed no genomic evidence of its presence in the gut of honey bee. However, it should be noted that some species of the genus Paenibacillus have been reported including P. larvae as a representative pathogen of honeybee, and P. apis and P. intestini isolated from the intestine of Apis mellifera (Ebeling et al. 2016; Yun et al. 2017). The further research on metagenomic analysis of the honey bee gut will be needed to track the genomic evidence for the plausible presence of strain MBLB2552T in the gut of honey bee.

Flower plot indicating core and strain-specific POGs of novel strains MBLB2552T, and six closely related taxa

Phylogenomic tree based on TYGS results showing the relationship between strain MBLB2552T with related type strains in genus Paenibacillus. The whole-genome sequence-based tree was inferred with FastME 2.1.6.1 (Lefort et al. 2015) from GBDP distances. calculated from genome sequences. The branch lengths are scaled in terms of GBDP distance formula d5
Phenotype, biochemical, and chemotaxonomic characteristics
Strain MBLB2552T cells were Gram stain positive, motile, spore forming, and rod shaped, 1.0‒2.0 µm in width by 5.0‒10.7 µm in length (Supplementary Fig. S5). Strain MBLB2552T was resistant to erythromycin and penicillin G, whereas it was sensitive to ampicillin, cephalothin, ciprofloxacin, gentamicin, kanamycin, lincomycin, neomycin, norfloxacin, novobiocin, polymyxin B, streptomycin, and tetracycline. The detailed results of physiological and biochemical characteristics are provided in Table 2, Supplementary Table S4, and the species description.
The cellular fatty acid profile of strain MBLB2552T was analyzed to anteiso-C15:0 (54.7%), C16:0 (11.2%), and iso-C16:0 (10.4%) as major fatty acids (> 10%). The major fatty acid of strain MBLB2552T were similar to those of the type strains of the genus Paenibacillus except for minor qualitative and quantitative differences. Compared with the data of other reference strains, iso-C15:0 was the major fatty acid of P. timonensis 2301032 T, but minor fatty acid (< 10%) of others (Supplementary Table S5).
The major polar lipids in strain MBLB2552T were identified as diphosphatidylglycerol (DPG), phosphatidylglycerol (PG), phosphatidylcholine (PC), phosphatidylserine (PS), unidentified polar lipids (PL), unidentified glycolipids (GL), and unidentified lipids (L) (Supplementary Fig. S6). According to available polar lipid data, DPG, PG, and phosphatidylethanolamine (PE) are the major polar lipids in the genus Paenibacillus. However, the polar lipid profile of isolate MBLB2552T was distinguished from other related genus Paenibacillus strains by differences in the minor proportion of PE (Supplementary data Fig. S6) (Jiang et al. 2015; Li et al. 2016). The major respiratory quinone analyzed was MK-7, which is the major respiratory quinone found in the genus Paenibacillus strains (Clermont et al. 2015; Li et al. 2016). The diagnostic diamino acid in the cell wall was meso-diaminopimelic acid. In summary, the combination of morphological, physiological, chemotaxonomic, phylogenetic, and genomic characteristics was typical of the genus Paenibacillus with a slight difference, which indicates that MBLB2552T belongs to the genus Paenibacillus. Therefore, we conclude that strain MBLB2552T represents a novel genus Paenibacillus species, for which the name Paenibacillus mellifer sp. nov. is proposed.
Description of Paenibacillus mellifer sp. nov.
Paenibacillus mellifer (mel’li.fer. L. masc. adj. mellifer, honey-producing, referring to the origin from the stomach and digestive tract of honey bees Apis mellifera).
Cells are Gram-positive, facultative anaerobic, motile, spore-forming, rod-shaped (1.0‒2.0 × 5.0‒10.7 µm) bacteria. Colonies on TSA are non-pigmented, circular, convex, bright and cream colored. Grows at 20‒45 °C, in 0‒2.0% (w/v) NaCl, and at pH 6.0‒9.0, with optimal growth at 30 °C, pH 8.0, and 0.5% (w/v) NaCl. Positive for catalase and oxidase. Hydrolysis of casein and starch are positive, but Tweens 20, and 80 and gelatin are negative. H2S production is positive. Using API 20NE kit, activity of nitrate reduction, β-glucosidase, β-galactosidase, utilization of glucose, arabinose, mannose, mannitol, N-acetyl-glucosamine, maltose and gluconate is positive. The API 50CH test shows positive for the utilization of D-arabinose, L-arabinose, D-xylose, D-galactose, D-glucose, D-fructose, D-mannose, L-rhamnose, D-mannitol, methyl-α-D-glucoside, N-acetylglucosamine, amygdalin, arbutin, esculin, salicin, D-cellobiose, D-maltose, D-lactose, D-melibiose, sucrose, D-trehalose, inulin, D-raffinose, starch, gentiobiose, D-turanose, D-lyxose, D-tagatose, D-fucose, L-fucose, D-arabitol, L-arabitol, and gluconate. The predominant fatty acid components are anteiso-C15:0, C16:0, and iso-C16:0. The major polar lipids are diphosphatidylglycerol (DPG), phosphatidylglycerol (PG), phosphatidylcholine (PC), phosphatidylserine (PS), unidentified polar lipids (PL), unidentified glycolipid (GL), and unidentified lipids (L). The diagnostic diamino acid in the cell wall was meso-diaminopimelic acid. The MK-7 is the predominant menaquinone. The DNA G + C content of the type strain is 52.4 mol%.
The type strain is MBLB2552T (= JCM 35371 T = KCTC 43386 T), isolated from the honey bee gut. The NCBI GenBank accession numbers for the 16S rRNA gene sequence and complete genome are OL468736 and JALPRK000000000, respectively.
References
Ash C, Priest FG, Collins MD (1993) Molecular identification of rRNA group 3 bacilli (Ash, Farrow, Wallbanks and Collins) using a PCR probe test. Proposal for the creation of a new genus Paenibacillus. Antonie Van Leeuwenhoek 64:253–260
Batra SWT (1995) Bees and pollination in our changing environment. Apidologie 26:361–370
Bauer AW, Kirby WM, Sherris JC, Turck M (1966) Antibiotic susceptibility testing by a standardized single disk method. Am J Clin Pathol 45:493–496
Benson HJ (2002) Microbiological applications: a laboratory manual in general microbiology. McGraw-Hill Book Company, New York
Chaudhari NM, Gupta VK, Dutta C (2016) BPGA- an ultra-fast pan-genome analysis pipeline. Sci Rep 6:24373
Chun J, Oren A, Ventosa A, Christensen H, Arahal DR, da Costa MS, Rooney AP, Yi H, Xu XW, De Meyer S, Trujillo ME (2018) Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int J Syst Evol Microbiol 68:461–466
Clermont D, Gomard M, Hamon S, Bonne I, Fernandez JC, Wheeler R, Malosse C, Chamot-Rooke J, Gribaldo S, Boneca IG, Bizet C (2015) Paenibacillus faecis sp. nov., isolated from human faeces. Int J Syst Evol Microbiol 65:4621–4626
Collins MD, Jones D (1981) Distribution of isoprenoid quinone structural types in bacteria and their taxonomic implication. Microbiol Rev 45:316–354
Dedej S, Delaplane KS (2003) Honey bee (Hymenoptera: Apidae) pollination of rabbiteye blueberry Vaccinium bashei var. ‘Climax’ is pollinator density-dependent. J Econ Entomol 96:1215–1220
Ebeling J, Knispel H, Hertlein G, Fünfhaus A, Genersch E (2016) Biology of Paenibacillus larvae, a deadly pathogen of honey bee larvae. Appl Microbiol Biotechnol 100:7387–7395
Elzeini HM, Ali ARAA, Nasr NF, Elenany YE, Hassan AAM (2021) Isolation and identification of lactic acid bacteria from the intestinal tracts of honey bees, Apis mellifera L., in Egypt. J Apic Res 60:349–357
Felsenstein J (1981) Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 17:368–376
Gilliam M, Lorenz BJ, Wenner AM, Thorp RW (1997) Occurrence and distribution of Ascosphaera apis in North America: chalkbrood in feral honey bee colonies that had been in isolation on Santa Cruz Island, California for over 110 years. Apidologie 28:329–338
Goulson D, Nicholls E, Botías C, Rotheray EL (2015) Bee declines driven by combined stress from parasites, pesticides, and lack of flowers. Science 347:1255957
Grady EN, MacDonald J, Liu L, Richman A, Yuan Z-C (2016) Current knowledge and perspectives of Paenibacillus: a review. Microb Cell Fact 15:203
Huerta-Cepas J, Szklarczyk D, Forslund K, Cook H, Heller D, Walter MC, Rattei T, Mende DR, Sunagwa S, Kuhn M, Jensen LJ, Mering C, Bork P (2015) eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res 44:286–293
Inglis GD, Yanke LJ, Goettel MS (1998) Anaerobic bacteria isolated from the alimentary canals of alfalfa leafcutting bee larvae. Apidologie 29:327–332
Jiang B, Zhao X, Liu J, Fu L, Yang C, Hu X (2015) Paenibacillus shenyangensis sp. nov., a bioflocculant-producing species isolated from soil under a peach tree. Int J Syst Evol Microbiol 65:220–224
Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30
Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066
Kimura M (1983) The Neutral Theory of Molecular Evolution. Cambrige University Press, Cambrige
Kluge AG, Farris JS (1969) Quantitative phyletics and the evolution of anurans. Syst Biol 18:1–32
Komagata K, Suzuki K (1987) Lipids and cell-wall analysis in bacterial systematics. Methods Microbiol 19:161–207
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Kwak MJ, Choi SB, Ha SM, Kim EH, Kim BY, Chun J (2020) Genome-based reclassification of Paenibacillus jamilae Aguilera et al. 2001 as a later heterotypic synonym of Paenibacillus polymyxa (Prazmowski 1880) Ash et al. Int J Syst Evol Microbiol 70:3134–3138
Lane DJ (1991) 16S/23S rRNA sequencing. In: Stackbrandt E, Goodfellow M (eds) Nucleic acid techniques in bacterial systematics. Wiley, New York, pp 115–117
Lee I, Kim YO, Park SC, Chun J (2015) OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int J Syst Evol Microbiol 66:1100–1103
Lee I, Chalita M, Ha SM, Na SI, Yoon SH, Chun J (2017) ContEst16S: an algorithm that identifies contaminated prokaryotic genomes using 16S RNA gene sequences. Int J Syst Evol Microbiol 67:2053–2057
Lefort V, Desper R, Gascuel O (2015) FastME 2.0: a comprehensive, accurate, and fast distance-based phylogeny inference program. Mol Biol Evol 32:2798–2800
Li P, Lin W, Liu X, Li S, Luo L, Lin WT (2016) Paenibacillus aceti sp. nov., isolated from the traditional solid-state acetic acid fermentation culture of Chinese cereal vinegar. Int J Syst Evol Microbiol 66:3426–3431
Meier-Kolthoff JP, Go¨ker M, (2019) TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat Commun 10:2182
Meier-Kolthoff JP, Auch AF, Klenk HP, Goker M (2013) Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform 14:60
Miller LT (1982) Single derivatization method for routine analysis of bacterial whole-cell fatty acid methyl esters, including hydroxy acids. J Clin Microbiol 16:584–586
Minnikin DE, O’Donnell AG, Goodfellow M, Alderson G, Athalye M, Schaal A, Parlett JH (1984) An integrated procedure for the extraction of bacterial isoprenoid quinones and polar lipids. J Microbiol Methods 2:233–241
Mohr KI, Tebbe C (2006) Diversity and phylotype consistency of bacteria in the guts of three bee species (Apoidea) at an oilseed rape field. Environ Microbiol 8:258–272
Osman S, Satomi M, Venkateswaran K (2006) Paenibacillus pasadenensis sp. nov. and Paenibacillus barengoltzii sp. nov., isolated from a spacecraft assembly facility. Int J Syst Evol Microbiol 56:1509–1514
Roh SW, Sung Y, Nam Y-D, Chang H-W, Kim K-H, Yoon J-H, Jeon CO, Oh H-M, Bae J-W (2008) Arthrobacter soli sp. nov., a novel bacterium isolated from wastewater reservoir sediment. J Microbiol 46:40–44
Roux V, Raoult D (2004) Paenibacillus massiliensis sp. nov., Paenibacillus sanguinis sp. nov. and Paenibacillus timonensis sp. nov., isolated from blood cultures. Int J Syst Evol Microbiol 54:1049–1054
Saccà ML, Lodesani M (2020) Isolation of bacterial microbiota associated to honey bees and evaluation of potential biocontrol agents of Varroa destructor. Benef Microbes 11:641–654
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sasser M (1990) Identification of bacteria by gas chromatography of cellular fatty acids. MIDI Technical Note 101. MIDI Inc., Newark, DE
Sneath PHA, Sokal RR (1973) Numerical taxonomy. The principles and practice of numerical classification. Freeman, San Francisco, CA
Staneck JL, Roberts GD (1974) Simplified approach to identification of aerobic actinomycetes by thin-layer chromatography. Appl Microbiol 28:226–231
Tatusov RL, Galperin MY, Natale DA, Koonin EV (2000) The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28:33–36
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Yoon SH, Ha SM, Kwon S, Lim J, Kim Y, Seo H, Chun J (2017) Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int J Syst Evol Microbiol 67:1613–1617
Yun JH, Lee JY, Kim PS, Jung MJ, Bae JW (2017) Paenibacillus apis sp. nov. and Paenibacillus intestini sp. nov., isolated from the intestine of the honey bee Apis mellifera. Int J Syst Evol Microbiol 67:1918–1924
Zhou Y, Gao S, Wei DQ, Yang L-L, Huang X, He J, Zhang Y-J, Li T-K, W-J (2012) Paenibacillus thermophilus sp. nov., a novel bacterium isolated from a sediment of hot spring in Fujian province. China Antonie Van Leeuwenhoek 102:601–609
Zhou X, Guo GN, Bai SL, Li CL, Yu R, Li YH (2015) Paenibacillus physcomitrellae sp. nov., isolated from the moss Physcomitrella patens. Int J Syst Evol Microbiol 65:3400–3406
Funding
This work was carried out with the support of Cooperative Research Program for Agriculture Science and Technology Development (Project No. PJ015755022022). This work was also supported by Research Assistance Program (2021) in the Incheon National University.
Author information
Authors and Affiliations
Contributions
ESC and CYH performed all the experiments, analyzed the data, and drafted the manuscript. HWK acquired funding. MJS designed all the experiments and supervised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Additional information
Communicated by Erko Stackebrandt.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Cho, ES., Hwang, C.Y., Kwon, H.W. et al. Paenibacillus mellifer sp. nov., isolated from gut of the honey bee Apis mellifera. Arch Microbiol 204, 558 (2022). https://doi.org/10.1007/s00203-022-03178-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00203-022-03178-0