
Abstract
A Gram-negative, strictly aerobic, non-motile, rod-shaped bacterial strain CAU 1614T was isolated from a marine sediment sample collected in the Republic of Korea. Optimal growth of strain CAU 1614T proceeded at 30 °C, pH 7.0, and 2% (w/v) NaCl. 16S rRNA gene similarity was lower than 94.5% with genera Aureisphaera, Marinirhabdus, Aureitalea, Gilvibacter, Ulvibacter, and Jejudonia. The highest similarity was with Aureisphaera galaxeae 04OKA003-7T (94.5%). The major cellular fatty acids were iso-C15:0, iso-C16:0, iso-C15:1 G, iso-C16:0 3-OH, and iso-C17:0 3-OH and the predominant menaquinone was MK-6. The polar lipids were phosphatidylethanolamine, phosphoglycolipid, an unidentified lipid, two unidentified aminolipids, and an unidentified glycolipid. The draft genome of strain CAU 1614T was 3.9 Mb and DNA G+C content was 36.0 mol%. On the basis of the phenotypic, chemotaxonomic, and genomic data, strain CAU 1614T presents a novel genus in the family Flavobacteriaceae, for which the name Halomarinibacterium sedimenti gen. nov., sp. nov. is proposed. The type strain is CAU 1614T (= KCTC 82457T = MCCC 1K06083T).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The family Flavobacteriaceae is a member of the order Flavobacteriales initially proposed by Reichenbach (1992) and emendated by Bernardet et al. (1996, 2002) and, more recently, García-López et al. (2019) based on genome information. According to the List of Prokaryotic names with Standing in Nomenclature (LPSN), Flavobacteriaceae comprises 151 validly published genera (https://lpsn.dsmz.de/family/flavobacteriaceae). Flavobacteriaceae is characterized as Gram-negative rod-shaped cells and MK-6 as the predominant respiratory quinone (Bernardet et al. 2002). Strain CAU 1614T was isolated from a marine sediment sample during an investigation of the marine novel bacterial diversity in the Republic of Korea. The purpose of this study was to examine its taxonomic position and characterization via phenotypic, chemotaxonomic, and genome-based approaches.
Materials and methods
Bacterial strain and culture conditions
Strain CAU 1614T was isolated from a marine sediment sample collected from Ayajin, Gangwon-do (38° 16′ 32.0″ N 128° 33′ 12.0″ E), the Republic of Korea. The sample was serially diluted with sterilized 0.85% NaCl solution several times and 100 µl of the diluent was spread onto marine agar 2216 (MA; BD Difco, Sparks, MD, USA) plate. After 7 days of incubation under aerobic conditions at 30 °C, yellow colonies of strain CAU 1614T were harvested and purified by re-streaking onto fresh MA plates more than three times. The purified culture of strain CAU 1614T was harvested and preserved in marine broth 2216 (MB) supplemented with 25% (v/v) glycerol at – 80 °C. The type strains of the species in closely related genera Aureisphaera, Marinirhabdus, Aureitalea, Gilvibacter, Ulvibacter, and Jejudonia were used as reference strains. Aureisphaera galaxeae KCTC 32993T, Aureisphaera salina KCTC 42975T, Marinirhabdus citrea KCCM 43216T, Aureitalea marina KCTC 23434T, Gilvibacter sediminis NBRC 101626T, Ulvibacter antarcticus DSM 23424T, and Jejudonia soesokkakensis KCTC 32325T were obtained from the Korean Collection for Type Cultures (KCTC; Jeollabuk-do, Korea), the Korean Culture Center of Microorganisms (KCCM; Seoul, Korea), the National Institute of Technology and Evaluation (NITE) Biological Resource Center (NBRC; Tokyo, Japan), and the Deutsche Sammlung von Mikroorganismen und Zellkulturen GmBH (DSMZ; Braunschweig, Germany).
Phylogenetic analysis
Genomic DNA from strain CAU 1614T was prepared using a bacterial genomic DNA extraction kit (iNtRON, Seongnam, Korea), and 16S rRNA gene fragments were amplified via polymerase chain reaction (PCR) (Nam et al. 2004). The sequence was determined using a BigDye Terminator Cycle Sequencing Kit and a 3730 Automated DNA Sequencer (Applied Biosystems, Foster City, CA, USA). The 16S rRNA gene sequence similarities between strain CAU 1614T and closely related strains were determined by referencing the NCBI GenBank database (https://www.ncbi.nlm.nih.gov/genbank/). Multiple alignments and phylogenetic tree construction with neighbor-joining (Saitou and Nei 1987), maximum-parsimony (Fitch 1971), and maximum-likelihood (Felsenstein 1981) methods were performed using MEGA7 software. Bootstrap analysis resampling with 1000 replicates was conducted to estimate the branch support (Felsenstein 1985).
Whole-genome sequence analysis
The whole genome of strain CAU 1614T was extracted using a TruSeq DNA PCR-Free kit (Illumina, San Diego, CA, USA). The sequence data were obtained using an Illumina Hiseq sequencer (Illumina). Assembling the sequences was performed via SPAdes version 3.13.0 (http://cab.spbu.ru/software/spades) and K-mer analysis was performed via Jellyfish (version 2.2.3) (http://www.genome.umd.edu/jellyfish.html) and GenomeScope (http://qb.cshl.edu/genomescope). Any contamination and the authenticity of the 16S rRNA gene sequence with PCR amplification were determined using the Basic Local Alignment Search Tool (BLAST; https://blast.ncbi.nlm.nih.gov/Blast.cgi). The digital DNA–DNA hybridization (dDDH), average nucleotide identity (ANI), and average amino acid identity (AAI) values between strain CAU 1614T and the reference strains with genome data on the National Center for Biotechnology Information (NCBI) database were calculated using the Genome-to-Genome Distance Calculator (GGDC; http://ggdc.dsmz.de/ggdc.php), OrthoANI program (http://www.ezbiocloud.net/sw/oat), and AAI calculator (http://enve-omics.ce.gatech.edu/aai/), respectively. DNA–DNA hybridization (DDH) between strain CAU 1614T and most closely related strain without genomic data on NCBI was performed as described previously (Ezaki et al. 1989). The G+C content was calculated based on the genome sequence of strain CAU 1614T. Proteome comparison was verified using the PATRIC webserver (www.patricbrc.org). The whole-genome sequence of strain CAU 1614T was annotated through the Rapid Annotation Using Subsystem Technology (RAST) webserver (http://rast.nmpdr.org/rast.cgi). Various secondary metabolite-related biosynthetic gene clusters (BGCs) were identified via antiSMASH version 6.0.1 (https://antismash.secondarymetabolites.org). The phylogenetic tree based on core genes constructed using UBCG version 3.0 (https://www.ezbiocloud.net/tools/ubcg).
Physiological, morphological, and biochemical analysis
The morphological characteristics of colonies of strain CAU 1614T, including color, texture, shape, and size, were examined after 3 days on MA at 30 °C. Morphological analysis of cells was conducted under a DM 1000 light microscope (Leica, Wetzlar, Germany). The presence of flagella was determined via a JEM 1010 transmission electron microscopy (JEOL, Tokyo, Japan). The gliding motility was examined using the hanging-drop method described by Bowman (2000). Gram staining was performed with a Gram staining kit (bioMérieux, Craponne, France). CAU 1614T was grown in MB at various temperatures (4, 10, 15, 20, 25, 30, 37, and 45 °C) and pH values (4.5–11.5 at 0.5 pH unit intervals and adjusted with 1 M HCl or 1 M NaOH) to determine the optimal growth conditions. Likewise, cells were grown in NaCl-free MB formula broth at various concentrations of NaCl (0–15% (w/v); at 1% (w/v) intervals) to establish the optimal NaCl concentration. The turbidity of the broth was measured after 72 h. Growth under anaerobic conditions was examined in a BACTRON anaerobic chamber (Sheldon Manufacturing, Cornelius, USA). MA, glucose yeast extract agar (GYE), nutrient agar (NA; Difco), brain heart infusion agar (BHI; Difco), tryptic soy agar (TSA; Difco), and Luria agar (LA; Difco) were used to explore its growth on a variety of media. Oxidase and catalase activity were tested with 1% (w/v) tetramethyl-p-phenylenediamine and 3% (v/v) hydrogen peroxide solution, respectively (Cappuccino and Sherman 2010). Various enzyme activity and biochemical tests were performed using API 50CH, API 20NE, and API ZYM kits (bioMérieux). Hydrolyses of casein and starch were examined according to the method of Smibert and Krieg (1994).
Chemotaxonomic characterization
Polar lipids were extracted and separated using two-dimensional thin-layer chromatography with a silica gel plate (60 F254; Merk, NJ, Kenilworth, USA) using the protocol according to Minnikin et al. (1984) plates were sprayed with 10% ethanolic molybdophosphoric acid for total lipids, ninhydrin for aminolipids, α-naphthol reagent for glycolipids, molybdenum blue for phospholipids, and Dragendorff’s reagent for choline, respectively (Kim et al. 2015). Respiratory quinones were extracted and analyzed as described previously (Sasser 2006). Cellular fatty acids were extracted according to the standard Microbial Identification System (MIDI) protocol and separated by 6890 N gas chromatography (Agilent, Santa Clara, CA, USA). The peaks were analyzed using microbial identification software package MMORE library (MIDI database TSBA6).
Results and discussion
Phenotypic and biochemical characteristics
Strain CAU 1614T was a Gram-negative rod-shaped, aerobic, non-motile, and non-flagellated (Fig. S1). Colonies of strain CAU 1614T were yellow, circular, and opaque with a smooth texture after incubation at 30 °C for 3 days. The growth of strain CAU 1614T occurred in a temperature range of 20 to 37 °C, a pH range of pH 6.0–8.0, and an NaCl concentration range of 0–2% (w/v) with optima of 30 °C, pH 7.0, and 2%w/v NaCl, respectively. Strain CAU 1614T grew well on an MA plate but not on NA, GYE, TSA, BHI, or LA plates. The detailed morphological, physiological, and biochemical characteristics of strain CAU 1614T and related strains of the family Flavobacteriaceae are reported in Table 1. Strain CAU 1614T was oxidase- and catalase-positive but cannot hydrolyze casein, starch, or urea. Esculin and potassium 5-ketogluconate were utilized as carbon sources. The narrow NaCl tolerance range (0–2.0% (w/v)) distinguished strain CAU 1614T from the genera Aureisphaera (0–5.5% (w/v)), Marinirhabdus (0.5–6.0% (w/v)), Aureitalea (0–4.5% (w/v)), Gilvibacter (0.5–6.0% (w/v)), Ulvibacter (1–3.0% (w/v)), and Jejudonia (1.0–5.0% (w/v)). Moreover, strain CAU 1614T differed from the most closely related strain Aureisphaera galaxeae 04OKA003-7T by being positive for esterase (C4), esterase lipase (C8), cysteine arylamidase, trypsin, and α-chymotrypsin.
Phylogenetic and genome characterization
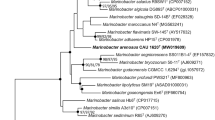
The 16S rRNA gene sequence of strain CAU 1614T (1450 bp) was obtained and compared with closely related species from the NCBI database (access March 2022). Strain CAU 1614T shows the closest similarity to Aureisphaera galaxeae 04OKA003-7T (94.5%), followed by Aureisphaera salina A6D-50T (94.3%) and Marinirhabdus citrea MEBiC09412T (93.5%), all of which belong to the family Flavobacteriaceae. The phylogenetic tree constructed based on the 16S rRNA gene showed the clusters of strain CAU 1614T and related strains in Flavobacteriaceae (Fig. 1). The phylogenetic tree between it and the members of Flavobacteriaceae constructed based on core genes indicates that it is a novel strain from a new genus in Flavobacteriaceae (Fig. 2). The results of the phylogenetic analysis of the 16S rRNA gene show that strain CAU 1614T should be distinguished as a novel taxon in Flavobacteriaceae.

The neighbor-joining tree based on nearly complete 16S rRNA gene sequence of strain CAU 1614T and closely related strains showing the relationship between strains. Bootstrap value > 70% based on 1000 resampling to neighbor-joining, maximum-likelihood and maximum-parsimony analyses (NJ/ML/MP) given. Bar, 0.02 substitutions per position. Flammeovirga aprica NBRC 15941T (AB247553) was used as outgroup species

The phylogenetic tree based on core genes of strain CAU 1614T and closely related strains constructed using UBCG program. Bootstrap value > 70% are shown. Myroides odoratus DSM 2801T (AHKQ01000000) was used as outgroup species
The whole genome of strain CAU 1614T (3.9 Mb in size) contained 15 contigs with an average length of 197,169 bp. The N50 value was 580,342 bp and the K-mer coverage was 175x. Strain CAU 1614T included 2679 protein-coding genes, 3 rRNAs (5S, 16S, and 23S), and 35 tRNAs in the draft genome. The DNA G+C content calculated based on the whole genome was 36.0 mol%, which is in the range of 30–42% reported for the family Flavobacteriaceae. The genome of strain CAU 1614T was not contaminated (the 16S rRNA gene sequence similarity with the PCR amplification result was 99.9%). The DDH value of strain CAU 1614T between Aureisphaera galaxeae 04OKA003-7T was 43.9%. Ortho ANI, dDDH, and AAI values between strain CAU 1614T and Aureitalea marina S1-66T were 68.4%, 19.7%, and 65.4%, respectively, while those between strain CAU 1614T and Ulvibacter antarcticu DSM 23424T were 70.9%, 18.6%, and 67.92%, respectively (Table S1). All of the values were below the thresholds proposed by Meier-Kolthoff et al. (2013) and Lee et al. (2016) for describing novel species. The proteome comparison results show that strain CAU 1614T and related strains share proteins with 10–90% similarity (Fig. S2). The annotated functional genes in the genome of strain CAU 1614T were 917 (Fig. S3). The most (> 100) were categorized as cofactors, vitamins, prosthetic groups, pigments (117 genes), protein metabolism (138 genes), amino acids and derivatives (181 genes), and carbohydrates (106 genes). It contained one secondary metabolite gene cluster verified as a terpene and showed 28% similarity with the carotenoid biosynthetic gene cluster in Algoriphagus sp. KK10202C (Table S2). The carotenoid biosynthesis is one of the characteristics of the family Flavobacteriaceae for yellow pigment (Bernadet et al. 2002) and strain CAU 1614T contained carotenoid biosynthetic gene cluster and produce yellow pigment. The genome of strain CAU 1614T was deposited in the GenBank/EMBL/DDBJ under accession number JAHWDP000000000.
Chemotaxonomic characterization
The major fatty acids (> 10%) identified in strain CAU 1614T were iso-C15:0 (19.5%), iso-C16:0 (11.2%), iso-C15:1 G (11.0%), iso-C16:0 3-OH (17.5%), and iso-C17:0 3-OH (11.8%). The fatty acid profiles of strain CAU 1614T and related type strains in the family Flavobacteriaceae provided in Table S3 show that their major fatty acid compositions are mostly similar. However, iso-C17:0 3-OH was observed in strain CAU 1614T and all of the reference strains except for U. antarcticus DSM 23424T, which only has C18:1 ω7c 11-methyl (10.1%) and summed feature 8 (C18:1 ω6c and/or C18:1 ω7c) (72.5%). The polar lipids detected in strain CAU 1614T were phosphatidylethanolamine, phosphoglycolipid, unidentified lipid, two unidentified aminolipids, and unidentified glycolipid (Fig. S4). The results show that although strain CAU 1614T and the reference strains had the same major polar lipid, strain CAU 1614T differed from other members in the family Flavobacteriaceae due to the presence of phosphoglycolipid (Table 1). The predominant quinone in strain CAU 1614T was menaquinone 6 (MK-6).
Taxonomic conclusion
The results of the phylogenetic analysis based on genome data evidence show the genera and phenotype clusters for strain CAU 1614T, and the chemotaxonomic data support these results. Therefore, we determined that strain CAU 1614T is a novel species in a novel genus, which we have named Halomarinibacterium sedimenti gen. nov. sp. nov.
Description of Halomarinibacterium gen. nov.
Halomarinibacterium (Ha.lo.ma.ri.ni.bac.te'ri.um. Gr. masc. n. hals, -halos, salt; L. adj. marinus, of the sea, marine; N.L. neut. n. bacterium, a small rod; N.L. neut. n. Halomarinibacterium, a halophilic marine rod.) (Table 2).
Cells are Gram-negative, non-motile, rod-shaped, and strictly aerobic, positive for oxidase and catalase. The only menaquinone is MK-6 and the predominant fatty acids are iso-C15:0, iso-C16:0, iso-C15:1 G, iso-C16:0 3-OH, and iso-C17:0 3-OH. The major polar lipids are a phosphoglycolipid and phosphatidylethanolamine.
Description of Halomarinibacterium sedimenti sp. nov.
Halomarinibacterium sedimenti sp. nov. (sed.i.men’ti. L. gen. n. sedimenti, sediment, from which the type strain was isolated).
Cells are Gram-negative, strictly aerobic, non-motile, and rod-shaped, approximately 0.2–0.3 µm in width and 3.0–2.0 µm in length. Colonies are yellow-colored, circular, opaque with a smooth texture, and 0.5–0.8 mm in diameter on MA after 3 days incubation at 30 °C. Optimal growth conditions are 30 °C, pH 7.0, and 2%w/v NaCl. Catalase and oxidase are present. Casein, starch, and urea hydrolysis are not present. Esculin and potassium 5-ketogluconate are used as carbon sources. Enzyme activity is positive for esterase (C4), esterase lipase (C8), alkaline phosphatase, cysteine arylamidase, leucine arylamidase, valine arylamidase, trypsin, acid phosphatase, naphthol-AS-BI-phosphohydrolase, α-chymotrypsin, α-galactosidase, α-glucosidase, and β-glucosidase. The major polar lipids are a phosphoglycolipid and phosphatidylethanolamine. iso-C15:0, iso-C16:0, iso-C15:1 G, iso-C16:0 3-OH, and iso-C17:0 3-OH are major fatty acids.
The type strain CAU 1614T (= KCTC 82457T = MCCC 1K06083T) was isolated from a marine sediment sample from Ayajin, Gangwon-do, Republic of Korea.
Data availability
The 16S rRNA sequence and whole-genome sequence of strain CAU 1614T were obtain to the GenBank/EMBL/DDBJ as MW012854 and JAHWDP000000000, respectively.
References
Bernardet J, Segers P, Vancanneyt M, Berthe F, Kersters K et al (1996) Cutting a Gordian knot: emended classification and description of the genus Flavobacterium, emended description of the family Flavobacteriaceae, and proposal of Flavobacterium hydatis nom. nov. (basonym, Cytophaga aquatilis Strohl and Tait 1978). Int J Syst Evol Microbiol 46:128–148
Bernardet JF, Nakagawa Y, Holmes B (2002) Subcommittee on the taxonomy of Flavobacterium and Cytophaga-Like bacteria of the international committee on systematics of prokaryotes. Proposed minimal standards for describing new taxa of the family Flavobacteriaceae and emended description of the family. Int J Syst Evol Microbiol 52:1049–1070
Bowman JP (2000) Description of Cellulophaga algicola sp. nov., isolated from the surfaces of Antarctic algae, and reclassification of Cytophaga uliginosa (ZoBell and Upham 1944) Reichenbach 1989 as Cellulophaga uliginosa comb. nov. Int J Syst Evol Microbiol 50:1861–1868
Cappuccino JG, Sherman N (2010) Microbiology: a laboratory manual, 9th edn. Benjamin Cummings, San Francisco, USA
Choi TH, Lee HK, Lee K, Cho JC (2007) Ulvibacter antarcticus sp. nov., isolated from Antarctic coastal seawater. Int J Syst Evol Microbiol 57:2922–2925
Ezaki T, Hashimoto Y, Yabuuchi E (1989) Fluorometric deoxyribonucleic acid-deoxyribonucleic acid hybridization in microdilution wells as an alternative to membrane filter hybridization in which radioisotopes are used to determine genetic relatedness among bacterial strains. Int J Syst Evol Microbiol 39:224–229
Felsenstein J (1981) Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 17:368–376
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Fitch WM (1971) Toward defining the course of evolution: minimum change for a specific tree topology. Syst Zool 20:406–416
García-López M, Meier-Kolthoff JP, Tindall BJ, Gronow S, Woyke T et al (2019) Analysis of 1,000 type-strain genomes improves taxonomic classification of Bacteroidetes. Front Microbiol 23:2083
Khan ST, Nakagawa Y, Harayama S (2007) Sediminibacter furfurosus gen. nov., sp. nov. and Gilvibacter sediminis gen. nov., sp. nov., novel members of the family Flavobacteriaceae. Int J Syst Evol Microbiol 57:265–269
Kim JH, Ward AC, Kim W (2015) Kangiella chungangensis sp. nov. isolated from a marine sand. Antonie Van Leeuwenhoek 107:1291–1298
Lee I, Kim YO, Park SC, Chun J (2016) OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int J Syst Evol Microbiol 66:1100–1103
Meier-Kolthoff JP, Auch AF, Klenk HP, Göker M (2013) Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform 14:60
Minnikin DE, O’Donnell AG, Goodfellow M, Alderson G, Athalye M et al (1984) An integrated procedure for the extraction of bacterial isoprenoid quinones and polar lipids. J Microbiol Methods 2:233–241
Nam SW, Kim W, Chun J, Goodfellow M (2004) Tsukamurella pseudospumae sp. nov., a novel actinomycete isolated from activated sludge foam. Int J Syst Evol Microbiol 54:1209–1212
Park S, Yoshizawa S, Inomata K, Kogure K, Yokota A (2012) Aureitalea marina gen. nov., sp. nov., a member of the family Flavobacteriaceae, isolated from seawater. Int J Syst Evol Microbiol 62:912–916
Park S, Lee JS, Lee KC, Yoon JH (2013) Jejudonia soesokkakensis gen. nov., sp. nov., a member of the family Flavobacteriaceae isolated from the junction between the ocean and a freshwater spring, and emended description of the genus Aureitalea Park et al. 2012. Antonie van Leeuwenhoek 104:139–147
Reichenbach H (1992) The order Cytophagales. In: Balows A, Trüper HG, Dworkin M, Schleifer KH, Harder W (eds) The prokaryotes. Springer, New York, pp 3631–3675
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sasser M (2006). Bacterial identification by gas chromatographic analysis of fatty acids methyl esters (GC-FAME). Technical Note 101, Newark, DE: Microbial ID Inc.
Smibert RM, Krieg NR (1994) Phenotypic characterization. In: Gerhardt P, Murray RGE, Wood WA, Krieg NR (eds) Methods for general and molecular bacteriology. American Society for Microbiology, Washington, pp 607–654
Yang SH, Oh JH, Seo HS, Lee JH, Kwon KK (2018) Marinirhabdus citrea sp. nov., a marine bacterium isolated from a seaweed. Int J Syst Evol Microbiol 68:547–551
Yoon J, Yasumoto-Hirose M, Kasai H (2015) Aureisphaera galaxeae gen. nov., sp. nov., a marine member of the family Flavobacteriaceae isolated from the hard coral Galaxea fascicularis. Antonie Van Leeuwenhoek 107:1379–1386
Yoon J, Adachi K, Kasai H (2016) Aureisphaera salina sp. nov., a member of the family Flavobacteriaceae isolated from an ascidian. Int J Syst Evol Microbiol 66:2999–3004
Acknowledgements
We thank Prof. Bernhard Schink and Prof. Aharon Oren for reviewing the nomenclature.
Funding
This work was supported by a grant from the National Institute of Biological Resources (NIBR), funded by the Ministry of Environment (MOE) of the Republic of Korea (NIBR202002203).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: W.K., Conducted the experiments: J.J., V.W., Y.L., Analyzed the data: J.-H.K., A.S., K.K., Contributed reagents, materials, and analysis tools: W. K., Wrote the paper: J.J., K.K., W.K.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Erko Stackebrandt.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Jeong, J., Weerawongwiwat, V., Lee, Y. et al. Halomarinibacterium sedimenti gen. nov., sp. nov., a carotenoid pigment-producing bacterium isolated from marine sediment. Arch Microbiol 204, 517 (2022). https://doi.org/10.1007/s00203-022-03140-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00203-022-03140-0