
Abstract
Continuous observation of wastewater treatment plants is very crucial to keep them safe for proper use and protection from pathogenic contamination. Illumina sequencing technology was used for microbiome structuring from various samples taken from different portions of the wastewater treatment plant, including influent, activated, return sludge and effluent, where different microbial compositions were found. The effluent section was found to have pathogenic microbes such as viruses, Alpha- and deltaproteobacteria, Actinobacteria, Bacteroidetes, clostridia, and bacilli in various concentrations. The presence of viruses, Mycobacterium sp., Mycobacterium fortuitum, bacteroidia, and bacilli was investigated. The species Mycobacterium was found to be higher in quantity in the effluent section. Viruses, including hepatitis A and E, were detected in higher quantity in the effluent part of the sludge in comparison with the influent part of the plant. Our discovery reveals the significance and observation of wastewater treatment plants for the existence of water-borne pathogens in the effluent, principally due to the effect on humans while reusing the water.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Removal of organic waste, including nitrogen, sulfur, phosphorus, and carbon from wastewater treatment, is considered a hot issue recently using specific microorganisms. The bacterial structure exists as inactivated microbial fluid important in the treatment of wastewater treatment plant decides the success potential of the treatment process, which was investigated formerly using several methodologies such as culture-dependent and culture-independent. The structure and framework of microorganisms have been recently sequenced and analyzed via various technologies including 16S rRNA gene, 454 pyrosequencing, and metagenomic sequencing, and these are considered effective tools for microbial evaluation from different wastewater treatment plants (Ye et al. 2012; Sanchez et al. 2013; Ye and Zhang 2013).
Besides, the clone library sequencing approach may produce non-accurate results due to their inherent bias of elaboration (Aird et al. 2011; Ye et al. 2012). The technologies discussed above, including Illumina sequencing technology that was recently verified as a contemporary and inventive practice to unveil microbial genome, is challenging and convoluted (Albertsen et al. 2006; Bragg and Tyson 2014). Most of the published works were previously conducted on the anaerobic microbial structure using 454 pyrosequencing (Wong et al. 2013; Li et al. 2013; Sundberg et al. 2013). The Illumina sequencing technology has reported superior importance regarding low-cost and effective mechanism as compared with 454 pyrosequencing to investigate the microbial community structure (Mardis 2008; Glenn 2011). This technology has been utilized to investigate microbial structure from soil samples (Mackelprang et al. 2011), samples from the ocean (Mason et al. 2014), from gut of human (Qin et al. 2010) and anaerobic digested sludge (Albertsen et al. 2006; Ju et al. 2014). However, only one little study has attempted to analyze complete data of microbiome from digested sludge samples (Yang et al. 2014). Researchers’ reports revealed that it is only bacteria that are involved in the treatment process in the various plants, although the complete genomic studies did not investigate whether the presence of pathogenic microbiome has an impact on the health of humans exposed to it. In this study, we dissect the microbiome structuring from a few sludge samples in the wastewater treatment plant in Beijing city with estimated population of more than 22.0 million.
Generally, a fecal signal such as Escherichia coli or enterococci is observed continuously in the effluent part of wastewater samples, although several other disease-causing agents, including bacteria and viruses may exist in fecal indicators and are transmittable to the environment via several carriers. These disease-causing agents’ expulsion is crucial for achievable consecutive reutilization of water along with the prevention measurement of pathogenic microorganisms in the environment (Varela and Manaia 2013). The presence of prominent water carrier pathogenic bacteria “Mycobacteria” has never been observed and monitored to take precautionary measures for human and animal infection by wastewater. Researchers from European countries have quantified it in the wastewater treatment plant (Radomski et al. 2011). They have reported that these bacteria were not expected to be detected by known parameters. Besides pathogenic bacteria, viruses were also reported to be non-predictable in treated wastewater by given parameters (Savichtcheva and Okabe 2006). In addition, their occurrence might be devalued for the difficulty in concentrating procedures.
Viruses are riskier regarding animals and human health, as they spread easily on contamination (Carducci et al. 2008; Rosa et al. 2010). Although a lot of investigation has been conducted on microbiome dissection from wastewater treatment plants previously, essential information is still missing in the published reports. The purpose of these analyses was to dissect the microbiome structure from various parts, including influent, activated sludge, return sludge, and effluent in a wastewater treatment plant in Beijing to discriminate against the expected phenomena and detect the concentration of viruses, mycobacteria, and other pathogenic microorganisms via metagenomic sequencing technology. The current assay is deeply concerned with the dominant pathogenic microbial species in various parts of the digesting sludge.
Materials and methods
Samples’ collection
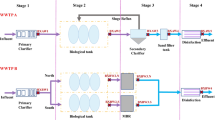
Various parts of the wastewater treatment plant were chosen for sample collection, including influent (R0), activated sludge (R1), return sludge (R2) and effluent (R3) for 24 h after every 30 min. The effluent is the wastewater sample that is clean for reutilizing purposes, while activated and return sludge are undigested and digested sludge, respectively, as shown in Fig. 1. Return sludge is utilized sludge that is reused for another round of influent processing. Besides, the effluent specimen, all the other samples were centrifuged for 10 min at 6000g to make them concentrated. A total of 250 mg of pellets were used for DNA extraction. A 0.22 µm filter injection was used for the filtration of effluent for discovering the structure of the microbiome and the occurrence of pathogens. The filtered effluent sample was used for the diagnosis of pathogenic viruses present as detected in published papers (Maunula et al. 2013).

Schematic diagram of the full-scale wastewater treatment plant and the sampling point
DNA extraction and library preparation
A total of 250 mg of pellets were used for DNA extraction using the Power Soil DNA Isolation kit (MoBio, USA), a market available kit, as reported previously (Kaevska et al. 2011). The quality of the DNA standard was analyzed using gel electrophoresis. Qubit Fluorometer (Thermo, USA) was used to find out the concentration of DNA, which was detected at 740 ng/μL. All the experimental analyses for Illumina HiSeq 2000 were conducted at the School of Medicine, Tsinghua University, Beijing, China, for metagenomic sequencing. The extracted DNA was diluted to 200–300 ng/μL, for further experimental use (Ali et al. 2019a). DNA library was constructed according to the procedure reported by Campanaro et al. 2016. The short reads were fragmented using a minimum 30 quality score, a least read length of 32 bp, and allowing no ambiguous nucleotides. The given parameters were adopted for overlapping: about 20 nucleotide length of the overlap region was needed and allowed at least two mismatches (Ali et al. 2019b).
Bioinformatic analysis
A prediction server (MG-RAST) for metagenomic computational annotation of non-assembled DNA was utilized from the database of Tsinghua University. This prediction server revealed the metabolic pathways and taxonomic affiliation and was executed to analyze protein similarities, as reported in previous reports (Meyer et al. 2008). About 10−5E value cutoff was considered for investigation of taxonomic affiliation on the basis of MG-RAST databases (Tatusov et al. 2000; Kanehisa et al. 2006; Mitra et al. 2011; Huson et al. 2011). The taxonomic affiliation, such as phyla, orders, families, and genus, was determined for annotations. Hierarchical classification at E value cutoff of 10−5 was used for gene annotation of functional profile (Yang et al. 2014). The majority of the genes were classified successfully into the hierarchical metabolic groups. The entire reads were decoded across the bibliography of the Kyoto Encyclopedia of Genes and Genomes (KEGG) databases using BLASTP with a cutoff E value of 10−5 to evaluate the characteristics of the gene engaged in the microbial structure of treatment plant (data not shown).
Results and discussion
About 29,000 active sequences of abundant quality were collected for further evaluation. Similar sequences were found in four parts of the sludge with different quantities. A comparable concentration of active sequences was evaluated in the previous study (Kaevska et al. 2016). The highest quantity of microbes, including viruses, was found in the effluent part of the activated sludge (2515) and fewer in the influent sample (1123), as shown in Supplementary Fig. S1, 2. The return sludge sample was reported in about 1954 discovered species, while the effluent part of the sludge was discovered in about 1743 in class-level classification. A similar report was revealed by Ye and his group in the previous study from a sludge (Ye and Zhang 2013). A group of researchers also noticed that diversification is higher in activated sludge in comparison with the rest of the sludge part (Lee et al. 2015), which is an agreement to our investigation according to the statistical data. The return sludge samples were found to be majorly like the samples from activated sludge as expected. There was found a comparable variance in the microbial structure of influent, activated sludge, return sludge and the effluent part of the sludge.
Bacteria were found to be a major dominance, accounting for 95.14% in the influent part of digesting sludge. In addition, the noticeable concentration ratio of archaea, eukaryote, and viruses was detected in the same part of the sludge. A comparable concentration was reported in previously published studies (Yang et al. 2014). A higher concentration of viruses reported in effluent part of the digesting sludge, accounting for 2.71%, was about three times higher than the influent part of the sludge. A smaller bacterial concentration, but more diversification was noticed in the effluent (R3). The entire domain percentage analysis details are shown in Fig. 2.

Taxonomic profiling at the domain level of the studied anaerobic digestion sludge. Total DNA sequences were assigned to bacteria, eukaryote, archaea, viruses, and other sequences
To dissect and uncover the whole microbial structure and the functional profiling of the digestion sludge, the total short reads were analyzed according to the KEGG category database (Fig. 3). The phyla Proteobacteria, Bacteroidetes, and Firmicutes-associated bacteria were found to be the dominant bacteria in the influent part of the sludge, as shown in Table 1. The discovery correlated with the previous report determined by Lee’s group (Lee et al. 2015). McLellan’s group found a huge number of Actinobacteria, an agreement with our investigation (McLellan et al. 2010). Alpha-, beta-, and deltaproteobacteria were reported to be in significant concentration in all parts of the sludge, while Epsilon- and Gammaproteobacteria were reported to be in less concentration. These findings prove the agreement with the investigation done by Lee and his group (Lee et al. 2015). However, the previous report found Gammaproteobacteria and Clostridiales to be the dominant classes (Ye and Zhang 2013). Many parameters’ variance may cause a difference in the microbial composition of the plant, including climate zones, industry type, and operational conditions (McLellan et al. 2010).

The interactive Krona chart of the full taxonomy
Regarding class-based classification, viruses were found to be in a large concentration of 7% abundance in the effluent part of the sludge, while less concentration was found in the rest of the samples. Hu and their group detected a minute concentration of viruses in their previous investigation (Wagner et al. 2002; Hu et al. 2012). Besides, viruses and proteobacterial classes, clostridia, actinomycetes, Pseudomonades, Burkholderiales, and Bifidiobacteriales were also reported to be in a higher concentration in the effluent part as compared with the other parts of the sludge as shown in Fig. 4. Several species belonging to Pseudomonades are opportunistic disease-causing agents in humans, animals, and plants.

Class-level classification of microbes distributed in wastewater treatment plant
Majorly, Burkholderia bacterial species are vital to both humans and plants. B. pseudomallei is an important pathogen of melioidosis, a certain kind of disease that affects humans, animals as well as septicemia, and pneumonia in susceptible individuals (Godoy et al. 2003). Bifidobacterium is a reported Gram-positive, branched, and immovable anaerobic bacteria. They are ubiquitous inhabitants of the gastrointestinal tract, vagina, and mouth of mammals, including humans (Schell et al. 2002; Mayo and Sinderen 2010). Some species are found useful for the human body, using probiotics.
The species-level classification was executed for better understanding to know the complete microbiota of all parts of the digester. Several species such as Pedobacter, Zunongwangia, Spirosoma, Slackia were found to be in an enhanced concentration in an effluent part of the digester as compared with the rest of the sample, as shown in Fig. 5. In previously published reports, the same concentration of said species was reported in previously published literature (Ye and Zhang 2013). A group of researchers used a FISH technology to reveal that Actinobacteria and Betaproteobacteria are primary bacterial composition in effluent part of the digester (Muszynski et al. 2015). Around 27% of the species that exist in the digester were not completely revealed. Many published data reported the dominant occurrence of Betaproteobacteria and Gammaproteobacteria in activated sludge samples (Kwon et al. 2010). A variance in the bacterial structure of various wastewater treatment plants based on locality, technology, size, and shape of the treatment plant may severely affect the efficiency of the treatment. A huge investigation under process to reveal the variance cofactor of the differences of the bacterial community was conducted (Helbling et al. 2015; Johnson et al. 2015). The wide range of bacterial species exists in activated sludge and effluent parts of the plant, which may have no critical role in the treatment procedure of the water as reported by several researchers (Wagner et al. 2002; Hu et al. 2012). The dominant sequences reported in the current study belonged to the phylum Proteobacteria and has no role in activated sludge fermentation. This study proved an interesting discovery for a comprehensive pathogenic detection in various parts of the digesting sludge. The activated sludge and effluent samples had a similar composition; however, a less concentration of Actinobacteria species was reported. The diversification and pathogenicity of effluent were reported to be higher in comparison with the other parts of the samples. This result revealed that the sedimentation procedure is not complete entirely, and microbiota exists in activated sludge transfer in the effluent section and subsequently in the surface water. Our findings were in agreement with a previously published study, regarding the concentration of pathogenic species such as Mycobacterium and Vibrio species in the effluent (Bibby et al. 2010; Ye and Zhang 2013). Illumina sequencing technology was used in the current study that proved to be a potential method to detect the pathogenic microbes, including Mycobacterium species and viruses.

Species-level classification of microbes distributed in wastewater treatment plant
The pathogenic members belong to Mycobacterium were reported in activated sludge and effluent samples. This is in agreement with the previous investigation observing the occurrence of Mycobacteria in wastewater treatment plants that suggest a safe way of removal by designing a superior technology to reduce the maximum alarming situations (Radomski et al. 2011). In addition, investigation about the removal of such disease-causing agents from the effluent part of the digester is rarely found. Several pathogenic microbiomes, including Mycobacteria, Treponema, Legionellam, and Clostridia, were reported via pyrosequencing technology (Ibekwe et al. 2013). The samples of RNA were detected for the existence virus group in the effluent part of the digester. The RNA samples were found for the occurrence of hepatitis A and E viruses. Our investigation is an agreement with the previous study, where Steyer and his group detected Norovirus and other several enteric virus groups in the effluent section of the digester plant (Steyer et al. 2015). This investigation reveals the significance of observing, supervision, and controlling the presence of dangerous pathogens in various parts of the digested plant, mainly the effluent part due to the spreading danger and persistence in the environment, which could be vital during the reuse of water. Besides, their spread in the environment may cause dangerous pathogenic endeavors.
There are some limitations to the active pathogen detections limit in this study. The concentration of bacteria in the active sludge part is around 107–109 cell mL−1 (Ewert and Paynter 1980); therefore, only a specific amount of this concentration could be detected using Illumina sequencing technology. The detection limits of qPCR were 10 fg DNA (Lee et al. 2006). Only half of the concentration of Escherichia coli cells could be detected. It is recommended that pathogens should be detected with all aspects, including metagenomics, meta-transcriptomics, and proteomic levels, for 100% detection to avoid chances of being spread and persistence in the environment. Only the metagenomic-level detection has faced specific limitations. There are thousands of same species in a specific part of the sludge, but only a few could be pathogenic. For example, there are more than fifty Bacillus species in the current study, but only three to four are pathogens according to the data bank. Besides, in the case of homo species, some could be beneficial, and some could be pathogenic. An example is E. coli, where few are useful for humans (Grozdanov et al. 1917), while some are pathogenic (Karch et al. 2005). However, Illumina sequencing technology is still an effective methodology to screen pathogens in the environment, mainly in the wastewater treatment plant, to avoid severe diseases.
Conclusion
This study investigates the significance of monitoring, observation, and precautionary measurement for the prevalence of disease-causing agents in effluent part of wastewater treatment plants, especially for reuse of water. In addition, it is not possible to detect the existence of pathogenic microbes in wastewater treatment plants only by observation of fecal pollution indicators, as described by several published reports. This study concluded that Ilumina sequencing technology is a potential and robust way to investigate the whole microbial structure affiliated with individual taxonomic groups that occur in the wastewater treatment procedure. In future correspondence, a strategic approach is required to investigate the point that could remove and eliminate the pathogens from the wastewater treatment process. However, Illumina sequencing technology among all the rest of the methods is a powerful technology to detect disease-causing agents in all parts of the wastewater treatment plant that can also quantify the concentration of nucleic acid that occurs in the samples.
References
Aird D, Ross MG, Chen WS, Danielsson M, Fennell T, Russ C (2011) Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol 12:18
Albertsen M, Hansen LBS, Saunders AM, Nielsen PH, Nielsen KL (2006) A metagenome of a full-scale microbial community carrying out enhanced biological phosphorus removal. ISME J 6:1094–1106
Ali N, Gong H, Giwa AS, Yuan Q, Wang KJ (2019a) Metagenomic analysis and characterization of acidogenic microbiome and effect of pH on organic acid production. Arch Microbiol 201:1163–1171
Ali N, Gong H, Liu X, Giwa AS, Yuan Q, Wang KJ (2019b) Evaluation of bacterial association in methane generation pathways of an anaerobic digesting sludge via metagenomic sequencing. Arch Microbiol. https://doi.org/10.1007/s00203-019-01716-x
Bibby K, Viau E, Peccia J (2010) Pyrosequencing of the 16S rRNA gene to reveal bacterial pathogen diversity in biosolids. Water Res 44:4252–4260
Bragg L, Tyson GW (2014) Metagenomics using next-generation sequencing. In: Paulsen IT, Holmes AJ (eds) Environmental microbiology: methods and protocols, methods in molecular biology, vol 1096, 2nd edn. Humana Press, New York, pp 183–201
Campanaro S, Treu L, Kougias PG, De Francisci D, Valle G, Angelidaki I (2016) Metagenomic analysis and functional characterization of the biogas microbiome using high throughput shotgun sequencing and a novel binning strategy. Biotechnol Biofuels 9:26
Carducci A, Morici P, Pizzi F, Battistini R, Rovini E, Verani M (2008) Study of the viral removal efficiency in an urban wastewater treatment plant. Water Sci Technol 58(4):893–897
Ewert DL, Paynter M (1980) Enumeration of bacteriophages and host bacteria in sewage and the activated-sludge treatment process. Appl Environ Microbiol 39(3):576–583
Glenn TC (2011) Field guide to next-generation DNA sequencers. Mol Ecol Resour 11:759–769
Godoy D, Randle G, Simpson AJ, Aanensen DM, Pitt TL, Kinoshita R, Spratt BG (2003) Multilocus sequence typing and evolutionary relationships among the causative agents of melioidosis and glanders, Burkholderia pseudomallei and Burkholderia mallei. J Clin Microbiol 41(5):2068–2079
Grozdanov L, Raasch C, Schulze J, Sonnenborn U, Gottschalk G, Hacker J, Dobrindt U (1917) Analysis of the genome structure of the nonpathogenic probiotic Escherichia coli strain Nissle. J Bacteriol 186(16):5432–5441
Helbling DE, Johnson DR, Lee TK, Scheidegger A, Fenner K (2015) A framework for establishing predictive relationships between specific bacterial 16S rRNA sequence abundances and biotransformation rates. Water Res 70:471–484
Hu M, Wang XH, Wen XH, Xia Y (2012) Microbial community structures in different wastewater treatment plants as revealed by 454-pyrosequencing analysis. Bioresour Technol 117:72–79
Huson D, Mitra S, Ruscheweyh H, Weber N, Schuster S (2011) Integrative analysis of environmental sequences using MEGAN4. Genome Res 21:1552–1560
Ibekwe AM, Leddy M, Murinda SE (2013) Potential human pathogenic bacteria in a mixed urban watershed as revealed by pyrosequencing. PLoS ONE 8(11):79490
Johnson DR, Lee TK, Park J, Fenner K, Helbling DE (2015) The functional and taxonomic richness of wastewater treatment plant microbial communities are associated with each other and with ambient nitrogen and carbon availability. Environ Microbiol 17(12):4851–4860
Ju F, Guo F, Ye L, Xia Y, Zhang T (2014) Metagenomic analysis on seasonal microbial variations of activated sludge from a full-scale wastewater treatment plant over 4 years. Environ Microbiol Rep 6:80–89
Kaevska M, Slana I, Kralik P, Reischl U, Orosova J, Holcikova A, Pavlik I (2011) ‘‘Mycobacterium avium subsp hominissuis’’ in neck lymph nodes of children and their environment examined by culture and triplex quantitative real-time PCR. Clin Microbiol 49(1):167–172
Kaevska M, Videnska P, Vasickova P (2016) Changes in microbial composition of wastewater during treatment in a full-scale plant. Curr Microbiol 72:128–132
Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S (2006) From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res 34:354–357
Karch H, Tarr PI, Bielaszewska M (2005) Enterohaemorrhagic Escherichia coli in human medicine. Int J Med Microbiol 295(6–7):405–418
Kwon S, Kim TS, Yu GH, Jung JH, Park HD (2010) Bacterial community composition and diversity of a full-scale integrated fixed-film activated sludge system as investigated by pyrosequencing. J Microbiol Biotechnol 20(12):1717–1723
La Rosa G, Pourshaban M, Iaconelli M, Muscillo M (2010) Quantitative real-time PCR of enteric viruses in influent and effluent samples from wastewater treatment plants in Italy. Ann Ist Super Sanita 46(3):266–273
Lee DY, Shannon K, Beaudette LA (2006) Detection of bacterial pathogens in municipal wastewater using an oligonucleotide microarray and real-time quantitative PCR. Microbiol Methods 65(3):453–467
Lee SH, Kang HJ, Park HD (2015) Influence of influent wastewater communities on temporal variation of activated sludge communities. Water Res 73:132–144
Li A, Chu Y, Wang X, Ren L, Yu J, Liu X (2013) A pyrosequencing-based metagenomic study of methane-producing microbial community in solid-state biogas reactor. Biotechnol Biofuels 6:3
Mackelprang R, Waldrop MP, DeAngelis KM, David MM, Chavarria KL, Blazewicz SJ (2011) Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature 480:368–U120
Mardis ER (2008) The impact of next-generation sequencing technology on genetics. Trends Genet 24:133–141
Mason OU, Scott NM, Gonzalez A, Robbins-Pianka A, Baelum J, Kimbrel J (2014) Metagenomics reveals sediment microbial community response to deepwater horizon oil spill. ISME 8:1464–1475
Maunula L, Kaupke A, Vasickova P, Soderberg K, Kozyra I, Lazic S, van der Poel WHM, Bouwknegt M, Rutjes S, Willems KA, Moloney R, D’Agostino M, Husman AMD, von Bonsdorff CH, Rzezutka A, Pavlik I, Petrovic T, Cook N (2013) Tracing enteric viruses in the European berry fruit supply chain. Int J Food Microbiol 167(2):177–185
Mayo B, Sinderen D (2010) Bifidobacteria: genomics and molecular aspects. Caister Academic Press, Wymondham (ISBN 978-1-904455-68-4)
McLellan SL, Huse SM, Mueller-Spitz SR, Andreishcheva EN, Sogin ML (2010) Diversity and population structure of sewage-derived microorganisms in wastewater treatment plant influent. Environ Microbiol 12(2):378–392
Meyer F, Paarmann D, D’Souza M, Olson R, Glass EM, Kubal M (2008) The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform 9:386
Mitra S, Rupek P, Richter DC, Urich T, Gilbert JA, Meyer F (2011) Functional analysis of metagenomes and metatranscriptomes using SEED and KEGG. BMC Bioinform 12:21
Muszynski A, Tabernacka A, Milobedzka A (2015) Long-term dynamics of the microbial community in a full-scale wastewater treatment plant. Int Biodeterior Biodegrad 100:44–51
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C et al (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464:59–U70
Radomski N, Betelli L, Moilleron R, Haenn S, Moulin L, Cambau E, Rocher V, Goncalves A, Lucas FS (2011) Mycobacterium behavior in wastewater treatment plant, a bacterial model distinct from Escherichia coli and enterococci. Environ Sci Technol 45(12):5380–5386
Sanchez O, Ferrera I, Gonzalez JM, Mas J (2013) Assessing bacterial diversity in a seawater-processing wastewater treatment plant by 454-pyrosequencing of the 16S rRNA and amoA genes. Microbiol Biotechnol 6:435–442
Savichtcheva O, Okabe S (2006) Alternative indicators of fecal pollution: relations with pathogens and conventional indicators, current methodologies for direct pathogen monitoring and future application perspectives. Water Res 40(13):2463–2476
Schell MA, Karmirantzou M, Snel B, Vilanova D, Berger B, Pessi G, Zwahlen MC, Desiere F, Bork P, Delley M, Pridmore RD, Arigoni F (2002) The genome sequence of Bifidobacterium longum reflects its adaptation to the human gastrointestinal tract. Proc Natl Acad Sci USA 99(22):14422
Steyer A, Gutierrez-Aguirre I, Racki N, Beigot Glaser S, Brajer Humar B, Strazar M, Skrjanc I, Poljsak-Prijatelj M, Ravnikar M, Rupnik M (2015) The detection rate of enteric viruses and Clostridium difficile in a waste water treatment plant effluent. Food Environ Virol 07:164–172
Sundberg C, Al-Soud WA, Larsson M, Alm E, Yekta SS, Svensson BH (2013) 454 pyrosequencing analyses of bacterial and archaeal richness in 21 full-scale biogas digesters. FEMS Microbiol Ecol 85:612–626
Tatusov RL, Galperin MY, Natale DA, Koonin EV (2000) The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28:33–36
Varela AR, Manaia CM (2013) Human health implications of clinically relevant bacteria in wastewater habitats. Environ Sci Pollut Res Int 20(6):3550–3569
Wagner M, Loy A, Nogueira R, Purkhold U, Lee N, Daims H (2002) Microbial community composition and function in wastewater treatment plants. Antonie Van Leeuwenhoek 81(1–4):665–680
Wong MT, Zhang D, Li J, Hui RKH, Tun HM, Brar MS (2013) Towards a metagenomic understanding on enhanced biomethane production from waste activated sludge after pH 10 pretreatment. Biotechnol Biofuels 6:38
Yang Y, Yu K, Xia Y, Lau FTK, Tang DTW, Fung WC (2014) Metagenomic analysis of sludge from full-scale anaerobic digesters operated in municipal wastewater treatment plants. Appl Microbiol Biotechnol 98:5709–5718
Ye L, Zhang T (2013) Bacterial communities in different sections of a municipal wastewater treatment plant revealed by 16S rDNA 454 pyrosequencing. Appl Microbiol Biotechnol 97(6):2681–2690
Ye L, Zhang T, Wang T, Fang Z (2012) Microbial structures, functions, and metabolic pathways in wastewater treatment bioreactors revealed using high-throughput sequencing. Environ Sci Technol 46(24):13244–13252
Acknowledgements
This work was supported by Major Science and Technology Program for Water Pollution Control and Treatment of China (Grant no. 2017ZX07102-004), and National Natural Science Foundation of China (Grant no. 21206084). We are also grateful to University of Chinese Academy of Sciences, China for additional support.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declared that there is no conflict of interest. All the authors read and approved the final manuscript.
Additional information
Communicated by Erko Stackebrandt.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Giwa, A.S., Ali, N., Athar, M.A. et al. Dissecting microbial community structure in sewage treatment plant for pathogens’ detection using metagenomic sequencing technology. Arch Microbiol 202, 825–833 (2020). https://doi.org/10.1007/s00203-019-01793-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-019-01793-y