
Abstract
Clustered regularly interspaced short palindromic repeats (CRISPR) consists of a series of regular repeat-spacer sequences. It can not only act as a natural immune system in most prokaryotes, but also be utilized as the tool of newly developed genome modification and evolutionary researches. Streptococcus thermophilus is an important model organism for the study and application of CRISPR systems. In present study, the occurrence and diversity of CRISPR–Cas systems in the genomes of S. thermophilus were investigated including 4 new sequenced strains CS5, CS9, CS18, CS20, and other 23 strains downloaded from NCBI website. 66 CRISPR/Cas systems were identified among these 27 strains and could divided into four subsystems according to the arrangement of Cas proteins, notably I-E, II-A, II-C and III-A. Overall, 26 type II-C systems, 18 type II-A systems, 13 type III-A systems, 9 type I-E systems were identified. It was mentioned that CS20 contained two type II-C systems which had not been identified in the other 26 S. thermophilus strains. Overall, 1,080 spacers were analyzed and blasted. Sequence identity searches of spacers implied that most spacers derived from partial sequences of exogenous DNA, including various bacteriophages and plasmids. Of note, a large number of novel spacers were found in this study, indicating the unique phage environment they have undergone, especially CS20 strain. In addition, the analysis of the cas1 and cas9 genes revealed the genetic relationship among CRISPR–Cas system in these strains. Furthermore, the analysis of CRISPR spacers also indicated protospacer adjacent motif (PAM) sequences. Summary of PAM sequences could lay the foundations for the application of S. thermophilus CRISPR–Cas system. Our results suggested CS5 and CS18 can be used as model strains in the research of CRISPR–Cas system, and CS20 might have greater application potential in gene editing.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Streptococcus thermophilus is the only species regarded as food-grade microorganism among the genus Streptococcus. It is also wildly recognized as a probiotic which has a positive effect to maintain the balance of the human gastrointestinal flora, improves lactose intolerance as well as immunity (Cui et al. 2016; Fernandez et al. 2017; Freitas 2017; Uriot et al. 2017).
As the dairy starter and probiotic strain, S. thermophilus faces the challenge of virus infection from different environments, including the fermented milk and human gastrointestinal tract. Especially, the latter represents a huge environmental challenge for probiotic bacteria because of containing various phages (Stern et al. 2012). The phage infection causes the failure of milk fermentation and the loss of the probiotic ability of strain (Mills et al. 2010).
Clustered regularly interspaced short palindromic repeats-CRISPR associated proteins (CRISPR–Cas) locus, which constitutes the adaptive immune system, is an important mechanism against exogenous elements infection in bacteria and archaea (Barrangou et al. 2007). CRISPR–Cas system is very important for both dairy and starter culture industries to guard against phage infection. CRISPR–Cas system is hypervariable among distinct prokaryotes, reflecting the diversity of these immune systems (van der Oost et al. 2009; Marraffini and Sontheimer 2010; Hidalgo-Cantabrana et al. 2017).
CRISPRs, a series of regular sequences, consist of the conserved short repeat sequences (24–37 bp) and various spacers with similar lengths (Grissa et al. 2007a). After the long-term immune and evolution process, CRISPR/Cas loci in S. thermophilus present rich diversities (Horvath et al. 2008; Horvath and Barrangou 2010; Deng and Huo 2013). Four types of CRISPR/Cas loci were found in some S. thermophilus strains, named as CRISPR1, CRISPR2, CRISPR3 and CRISPR4 (Wu et al. 2014). Of note, the distributions of these four CRISPR modules in different strains are diverse, of which CRISPR1 is the most prevalent while CRISPR4 only exists in strains containing all four CRISPR loci (Carte et al. 2014). Researchers have analyzed three CRISPRs, including CRISPR1, CRSPR2 and CRISPR3, in eight S. thermophilus strains and the results indicated CRISPR4 was rare (Deng and Huo 2013).
What’s more, every CRISPR locus owns its specific set of Cas proteins and cas genes is located directly near the corresponding CRISPR loci, present both conservations and polymorphisms (Haft et al. 2005; Godde and Bickerton 2006). The diversity and functions of Cas proteins correspond to the functional diversity of the CRISPR systems.
At least 45 different protein families associated with the CRISPR system have been identified in the bacterial and archaeal genomes (Koo et al. 2012). Moreover, Cas1 is regarded as the core protein of Cas protein family and exists in all CRISPR-containing prokaryotes as well as Cas2 (He et al. 2013). It has been demonstrated that increased expression of cas1 and cas2 gene was indicative of higher activity in S. thermophilus LMD-9 during bacteriophage response (Goh et al. 2011). Therefore, the distribution of cas1 or cas2 gene in four CRISPR/Cas loci may confer their active roles in the defense system.
The CRISPR/Cas systems could be divided into three subtypes based on the type and homology of the Cas proteins, which are characterized by different effector complexes that mediate the binding of crRNA to target DNA or RNA. The signature protein of subtype I is Cas3; Cas9 for subtype II and Cas10 for subtype III (Hrle et al. 2014). They are the most common systems detected in S. thermophilus strains. Furthermore, according to the composition and structure of the Cas protein, the three most common subtypes of the CRISPR/Cas system were further divided into I-A, I-B, I-C, I-D, I-E, I-F, I-U; II-A, II-B, II-C; and III-A, III-B, III-C, III-D (Hrle et al. 2014).
In general, lactic acid bacteria (LAB) own a series of mechanisms to defend invasions of various phages and plasmids, including phage-abortive infection, restriction modification and adsorption barriers systems (Allison and Klaenhammer 1998; Chopin et al. 2005). However, there are few foregoing resistance mechanisms found in S. thermophilus (Ali et al. 2014). Instead, S. thermophilus develops various types of CRISPR/Cas systems. To provide immunity for the host cell, CRISPR/Cas system is able to cutoff exogenous DNA through spacer recognition (Stranges et al. 2013). So the spacer sequences are highly identical to exogenous genes, especially diverse Streptococcus species and S. thermophilus bacteriophages. Its immune ability is positively correlated to the ease of spacer acquisition. It has been found that new spacer integration was only detected in CRISPR1 and CRISPR3 when upon the infection of foreign DNA (Paezespino et al. 2015).
In our previous study, 22 S. thermophilus strains were isolated from traditional fermented products in China (Hu et al. 2018). CS5, CS9, CS18 and CS20 strains with excellent technological performances and application potential were used in this study and their genomes were identified. The occurrence and diversity of CRISPR loci in 27 S. thermophilus strains were analyzed.
Materials and methods
Bacterial strains
Streptococcus thermophilus CS5, CS9, CS18, and CS20 were obtained from traditional fermented milk in our previous study (Hu et al. 2018). The nucleotide sequences of CS5, CS9, CS18, and CS20 genomes were submitted to GenBank and assigned accession numbers CP028896, CP030927, CP030928, and CP030250.
CRISPR detection and identification
The 23 S. thermophilus genomes (Supplementary Table S1) in the GenBank database (NCBI) as of August 2018 and four new genomes (CS5, CS9, CS18 and CS20) were used to characterize the occurrence and diversity of CRISPR–Cas systems in S. thermophilus strains according to Bolotin et al. (2005) and Barrangou et al. (2007). The CRISPR Finder was used to find the repeats sequences (Grissa et al. 2007a, b). In addition, secondary structures were predicted through RNAfold web server (https://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) with the minimum free energy (MFE) calculated (Hofacker et al. 1994). The CLC sequence viewer 6 was used to compare the different DRs of the tested strains with the standard strains. Sequence logos were a graphical representation of a nucleic acid multiple sequence alignment developed by WebLogo (https://weblogo.berkeley.edu/logo.cgi, Crooks et al. 2004).
Then, the Cas proteins were predicted by the CRISPR–Cas–Finder (https://crispr.u-psud.fr/crispr/) (Grissa et al. 2007a, b). The CRISPR subtypes designation was performed according to the signature Cas proteins as previously reported (Makarova et al. 2011, 2015; Koonin et al. 2017). Phylogenetic trees based on alignments of cas1 and cas9 sequences in distinct S. thermophilus strains were constructed by the method of Maximum Likelihood using MEGA 7.0 (Kumar et al. 2016).
Spacers’ analyses
CRISPR spacers were analyzed using a custom Excel Macro tool (Horvath et al. 2008) to identify similarity among strains and their divergent evolution under DNA selective pressure. Additional studies were carried out to detect similarity among the CRISPR spacers detected in S. thermophilus strains, plasmids and prophages sequences present in S. thermophilus chromosomes, using BLASTn analyses at GenBank database (NCBI) (Altschul et al. 1997). The software HEMI Illustrator 1.0 was used to depict the heatmaps (Deng et al. 2014).
For the similarity BLAST of spacers, the query coverage and percent identity were both required to be greater than 90%, while the cutoff E-value was 1e-03. But to determine the prophage, the spacers needed to completely match the partial phase sequences, which means the query coverage and identity were both 100%, while the E-value was lower than 1e-06.
Protospacers and PAM (Deveau et al. 2008; Horvath et al. 2008; Mojica et al. 2009) were predicted based on the analysis of CRISPR spacers, and WebLogo server online was used to represent the PAM sequences based on a frequency chart were the height of each nucleotide represents the conservation of that nucleotide at each position (Crooks et al. 2004).
Results and discussion
CRISPR Loci characterization on S. thermophilus strains
The 27 S. thermophilus strains with complete genome sequences were analyzed for the occurrence and diversity of CRISPR–Cas systems by bioinformatics analysis (Table 1, Fig. 1). Among the 27 genomes analyzed, we observed a high rate of occurrence of CRISPR–Cas systems in the species S. thermophilus (96.3%) except strain ACA-DC2. Most strains lack at least one type of CRSIPR, especially CRISPR4. Moreover, four CRISPR loci have different spacer numbers and four different consensus sequences of direct repeats (DRs).

CRISPR loci in S. thermophilus. The CRISPR locus of each strain was annotated and depicted with cas genes in different colors. CRISPR repeats are represented in brackets of each locus (spacers are not represented). Numbers above CRISPR–Cas systems represent their position in the genome (or contig), the comments on right top of the repeat sequences and the number of spacers, respectively (a). Percentage of each subtypes located in all 66 S. thermophilus CRISPR/Cas systems (b)
The GC content of the CRISPR loci was analyzed for each strain and presented in Table 1. While different S. thermophilus strains genomes present a GC content of 39.0% in average, CRISPR loci have GC content between 33 and 35.9% in CRIPSR1 locus, between 38.4 and 40.2% in CRIPSR2 locus, between 36.4 and 39.6% in CRIPSR3 locus, and between 49.3 and 55.2% in CRIPSR4 locus.
Interestingly, CS5, CS18, ASCC 1275, KLDS SM, MN-BM-A 02, and DGCC 7710 strains possessed all four CRISPR loci and 22 CRISPR-associated protein (cas) genes (Table 1, Fig. 1). The diverse CRISPR/Cas loci in these strains suggest that they may have a better adaptive immunity against different bacteriophages compared with those in other sequenced S. thermophilus. This is important for industrial manufacturing of dairy products that use this organism. At the same time, it may well be that these strains have been exposed to more phages. Therefore, S. thermophilus CS5 and CS18, containing all CRISPR loci, can be used as model strains for the study of CRISPR diversity.
CRISPR1 is the most common CRISPR locus in 78% of known sequenced strains of S. thermophilus, except strains CS9, ND 07, ACA-DC 2, EPS, CS8 and S9. In particular, CRISPR1 locus has the highest numbers of DRs and spacers when compared with other three loci. This suggests CRISPR1 is the oldest CRISPR locus in S. thermophilus and a possible effective defense mechanism to integrate novel spacers in CRISPR1 when S. thermophilus is exposed to bacteriophages. At the same time, CRISPR1 is an ideal tool for gene editing because it can form a gRNA–Cas9 complex system (Hao et al. 2018). S. thermophilus CS20 contains two CRISPR1 loci, therefore the strain might have greater application potential for the evolution and transformation study of S. thermophilus (Fig. 1).
In general, CRISPR1, CRISPR3 and CRISPR4 are all located downstream of the cas gene, while CRISPR2 locus is located between the cas genes, separating cas1, cas2 from other cas genes, which may be related to its specific mechanism when facing exogenous DNA invasions. This is consistent with the previous study (Wu et al. 2014).
Diversity of CRISPR in S. thermophilus
The CRISPR subtypes designation was performed based on the signature cas genes and associated ones as previously reported for CRISPR/Cas systems classification (Makarova et al. 2011, 2015; Koonin et al. 2017). Except the strain ACA DC-2 without any CRISPR–Cas system, the type II-C was detected in the other 26 S. thermophilus strains, while type I-E systems were represented in only 9 strains (Table 1, Fig. 1). At the same time, 18 type II-A systems and 13 type III-A systems were identified. While two type II-C systems were detected in strain CS20, and this was not found in any other strains. Generally, CRISPR1 belongs to type II-C, CRISPR2 only appear in type III-A, CRISPR3 is included in type II-A while CRISPR4 exists in type I-E.
It was known Cas1 was the core protein which is widespread among the CRISPR/Cas systems. All of the 66 CRISPR/Cas systems detected in the 27 S. thermophilus strains harbored cas1 gene (Table 1, Fig. 1). The Cas9 also displayed high rate of the occurrence in S. thermophilus strains. Furthermore, the phylogenetic analyses performed with Cas1 and Cas9 protein are shown in Fig. 2a, b, respectively. Two clusters from Cas1 proteins (Fig. 2a) and Cas9 proteins (Fig. 2b) were not independent. The phylogenetic analysis based on Cas9 proteins indicated that Cas9 proteins from different strains had been divided into two groups, including group II-A and group II-C. The Cas9 proteins of the group II-A are from the CRISPR3 locus, while those of group II-C from CRISPR1 locus. Similarly, the Cas1 proteins from the CRISPR3 locus were clustered in group II-A, and the Cas1 proteins from the CRISPR1 locus were clustered in group II-C. The results indicated that Cas1 as core protein in all CRISPR loci, it was a partner of Cas9, which is a signature protein of subtypes II-A and II-C, and they are co-evolving. Furthermore, it was found that Cas1 evolved with Cas3 and Cas10 (data not shown).

CRISPR phylogenetic analyses in S. thermophilus. Phylogenetic tree based on the Cas1 (a) and Cas9 (b) of S. thermophilus strains. The evolutionary history was inferred using the Maximum Likelihood method by MEGA 7.0. The bootstrap consensus tree inferred from 1000 replicates is taken to represent the evolutionary history of the taxa analyzed. Branches corresponding to partitions reproduced in less than 50% bootstrap replicates are collapsed. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) is shown next to the branches
The results confirmed the co-evolutionary trends observed in CRISPR immune systems that the components of these systems co-evolve (Makarova et al. 2011; Chylinski et al. 2014). Interestingly, it could be found that part of the type II-C and I-E evolved from the same branch, while ND 07-2 CRISPR/Cas system is located in the same branch with I-E but belong to type II-A.
Secondary structure prediction and diversity analysis of DR sequences
As its name implies, an important feature of CRISPR DR sequences is the palindromic signature, which is demonstrated to be related to their functional RNA secondary structures. Experiment has indicated that DR sequences act through intermediate messenger RNAs (Tang et al. 2002). According to the summary of CRISPR repeats in LAB, DR sequences are rather various among these species, in both sequences (29–37 bp) and secondary structures (Horvath et al. 2008).
Four common repeat sequences (DR1, DR2, DR3, and DR4) and three non-common repeat sequences (DR*, DR, and
 ) were found in different S. thermophilus strains (Table 1, Fig. 3). The repeat sequences displaying in type II-C could be classified into two kinds, most are DR1, while DR* only existed in strains CS9, CS8, S9, EPS and ND 07. DR2 could be detected in almost all type III-A systems. The repeat sequences locating in type I-E and II-A of almost all strains were DR4 and DR3, respectively, except ND07 strain. It has been known that the most common repeat sequences locating in CRISPR/Cas systems are DR1, DR2, DR3 and DR4, which revealed that the appearance of other types of DR might be caused by mutation or metastasis of genes.
) were found in different S. thermophilus strains (Table 1, Fig. 3). The repeat sequences displaying in type II-C could be classified into two kinds, most are DR1, while DR* only existed in strains CS9, CS8, S9, EPS and ND 07. DR2 could be detected in almost all type III-A systems. The repeat sequences locating in type I-E and II-A of almost all strains were DR4 and DR3, respectively, except ND07 strain. It has been known that the most common repeat sequences locating in CRISPR/Cas systems are DR1, DR2, DR3 and DR4, which revealed that the appearance of other types of DR might be caused by mutation or metastasis of genes.

The centroid secondary structure prediction and the corresponding MFE values (a). Every single circle represented one base and MFE value below implied stabilities of these structures. The left color bar denoted dot plot containing the base pair probabilities. Atypical DR sequences in four types of CRISPRs and their frequency (b). The multiple alignment results are shown in upper half of every CRISPR part, and the sequence frequency logo are shown in lower half
DRs present both diversities and conservations. Except DR4 and
with 28 bp length, other types of DRs are all 36 bp lengths. First, the typical stem-loop secondary structures exist in all types of DR with distinct sizes of stem and loop (structured and unstructured regions) as well as different stability (Fig. 3a). The unstructured regions contribute a lot to the combination between target DNA and relevant Cas proteins together with partial recognition (Cusack 1999), which is an important embodiment of CRISPR functionality. In particular, the common conserved 3′ termini of (C/G)AAC in all DR clusters can further highlight this opinion (Godde and Bickerton 2006; Kunin et al. 2007).
However, the structured stem regions are responsible for stabilities of RNA secondary structures. It can be concluded that MFEs among all DR types are different (Fig. 3a). It is getting lower from DR1 to DR4, which implies a more stable structure. This is closely related to its stem length and G-C base pair amount in stem part. As G-C base pair could form more stable combination, the more G-C base pairs are included in the stem structure, the more stable DR structure can be (Fig. 3a). It can be calculated that the GC percentages of four types of DRs are 30.6%, 44.4%, 38.9% and 64.2%, respectively. Among them, there are less G-C base pairs but longer stem in DR3, thus it is more stable than DR2. Of note, compensatory G-U base pairs, the typical characteristic of RNA secondary structures, can be noticed in DR3 stem structure. In addition, CRISPR repeats tend to form more stable stem-loop structures than the random sequences (Kunin et al. 2007). This finding implies the importance of repeat stabilities in CRISPR/Cas system functioning. Compared with common repeat sequences (DR1, DR2, DR3, and DR4), three non-common repeat sequences (DR*, DR, and
) contain longer stem and additional loop.
What’s more, there are a few atypical repeats (Fig. 3b), which are associated with repeat degeneracy, existing in termini of CRISPR loci, for DR1 and DR2, in the 3′ region, while for DR4 and DR*, in 5′ region. And the appearance of partial 5′ atypical repeats may result from seizing nucleotide from PAM or new spacers (Datsenko et al. 2012). Normally, the atypical repeats are diverse and highly homologous to typical repeats with only one or two nucleotide missing, while atypical repeats and typical repeat (DR2) of CRISPR2 are less similar with lower 83.8% homology (Fig. 3b). In general, trifling repeat degeneracies are observed in DR1 and DR4, while the ratio of atypical repeats in DR2 is relatively high, which is consistent with the result of Horvath et al. (Horvath et al. 2008). At the same time, there are no atypical repeats in DR3.
CRISPR Spacers’ analyses
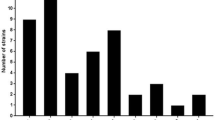
It can be concluded that spacer amounts in different CRISPR types or strains are diverse while spacer lengths with 33–35 bp are rather conservative (Fig. 4). Spacer number in CRISPR1 is the most diverse from 14 to 41, while 32 spacers are common in 7 strains. Strains CNRZ1066 and JIM8232 possessed the largest number of spacers (41). On the contrary, KLDS 3.1003 contained the least number of spacers (14).

Number of CRISPR spacers in strains. The x axis represents the number of CRISPR spacer. The y axis represents the number of strains containing the corresponding spacers. a CRISPR1 spacers, b CRISPR2 spacers, c CRISPR3 spacers, and d CRISPR4 spacers
Similarly, spacer amount in CRISPR2 locus ranged from 3 to 17 and there were 3 spacers in most strains of CRISPR2 locus. Surprisingly, the large number of spacers was in the CRISPR2 loci of KLDS 3.1003 and JIM 8232. Especially, with the developed CRISPR2-Cas but degraded CRISPR3-Cas9 system, KLDS 3.1003 is worthy to be studied further. Likewise, spacer amount in CRISPR3 locus ranged from 3 to 26, and 12 spacers were most common. Eventually, spacers in CRISPR4 locus ranged from 4 to 25, and the general amount of spacers in CRISPR4 locus was 12.
To sum up, spacer distributions in CRISPR loci present diversity. CRISPR1 includes the largest quantities of spacers, and the spacer numbers in CRISPR3 and CRISPR4 are similar, whereas there are only a few spacers in CRISPR2. The vitro experiments have demonstrated that spacers are inclined to integrate into CRISPR1 and CRISPR3 (both belonging to type II-A system), while spacer deletions tend to happen in CRISPR2 more frequently (Achigar et al. 2017). It is likely that CRISPR2 locus may have limited contribution to bacteriophage response because of a less numbers of spacers. As for CRISPR4, there was no novel spacer obtained but a significant increase in expression of Cas7 protein, implying an active immune process, during phage invasion (Sinkunas et al. 2013; Young et al. 2012).
Each unique spacer sequence is obtained from an invading foreign gene element, so the number of unique spacer sequences in CRISPR locus can reflect the activity of the CRISPR locus. The number of spacer in four CRISPR loci in different strains is shown in Fig. 4. Compared with other CRISPR loci, both maximum and average numbers of spacer sequences in the CRISPR1 locus are highest. Among four CRISPR loci, the average number of spacers in the CRISPR2 locus was the lowest, and the number of spacers in the majority of strains in the CRISPR2 locus was only three. Therefore it is speculated that the CRISPR1 locus is the most active in S. thermophilus strains as well as the activity of the CRISPR2 locus was the lowest.
At the same time, the number of spacers can reflect the ability of bacterial challenges against invasive foreign DNA (Hidalgo-Cantabrana et al. 2017). The number of spacers is higher, the ability is stronger. A high number of spacers may reflect higher bacterial challenges against invasive DNA, and these strains have been exposed to more phages. The lower number of spacers is detected in the CRISPR2 of most strains, and high number of spacers is detected in the CRISPR1 of strains CNRZ1066 and JIM8232.
The spacer arrangements of each CRISPR locus were displayed in Fig. 5. The spacer arrangements of CRISPR1 locus could be divided into 13 types. Strain CS5, CS18, CS20-1, ASCC1275, DGCC 7710, KLDS SM and MW-BM-A02 had the same 32 spacer sequences. LMD-09 and SMQ301 belong to the same group. The spacer 6 to the spacer 15 of LMD-09 matched the spacer 7 to the spacer 16 of SMQ301. Moreover, ND03 and APC151, MN-ZLW-002 and MN-BM-A01 owned the same spacer representation with the spacer number of 36 and 30, respectively.

CRISPR spacers arrangement comparison in four CRISPR loci of S. thermophilus. The CRISPR spacer representation was performed based on the length and nucleotide sequence of each spacer. The spacers are represented by a square, different numbers present different group, and each unique spacer sequence is indicated as a unique color. Each unique color combination is a unique spacer sequence while the internal number indicates the group of the spacer. a CRISPR1 spacers; b CRISPR2 spacers; c CRISPR3 spacers; d CRISPR4 spacers. Numbers on top of the spacers array indicate the spacer order. S. thermophilus strains names were displayed on the left
Similarly, the spacer arrangement of CRISPR3 in S. thermophilus CRISPR–Cas System was various and presented 13 different types. These six strains, CS5, CS18, ASCC1275, DGCC 7710, KLDS SM and MW-BM-A02 also had the consistent spacer arrangement. MN-ZLW-002 and MN-BM-A01 contained the same spacer sequences and arrangements with 26 spacers. Interestingly, strain JIM8232 had the shortest spacer arrangement with three spacers. This might be due to gene deletion during the long evolution.
Spacer arrangements in the CRISPR2 and CRISPR4 of S. thermophilus strains showed higher conservation. They might be from the common ancestor, despite the individual, spatial, and temporal differences in sampling, illustrating how stable these loci are (Hidalgo-Cantabrana et al. 2017).
Noteworthy, CRISPR spacer arrangements in CS5, CS18, KLDS SM, MN-BM-A02, ASCC1275, and DGCC 7710 are entirely the same. The results indicated that these strains had a close relationship. These strains all isolated from fermented milk, the first four strains from China, and the last two strains from the United States and Australia, respectively (Hatmaker et al. 2018; Li et al. 2017; Shi et al. 2015; Wu et al. 2014). They had similar genome size, and numbers of proteins. It was speculated that these strains exposed to similar phages environment and formed the same CRISPR–Cas system.
CRISPR Spacers homology to phage and plasmid sequences in S. thermophilus strains
CRISPR/Cas systems in bacteria were used against the infection of foreign DNA and RNA of phages and plasmids (Barrangou and Doudna 2016). In other words, the spacer is a trace of foreign genes’ infestation. The characteristics of the spacers may affect the ability of the strains to resist the infection by different bacterial phages (Barrangou and Horvath 2017).
To determine the origin of each spacer, the spacer sequences were blasted to find the similarity and identity with Streptococcus phages and plasmids, especially S. thermophilus strains. Sequences above 90%, both in query coverage and percent identity, as well as having an E-value at or below 1e−03, were picked. A total of 1080 spacers were blasted, including 635, 71, 274 and 100 spacers for CRISPR1 (type II-C), CRISPR2 (type III-A), CRISPR3 (type II-A) and CRISPR4 (type I-E), respectively.
In general, spacers between DR1 belonging to CRISPR1 locus showed the largest number of spacers targeted phages and plasmids DNA, accounting for 58.80% of the total spacers. CRISPR1 locus is the most widespread type in S. thermophilus strains and owns the largest number of spacers followed by CRISPR3. The spacers were obtained by means of host randomly integrate invader's DNA fragment through homologous recombination and horizontal gene transfer (Deveau et al. 2008). Accordingly, after exposure to phage invasion, host and phages would undergo coevolution (Sapranauskas et al. 2011). It seems that CRISPR1 and CRISPR3 have more chances to realize the “co-revolution” with foreign plasmids and phages DNA during the long process of defense (Bolotin et al. 2005). Among the 274 spacers of CRISPR3, 125 spacers (45.62%) showed similarity to prophage sequences. The numbers of the spacers matched foreign DNA in CRISPR2 and CRISPR4 were 14 (19.72%) and 36 (36%), respectively.
The number of spacer-matched phages and plasmids of each strain were represented by Fig. 6. The CRISPR–Cas systems of the strains CS20 and GABA showed the higher number of spacer-targeting phages and plasmid DNA. The results revealed that the new sequenced strain CS20 might have the higher chance of surviving during infection of prophages. Conversely, strains EPS and S9 presented the lower number of spacers that matched foreign DNA. Noteworthy, the strains CS5, CS18, ASCC1275, DGCC 7710, KLDS SM and MN-BM-002 presented the same 31 spacers matching the phages and plasmids.

The number of spacers matched phages and plasmids. Block color from red to blue represented the number value from large to small. Color gray indicates that the strain did not have this type of CRISPR gene
Results of homology comparison of spacers are listed in Table 2 and Supplementary Table S2. There are several conclusions that can be drawn from the statistical results. First of all, most spacers are homologous with the phage genomes. There are some common phages acting as spacer donators, such as S. thermophilus bacteriophages 20617, 7201, Sfi 19, Sfi 21, and Sfi 11. Especially, the bacteriophage 20617 genome is the most targeted prophage for S. thermophilus spacers. A total of 83 spacers distributed in different types of CRISPR loci had completely matched with 20617 (Table 2 and Supplementary Table S2). These spacers matched with some crucial function regions of the prophage 20617, such as the portal protein and the HNH endonuclease related to the major capsid protein and in the DNA packaging machinery components. The HNH endonuclease is an important component of the terminase packaging reaction (Kala et al. 2014) and the portal protein is a vital character in head assembly, genome packaging, tail attachment, and genome injection (Sun et al. 2015). Thus, the cleavage and insertion of these prophage critical components through CRISPR/Cas immune systems will prevent prophage replication. These S. thermophilus strains will then acquire immunity and survive during the infection process. But for CRISPR loci in S9, NTC 12958, JIM 8232, EPS, LMD-9 and SMQ-301, there is no spacer homologous to bacteriophage 20617 although their homologies with other phages are relatively high.
Remarkably, spacers of CRISPR2, with a few amounts, are also less homologous to foreign DNA. Among the 26 tested strains, these homologous exogenous genes belong to several specific phages including bacteriophages DT1, 7201, TP-778L, TP-J34, and 53. Especially, bacteriophage DT1 seems like a CRISPR2-specific phage, which is rarely found in other types, whereas, in several other strains named KLDS 3.1003, JIM 8232, LMG 18311 and CS9, this specificity of bacteriophage DT1 was visibly weakened. It can be concluded resulting from their varied evolutionary environment. Besides, bacteriophage DT1 had a limited host range (Tremblay and Moineau 1999). Thus, this may lead to the fact that information about its infection history is mostly retained in the degenerated CRISPR2 locus.
What’s more, almost only spacers in CRISPR1 are homologous with several different plasmids, notably pSt08, pt38, pND103, and pND03 (Table 2 and Supplementary Table S2). Intriguingly, they all belong to the same pC19/pUB1104 rolling-circle family even though their hosts are diverse S. thermophilus strains (Turgeon et al. 2004). This result is in accordance with the research carried out by Garneau (Garneau et al. 2010).
Specifically, it was found that the first spacer at 3′ end (tail end) spacers in SMQ-301 CRISPR3 locus presents high homologies with pSt08 and pND103 plasmid genomes along with several replication protein genes. We hypothesized that the ancestor of strain SMQ-301 was presumably an important host for many plasmids, although it is a host of the model cos-type phage DT1 now and can be infected by phages 73 (Achigar et al. 2017; Labrie et al. 2015).
Remarkably, there are many unique spacers distributed in four CRISPR loci of our 26 tested strains. In particular, a large number of spacers in CRISPR4 (64) have no homology to any known S. thermophilus strains, which depends heavily on the lack of CRISPR4 in genomes of S. thermophilus strains available in the public databases. At the same time, some unique spacers were found in the CRISPR2 (57).
In addition, it seems that strain CS20 is pretty special with two CRISPR1 loci, and all of its spacers belonging to the second CRISPR1 locus (CS20-3 CRISPR) are unique. Thus, there is a putative conclusion that it presents a more different phage environment together with the more distant relationship with the other S. thermophilus strains.
Leader and PAM mediate CRISPR adaptation
Among bacteria CRISPR systems, PAM as the undertaker of specificity identification, is critical to both adaptation and interference procedures. These short sequences exist in intrusion DNA rather than CRISPR system, and are located immediately adjacent to the protospacers, typically at the 5′ end for type I systems, and at the 3′ end for type II systems (Gasiunas et al. 2014). In S. thermophilus strains, CRISPR/Cas type II system is the most common model system, its gRNA–Cas9 complex is a traditional gene editing tool which could integrate the foreign DNA into the host's CRISPR by recognizing the PAM during the adaption phase. Detecting the PAM of type II could make better use of the CRISPR/Cas9 system. Compared to Streptococcus pyogenes, PAMs of S. thermophilus (Sth-PAM) seem longer and more restrictive. In addition, they can only be used for double-strand rather than single-strand cutting like S. pyogenes PAMs (Gasiunas et al. 2012; Jinek et al. 2012).
In this analysis, different PAM sequences were identified for each CRISPR type II subtypes present in S. thermophilus strains CS5 and CS20 (Fig. 7). For type II-A, the PAM was identified as 5′-NNAGAAW-3′ is located immediately downstream of the protospacer which was consistent with the previous study (Fujii et al. 2016). Whereas the PAM for type II-C was defined as 5′-GGNG-3′, located in one nucleotide downstream of the protospacer just as the description by Horvath which also reveals that each subtype contains a unique PAM that can serve as a sequence recognition pattern, specific to a particular Cas enzymatic machinery (Horvath et al. 2008).

PAM predictions for subtype II-A (a) and II-C (b). The figure on the left shows the protospacer sequence of the prophage 20617 matched by each spacer (underlined) located on the new sequenced S. thermophilus strains and the downstream region containing the Protospacer Adjacent Motif (PAM) colored red, whereas right displayed the consensus PAM represented with the frequency plot of WebLogo server
In terms of spacer adaptation of CRISPR–Cas systems in S. thermophilus, their chief undertaker can be described as the leader-repeat junction (Wei et al. 2015). As for leaders, the 100–500 bp sequences upstream the CRISPR arrays, their adaptation control functions are revealed in regulating new spacer integration through sequence information directing, especially the nearest conservative sequences of leader-repeat spanning region. These sequences rich in extremely conserved ATTTGA are essential for spacer nick formation during the adaptation process, while the distal region is influence-free for adaptation. In addition, partial core promoter sequences in leaders can also contribute to crRNA transcripts and CRISPR loci expression. In summary, leaders are essential for CRISPR system to recognize and memorize exogenous invasion DNA.
Discussion
CRISPR/Cas systems in four new sequenced S. thermophilus strains, CS5, CS, CS18 and CS20 were analyzed together with other 23 S. thermophilus strains from NCBI. There are several traits of these typical CRISPRs including diversities and conservations.
The distribution of CRISPR loci in S. thermophilus strains are various and different. Among 27 strains, only six strains have four types of CRISPR loci, two of them are strains CS5 and CS18. At the same time, CRISPR/Cas systems can be classified as different subtypes based on the arrangement of Cas protein (Hrle et al. 2014). Four different subtypes, type I-E, type II-A, type II-C and type III-A were identified in S. thermophilus strains among which the type II-C is the most extensive system among these strains. Interestingly, two type II-C systems were detected in S. thermophilus CS20. However, strain 20 does not have CRISPR3 locus, which is common in other strains. Phylogenetic analyses performed with Cas1 and Cas9 proteins revealed that the co-evolutionary trends in CRISPR immune systems in S. thermophilus strains. The results were consistent with the previous studies (Makarova et al. 2011; Chylinski et al. 2014).
When it comes to secondary structures of CRISPR repeats, the specific stem-loop structures not only act as bridges between Cas and the target fragment, but also are responsible for maintaining the stability of the structure. Moreover, better stability in these structures will be in favor of the foreign DNA resistance functions. It is the partial palindromic property of repeats and their transcribed single-strand fragments that mainly determine their special structures (Lillestøl et al. 2006; Kunin et al. 2007). In addition, there are great possibilities for interacted repeats to form stable secondary structures end to end (Horvath et al. 2008). Three non-common repeat sequences (DR*, DR, and
) contain longer stem and additional loop. Interestingly, all repeat sequences in CRISPR loci of strain ND 07 are non-common. Therefore ND 07 can be used as the model strain for the research of structure and function of DR.
Some obvious atypical repeats, closely related to sequence degeneracy and novel spacer acquisition, are observed from the terminal base sequences among four types of CRISPRs loci. The atypical repeats and typical repeat (DR2) of CRISPR2 are less similar with lower 83.8% homology. Based on the particularity of atypical CRISPR2 repeats, further conclusions can be drawn that CRISPR2 has undergone more degeneracy than others. This is confirmed by higher ratio of its atypical repeats.
Spacers, with 33–35 bp similar lengths, have relatively conservative amounts in distinct CRISPR loci. A total of 1080 spacers were identified in 27 strains, including 635, 71, 274 and 100 spacers for CRISPR1, CRISPR2, CRISPR3 and CRISPR4, respectively. CRISPR1 and CRISPR3 loci own high number of spacers. It suggests that these two types of CRISPR systems possessed the higher activity and can largely complete the gene exchange with foreign plasmids and phages DNA to fight against the threatening conditions.
The spacer arrangements of CRISPR1 and CRISPR3 presented diverse, and they could be divided into 13 types. Spacer arrangement in CRISPR2 is the most conservative. It is worth mentioning that CRISPR spacer arrangements in strains CS5, CS18, ASCC1275, DGCC 7710, KLDS SM and MN-BM-A02 are entirely the same. It was concluded that these strains exposed to similar surroundings for a long time and they have quite relative evolution relationship.
Spacers sequences in CRISPR loci are quite diverse but rules-based with great identity with phages genomes of S. thermophilus, including bacteriophages 20617, 7201, Sfi 19, Sfi 21, and Sfi 11. Further, spacers at the 5′ end appear to be more homologous with exogenous DNA and hypervariable. In fact, it has been reported that new spacer integrations are inclined to happen at this end, although novel spacers integrating into the CRISPR middle array were noticed after undergoing a phage challenge assay (Achigar et al. 2017; Hynes et al. 2016a). The latter phenomenon has been described as ectopic spacer integration (Hynes et al. 2016b; McGinn and Marraffini 2016). Remarkably, many unique spacers were found in four CRISPR loci of our 26 tested strains in this study. In particular, a large number of spacers in CRISPR2 (57) and CRISPR4 (64) have no homology to any known S. thermophilus strains.
It seems that strain CS20 is unique in both CRISPR distribution and sequence among these 26 strains, especially in spacers. Although no CRISPR3 locus was found in CS20, it contains two CRISPR1 loci. The strain CS20 own the highest number of spacers (85) and all of its spacers belonging to the second CRISPR1 locus are unique among 26 strains. Therefore it was speculated that CS20 exposed to surroundings with more phages.
Ultimately, the PAM sequence types and the irreplaceable role of leaders in S. thermophilus are also discussed in this paper, which will benefit a lot for the application and mechanism researches about S. thermophilus CRISPRs.
Furthermore, studies about selected CRISPR distributions in different strains will provide references for several important applications of this system, including searches for their evolution background and process, further advance of their anti-phage abilities, selection of another outstanding model CRISPR system together with both the genome modification in these strains using CRISPR/Cas system and utilization of selected CRISPR–Cas system in extensive gene editing.
References
Achigar R, Magadán AH, Tremblay DM, Pianzzola MJ, Moineau S (2017) Phage-host interactions in Streptococcus thermophilus: genome analysis of phages isolated in Uruguay and ectopic spacer acquisition in CRISPR array. Sci Rep 7:43438
Ali Y, Koberg S, Heßner S, Sun X, Rabe B, Back A, Neve H, Heller KJ (2014) Temperate Streptococcus thermophilus phages expressing superinfection exclusion proteins of the ltp type. Front Microbiol 5:98
Allison GE, Klaenhammer TR (1998) Phage resistance mechanisms in lactic acid bacteria. Int Dairy J 8:207–226
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Barrangou R, Doudna JA (2016) Applications of CRISPR technologies in research and beyond. Nat Biotechnol 34(9):933–941
Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712
Barrangou R, Horvath P (2017) A decade of discovery: CRISPR functions and applications. Nat Microbiol 2(7):17092
Bolotin A, Quinquis B, Sorokin A, Ehrlich SD (2005) Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151(8):2551–2561
Carte J, Christopher RT, Smith JT, Olson S, Barrangou R, Moineau S, Glover CV, Graveley BR, Terns RM, Terns MP (2014) The three major types of CRISPR–Cas systems function independently in CRISPR RNA biogenesis in Streptococcus thermophilus. Mol Microbiol 93:98–112
Chopin MC, Chopin A, Bidnenko E (2005) Phage abortive infection in lactococci: variations on a theme. Curr Opin Microbiol 8:473–479
Chylinski K, Makarova KS, Charpentier E, Koonin EV (2014) Classification and evolution of type II CRISPR–Cas systems. Nucleic Acids Res 42:6091–6105
Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Res 14:1188–1190
Cui Y, Xu T, Qu X, Hu T, Xu J, Zhao C (2016) New insights into various production characteristics of Streptococcus thermophilus strains. Int J Mol Sci 17(10):1701
Cusack S (1999) RNA–protein complexes. Curr Opin Struct Biol 9:66–73
Datsenko KA, Pougach K, Tikhonov A, Wanner BL, Severinov K, Semenova E (2012) Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat Commun 3:945
Deng K, Huo G (2013) Detection and homology analysis of CRISPR in Streptococcus thermophilus. Food Sci 34:153–157 (In Chinese)
Deng W, Wang Y, Liu Z, Cheng H, Xue Y (2014) Hemi: a toolkit for illustrating heatmaps. PLoS One 9(11):e111988
Deveau H, Barrangou R, Garneau JE, Labonte J, Fremaux C, Boyaval P, Romero DA, Horvath P, Moineau S (2008) Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol 190:1390–1400
Fernandez MA, Picard-Deland É, Le Barz M, Daniel N, Marette A (2017) Chap. 13 Yogurt and health. In: Frías J, Martínez-Villaluenga C, Peñas E (eds) Fermented foods in health and disease prevention. Academic Press, Cambridge, pp 305–338
Freitas M (2017) Chapter 24. The benefits of yogurt, cultures, and fermentation. In: Floch MH, Ringel Y, Walker WA (eds) The microbiota in gastrointestinal pathophysiology. Implications for human health, prebiotics, probiotics, and dysbiosis. Academic Press, Cambridge, pp 209–223
Fujii W, Kakuta S, Yoshioka S, Kyuwa S, Sugiura K, Naito K (2016) Zygote-mediated generation of genome-modified mice using Streptococcus thermophilus 1-derived CRISPR/Cas system. Biochem Biophys Res Commun 477(3):473–476
Garneau JE, Dupuis M, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadán AH, Moineau S (2010) The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468(7320):67–71
Gasiunas G, Barrangou R, Horvath P, Siksnys V (2012) Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci USA 109:15539–15540
Gasiunas G, Sinkunas T, Siksnys V (2014) Molecular mechanisms of CRISPR-mediated microbial immunity. Cell Mol Life Sci 71:449–465
Godde JS, Bickerton A (2006) The repetitive DNA elements called CRISPRs and their associated genes: evidence of horizontal transfer among prokaryotes. J Mol Evol 62:718–729
Goh YJ, Goin C, O’Flaherty S, Altermann E, Hutkins R (2011) Specialized adaptation of a lactic acid bacterium to the milk environment: the comparative genomics of Streptococcus thermophilus LMD-9. Microb Cell Fact 10:S22
Grissa I, Vergnaud G, Pourcel C (2007a) CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res 35:1–6
Grissa I, Vergnaud G, Pourcel C (2007b) The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinform 8:172–182
Haft DH, Selengut J, Mongodin EF, Nelson KE (2005) A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput Biol 1(6):e60
Hao M, Cui Y, Qu X (2018) Analysis of CRISPR–Cas system in Streptococcus thermophilus and its application. Front Microbiol 9:257
Hatmaker EA, Riley LA, O'Dell KB, Papanek B, Graveley B, Garrett SC, Wei Y, Terns MP, Guss AM (2018) Complete genome sequence of industrial dairy strain Streptococcus thermophilus DGCC 7710. Genome Announc 6(6):e01587–e1617
He Y, Cheng A, Wang M, Zhu D, Wang X, Zhang X (2013) Sequence analysis of the cas2 gene in riemerella anatipestifer. Adv Mater Res 647(3):570–576
Hidalgo-Cantabrana C, Crawley AB, Sanchez B, Barrangou R (2017) Characterization and exploitation of CRISPR loci in Bifidobacterium longum. Front Microbiol 8:1851
Hofacker IL, Fontana W, Stadler PF, Bonhoeffer LS, Tacker M, Schuster P (1994) Fast folding and comparison of RNA secondary structures. Monatsh Chem 125:167–188
Horvath P, Barrangou R (2010) CRISPR/Cas, the immune system of bacteria and archaea. Science 327:167–170
Horvath P, Romero DA, Coûté-Monvoisin AC, Richards M, Deveau H, Moineau S, Boyaval P, Fremaux C, Barrangou R (2008) Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J Bacteriol 190:1401–1412
Hrle A, Maier LK, Sharma K, Ebert J, Basquin C, Urlaub H, Marchfelder A, Conti E (2014) Structural analyses of the crispr protein csc2 reveal the RNA-binding interface of the type I-D cas7 family. RNA Biol 11(8):1072–1082
Hu T, Zhang Y, Cui Y, Zhao C, Jiang X, Zhu X, Wang Y, Qu X (2018) Technological properties assessment and two component systems distribution of Streptococcus thermophilus strains isolated from fermented milk. Arch Microbiol 200(4):567–580
Hynes AP, Labrie SJ, Moineau S (2016a) Programming native CRISPR arrays for the generation of targeted immunity. MBio 7:e00202–00216
Hynes AP, Lemay ML, Moineau S (2016b) Applications of CRISPR–Cas in its natural habitat. Curr Opin Chem Biol 34:30–36
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821
Kala S, Cumby N, Sadowski PD, Hyder BZ, Kanelis V, Davidson AR, Maxwell KL (2014) HNH proteins are a widespread component of phage DNA packaging machines. Proc Natl Acad Sci USA 111:6022–6027
Koo Y, Jung D, Bae E (2012) Crystal structure of Streptococcus pyogenes csn2 reveals calcium-dependent conformational changes in its tertiary and quaternary structure. PLoS One 7(3):e33401
Koonin EV, Makarova KS, Zhang F (2017) Diversity, classification and evolution of CRISPR–Cas systems. Curr Opin Microbiol 37:67–78
Kumar S, Stecher G, Tamura K (2016) Mega7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1897
Kunin V, Sorek R, Hugenholtz P (2007) Evolutionary conservation of sequence and secondary structures in CRISPR repeats. Genome Biol 8:1–7
Labrie SJ, Tremblay DM, Plante PL, Wasserscheid J, Dewar K, Corbeil J, Moineau S (2015) Complete genome sequence of Streptococcus thermophilus SMQ-301, a model strain for phage-host interactions. Genome Announc 3:e480–e1415
Li B, Ding X, Evivie SE, Jin D, Meng Y, Huo G, Liu F (2017) Short communication: genomic and phenotypic analyses of exopolysaccharides produced by, Streptococcus thermophilus KLDS SM. J Dairy Sci 101(1):106–112
Lillestøl RK, Redder P, Garrett RA, Brügger K (2006) A putative viral defence mechanism in archaeal cells. Archaea 2:59–72
Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, van der Oost J, Koonin EV (2011) Evolution and classification of the CRISPR–Cas systems. Nat Rev Microbiol 9:467–477
Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJ, Charpentier E, Haft DH, Horvath P, Moineau S, Mojica FJ, Terns RM, Terns MP, White MF, Yakunin AF, Garrett RA, van der Oost J, Backofen R, Koonin EV (2015) An updated evolutionary classification of CRISPR–Cas systems. Nat Rev Microbiol 13:722–736
Marraffini LA, Sontheimer EJ (2010) CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat Rev Genet 11:181–190
McGinn J, Marraffini LA (2016) CRISPR–Cas systems optimize their immune response by specifying the site of spacer integration. Mol Cell 64:616–623
Mills S, Griffin C, Coffey A, Meijer WC, Hafkamp B, Ross RP (2010) CRISPR analysis of bacteriophage insensitive mutants (BIMs) of industrial Streptococcus thermophilus implications for starter design. J Appl Microbiol 108(3):945–955
Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Almendros C (2009) Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155(Pt 3):733–740
Paezespino D, Sharon I, Morovic W, Stahl B, Thomas BC, Barrangou R, Banfield JF (2015) CRISPR immunity drives rapid phage genome evolution in Streptococcus thermophilus. Mbio 6(2):e00262–e315
Sapranauskas R, Gasiunas G, Fremaux C, Barrangou R, Horvath P, Siksnys V (2011) The Streptococcus thermophilus crispr/cas system provides immunity in Escherichia coli. Nucleic Acids Res 39(21):9275–9282
Shi Y, Chen Y, Li Z, Yang L, Chen W, Mu Z (2015) Complete genome sequence of Streptococcus thermophilus MN-BM-A02, a rare strain with a high acid-producing rate and low post-acidification ability. Genome Announc 3(5):e00979–e1015
Sinkunas T, Gasiunas G, Waghmare SP, Dickman MJ, Barrangou R, Horvath P, Siksnys V (2013) In vitro reconstitution of Cascade-mediated CRISPR immunity in Streptococcus thermophilus. Embo J 32:385–394
Stern A, Mick E, Tirosh I, Sagy O, Sorek R (2012) CRISPR targeting reveals a reservoir of common phages associated with the human gut microbiome. Genome Res 22:1985–1994
Stranges PB, Esvelt KM, Moosburner M (2013) Cas9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol 31:833–838
Sun L, Zhang X, Gao S, Rao PA, Padilla-Sanchez V, Chen Z, Sun S, Xiang Y, Subramaniam S, Rao VB, Rossmann MG (2015) Cryo-EM structure of the bacteriophage T4 portal protein assembly at near-atomic resolution. Nat Commun 6:7548
Tang TH, Bachellerie JP, Rozhdestvensky T, Bortolin ML, Huber H, Drungowski M, Elge T, Brosius J, Hüttenhofer A (2002) Identification of 86 candidates for small non-messenger rnas from the archaeon archaeoglobus fulgidus. Proc Natl Acad Sci USA 99(11):7536–7541
Tremblay DM, Moineau S (1999) Complete genomic sequence of the lytic bacteriophage DT1 of Streptococcus thermophilus. Virology 255(1):63–76
Turgeon N, Frenette M, Moineau S (2004) Characterization of a theta-replicating plasmid from Streptococcus thermophilus. Plasmid 51:24–36
Uriot O, Denis S, Junjua M, Roussel Y, Dary-Mourot A, Blanquet-Diot S (2017) Streptococcus thermophilus: from yogurt starter to a new promising probiotic candidate? J Funct Foods 37:74–89
van der Oost J, Jore MM, Westra ER, Lundgren M, Brouns SJ (2009) CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem Sci 34:401–407
Wei Y, Chesne MT, Terns RM, Terns MP (2015) Sequences spanning the leader-repeat junction mediate CRISPR adaptation to phage in Streptococcus thermophilus. Nucleic Acids Res 43:1749–1758
Wu Q, Tun HM, Leung CC, Shah NP (2014) Genomic insights into high exopolysaccharide-producing dairy starter bacterium Streptococcus thermophilus ASCC 1275. Sci Rep UK 4(7500):4974–4974
Young JC, Dill BD, Pan C, Hettich RL, Banfield JF, Shah M, Fremaux C, Horvath P, Barrangou R, Verberkmoes NC (2012) Phage-induced expression of CRISPR-associated proteins is revealed by shotgun proteomics in Streptococcus thermophilus. PLoS One 7:e38077
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant nos. 31471712; 31371827).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interests to declare.
Additional information
Communicated by Erko Stackebrandt.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hu, T., Cui, Y. & Qu, X. Characterization and comparison of CRISPR Loci in Streptococcus thermophilus. Arch Microbiol 202, 695–710 (2020). https://doi.org/10.1007/s00203-019-01780-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-019-01780-3