
Abstract
SYJ15 is a highly pathogenic Gram-positive Bacillus sp. with top bud spore newly isolated from dying soft shell turtle. 16SrDNA sequencing showed that it is highly homologous to B. cereus, B. thuringiensis and B. anthracis. Biochemical examinations showed that it belongs to B. cereus. To further study the new pathogen, we conducted whole-genome sequencing based on single-molecular sequencing technology from PacBio. Genome assembly analysis showed that the strain has a 5,296,886 bp chromosome, a 218,649 bp plasmid and a 5221 bp plasmid with GC content of 35.51%, 31.91% and 29.75%, respectively. The genome contains 5736 coding sequences and 6 CRISPR systems located in the chromosome as well as 11 genomic islands in the chromosome and the large plasmid. Genome function analyses were annotated by nr database, SwissProt, KEGG, COG, GO, PHI, VFDB, ARDB, Secretory_Protein and T3SS. In addition, 13 gene clusters of secondary metabolism were predicted by antiSMASH. Comparison of SYJ15 with B. subtilis, B. anthracis, B. cereus and B. thuringiensis identified 1031 core genes of the five strains and 816 genes specific to SYJ15. In addition, SYJ15 had the most common core genes with B. thuringiensis, and the least with B. subtilis. Phylogenetic analysis demonstrated that SYJ15 is between B. thuringiensis and B. cereus, suggesting that SYJ15 belongs to Bacillus cereus group. We designed a specific primer pair to distinguish SYJ15 from B. pumilus, B. licheniformis, B.subtilis, B. thuringiensis and B. cereus. In conclusion, information of SYJ15 genome will help to enhance our understanding of pathogenesis of SYJ15 and find effective treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chinese soft shell turtle (Pelodiscus sinensis) is a special freshwater aquaculture species with high nutritional and economical values in China (Chen et al. 1999). However, diseases due to viral and bacterial infections remain one of the important threats for turtle farming under commercial culture conditions. However, relatively few studies have been conducted on the pathogenesis of P. sinensis focusing on Aeromonas hydrophila, A. sobria, A. caviae, Edwardsiella tarda and other conditioned bacterial pathogens (Chen et al. 2013; Li et al. 2008; Lin et al. 2008; Pan et al. 2010; Xiao et al. 2011).
Some Bacillus species, including B. subtilis and B. lichen, B. Licheniformis, and B. cereus, have been widely used in aquaculture to improve culture environment and reduce disease occurrence (Yu et al. 2007). However, studies have also shown that Bacillus sp. could cause diseases in aquatic animals. For examples, Pychynskid et al. pointed out that B. cereus and B. subtilis can cause gill arch necrosis in common carp (Cyprinuscarpio) (Pychynski et al. 1981). Luo et al. found that Bacillus cereus HP1 can cause the skin ulcer syndrome of Stichopus japonicas (Luo et al. 2009). Goodwin et al. reported that B. mushroom caused torso muscle “ulcer” in speckle fork tail catfish (Ictaluruspunctatus) (Goodwin et al. 1994). Chen et al. verified that B. mycoides could lead to severe eyeball and peripheral subcutaneous ulcers in Chinese soft-shell turtle (Chen et al. 2011), similar to human corneal ulcer caused by B. mycoides (Luo et al. 1997).
Chen et al. (2014) reported high farm turtle mortality due to B. thrujing infection-resulted listless swimming, craned or twisted necks, rigid legs, hyperemic laryngeal mucosa, and intestinal hyperemia. In this study, we isolated a dominant pathogenic strain from dying turtle with the clinical signs similar to the above-mentioned syndromes caused by the novel strain named SYJ15, which belongs to Bacillus cereus group. To deepening our understanding of SYJ15, we conducted whole-genome sequencing and performed bioinformatic analyses. In addition, we also designed primers specific to SYJ15 to distinguish SYJ15 from other Bacillus by PCR.
Material and methods
Bacterial isolation and identification
In June 2015, mass animal deaths occurred in a Chinese soft shell turtle culture farm in Zhejiang Province, China. Their clinical signs were similar to those described by Chen et al. (2014). Dominant colonies with consistent morphological features characterized under a light microscope after Gram-stain and spore-stain were isolated from the intestinal lumen, liver, kidney, spleen, and lung of five dying turtles on tryptone soya agar plate at 28 °C for 18–24 h. PCR fragments of 16S rDNA gene from diseased turtle isolates were obtained by amplification using 27F and 1492R primers and sequenced. Nucleotide similarity analysis was conducted using BLAST Searching (NCBI).
Bacterial challenge trial
To evaluate the pathogenic potential of diseased turtle isolates, the isolate SYJ15 was selected for experimental infection. A total of 60 healthy juvenile Chinese soft-shelled turtles with weight of 100–150 g were randomly assigned into six groups with 10 per group. After acclimatized to 25 °C for seven days, five groups of turtles were intraperitoneally inoculated with 0.1 mL saline solutions containing 109, 10 8, 107, 106 and 105 CFU/mL saline. The other group of turtles receiving same amount of saline only was included as control. Necropsy of dying turtles was performed using aseptic techniques, and bacterial isolates were isolated from livers and kidneys of the turtles and identified by species-specific PCR. The 50% lethal dose (LD50) was calculated using the trimmed Spearman–Karber method on the basis of animal mortality data recorded during a 7-day post-injection period (Shen 2011). The animal experiments were approved by the Institutional Animal Care Committee and performed following the guidelines for the care and use of animals in research.
Bacterial genomic sequencing and characters analysis
The genomic DNA of SYJ15 was extracted and purified using a genomic DNA extraction and clean kit (Axygen, Hangzhou, China) following the protocol of the manufacturer. The total DNA obtained was subjected to quality control by agarose gel electrophoresis and quantified by Qubit. Sequencing was performed based on single molecular sequencing technology from PacBio at Beijing Novogene Bioinformatics Technology Co., Ltd. Low quality reads were filtered using SMRT Analysis 2.3.0 and the filtered reads were assembled to generate one contig without gaps.
Prediction of genes in SYJ15 genome assembly was performed using GeneMarkS (https://topaz.gatech.edu/) with integrated model, which combines the native parameters generated by GeneMarkS and parameters of Heuristic model. A whole-genome Blast search with E value < 1e−5 and minimal alignment length percentage > 40% was performed for functional classification against six databases including KEGG (Kyoto Encyclopedia of Genes and Genomes), COG (Clusters of Orthologous Groups), NR (Non-Redundant Protein Database databases), Swiss-Prot, GO (Gene Ontology) and TrEMBL, as well as for pathogenicity and drug resistance analysis against four databases including PHI (Pathogen–Host Interactions), VFDB (Virulence Factors of Pathogenic Bacteria), ARDB (Antibiotic Resistance Genes Database) and CAZy (Carbohydrate-Active enZYmes Database). Secretory proteins were identified using SignalP, proteins related to type I–VII secretion systems were extracted from all the annotation results, and effector proteins of type III secretion system were detected using EffectiveT3. Secondary metabolite gene clusters were predicted using antiSMASH. All annotation files were further combined into one table. Genome overview was created using Circos to show annotation information. Gene predictions and annotations were performed with Rapid Annotation using Subsystem Technology (RAST) database and NCBI Prokaryotic Genome Annotation Pipeline (PGAP) (https://www.ncbi.nlm.nih.gov/genome/annotation_prok/process/).
Genomic sequence comparison of SYJ15 and other Bacillus species
Genomic sequence comparison of SYJ15 and 4 other Bacillus species (B. subtilis, B. anthracis, B. cereus and B. thuringiensis) was performed using MUMmer and LASTZ alignment tools. Translocation/Trans, Inversion/Inv and Trans+Inv were found by alignment analyses. Core/Pan genes of SYJ15 and four other Bacillus strains were clustered using the CD-HIT rapid clustering of similar proteins software with a threshold of 50% pairwise identity and 0.7 length difference cutoff in amino acids. Gene family of SYJ15 and other 4 Bacillus strains is constructed using multi software. Blast was used to pairwise align all genes and eliminate the redundancy by solar and carry out gene family clustering treatment for the alignment results with Hcluster_sgsoftware. The phylogenetic tree was constructed by the TreeBeST using the method of PhyML, and the setting of bootstraps is 1000 with the orthologous genes detected from gene family analysis.
Specific PCR primers for SYJ15 identification
Although the gyrB gene is often used to identify the bacillus with closer genetic relationship, in order to distinguish the highly pathogenic strain SYJ15 from other bacillus, PCR primers specific to SYJ15 were designed for molecular diagnosis of SYJ15-induced diseases. Three primer pairs were designed using primer5.0 based on the large plasmid sequence of SYJ15 (Table 1). DNA samples were extracted from the cultured SYJ15, B. pumilus, B. licheniformis, B. subtilis, B. thuringiensis and B. cereus as PCR templates.
Results
The basic characters of SYJ15
Classic clinic symptoms were observed in ill Chinese soft shell turtles infected by SYJ15, including congestion of internal organs (Fig. 1). Meanwhile, SYJ15 as a dominant strain can be isolated from intestinal lumen, kidney, spleen, lung and liver of five dying turtles. Moreover, SYJ15 was confirmed to cause symptoms similar to those caused by natural infection in bacterial challenge trial. SYJ15 is identified to be a Gram-positive, rod-shaped, and spore-forming bacterium (Fig. 2) belonging to the family of Bacillus. The sequence analysis of PCR fragment of 16sDNA and identification with automatic bacterial test systems by BD Phoenix-100 showed that SYJ15 belongs to B. cerus. The lethal dose 50 (LD50) of SYJ15 was determined as 1.14 × 105 CFU/mL according to the data record of animal challenge trial, which is comparable to that of B. anthracis (Savransky et al. 2013).

Clinic symptoms of a dying turtle (a), intestine with hyperaemia (b) and enlargement lung with hyperaemia (c)

Morphological characters of SYJ15. a Isolates from tissues of sick turtles on TSA plate; b images of SYJ15 after spore staining, where spores were red (arrow) and thallus was blue
Bacterial genomic sequencing and characters analysis
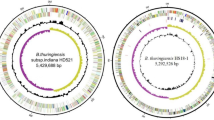
The genome of SYJ15 was sequenced using single molecule, real-time (SMRT) technology. About 67,877 raw sequence reads with mean quality score of 0.85 were generated and assembled into three contigs (N50 contig size, approximately 5.3 Mbp), with a total length of 5,520,756 bp, including a 5,296,886 bp chromosome, a 218,649 bp large plasmid and a 5221 bp small plasmid with GC content of 35.51%, 31.91% and 29.75%, respectively (Fig. 3).

Assembly of SYJ15 genome. a a 5,296,886 bp chromosome, b a 218,649 bp plasmid, and c a 5221 bp plasmid
Gene prediction using GeneMark software (https://topaz.gatech.edu/) indicates that the completed genome contains 5736 coding sequences, which accounts for 84.2% of the total genome. Analysis using CRISPRFinder (https://crispr.u-psud.fr/) identified 6 CRISPR located in chromosome. Analysis using IslandPath-DIOMB software identified 11 genomic islands, ten of which were located in the chromosome and one in the large plasmid.
Function annotation of SYJ15 genome was performed against multiple gene annotation databases. A total of 5,538 annotations were found in nr database (Non-Redundant Protein Database) database, 2700 in SwissProt, 2945 in KEGG (Kyoto Encyclopedia of Genes and Genomes) (Fig. 4), 3899 in COG (Cluster of Orthologous Groups of proteins), and 3756 in GO (Gene Ontology). Figure 5 shows the statistical results of annotated gene distribution in categories of molecular function, cellular component and biological process in GO database. In addition, 64 annotations were found in PHI (Pathogen–Host Interactions database), 86 in VFDB (Virulence Factors of Pathogenic Bacteria), 4 in ARDB (Antibiotic Resistance Genes Database), 151 in Secretory_Protein (SignalP) and 133 in T3SS (Type III secretion system Effector protein, EffectiveT3 software). Moreover, 13 gene clusters of secondary metabolism were predicted by antiSMASH, 10 of which are related to bacteriocin. Furthermore, 14 denovo 5S rRNA, 16S rRNA and 23S rRNA encoding regions were predicted by RNAmmer1.2 (9), 106 tRNAs were identified by tRNAscan-SE (10), 4 sRNAs were predicted by Rfam, 4 prophages were predicted by PHAST software, as well as 393 TRF (Tandem Repeat), 289 minisatellite DNAs and 3 microsatellite DNAs were predicted by RepeatMasker and TRF. Among the four predicted prophages, three are located in the chromosome and one in the large plasmid, and three are incomplete and one is intact.

KEGG metabolic pathway annotation of SYJ15 genome

Statistical results of GO annotation of SYJ15 genome
The genome sequence of SYJ15 has been deposited in GenBank under the accession number CP036354 (small plasmid), CP036355 (large plasmid) and CP036356 (Chromosome).
Genome comparative analysis between SYJ15 and other bacillus
The genome of SYJ15 was compared with that of 4 other Bacillus strains, including NC_000964.3 (B. subtilis subsp. subtilis str. 168), NC_003997.3 (B. anthracis str. Ames), NC_004722.1 (B. cereus ATCC 14,579) and NC_005957.1 (B. thuringiensis serovar konkukian str. 97–27). Of the identified genes, 1031 were the core genes of the five strains and 816 were specific to SYJ15. In addition, SYJ15 shares the most genes, i.e. 4131 genes, with B. thuringiensis (NC_005957.1) and the least genes with B. subtilis (NC_000964.3). Figure 6 shows the Venn diagrams of the identified core genes and pan genes. Average nucleotide identity analyses showed that the ANI values of SYJ15 against B. subtilis, B. cereus, B. thuringiensis and B. anthracis are 67.56%, 91.82%, 94.59% and 94.78%, respectively.

Venn diagrams of the identified core genes and pan genes of SYJ15 with the other 4 Bacillus species
Phylogenetic analysis based on whole genome demonstrated that SYJ15 is clearly distinguishable from other Bacillus species (Fig. 7). Based on the phylogenetic characteristics, SYJ15 is between B. thuringiensis and B. cereus.

Phylogenetic cluster of SYJ15 with NC_000964.3 (Bacillus subtilis subsp. subtilis str. 168), NC_003997.3 (Bacillus anthracis str. Ames), NC_004722.1 (Bacillus cereus ATCC 14,579), and NC_005957.1 (Bacillus thuringiensis serovar konkukian str. 97–27)
Identification of SYJ15 using PCR with specific primer pairs
Three primer pairs were selected for PCR to identify SYJ15. The results showed that a specific and single PCR fragment was amplified using the SYJ153-F and SYJ153-R primer pair with template of SYJ15 DNA, but not other bacillus DNA (Fig. 8). More fragments were amplified using the other two primer pairs from different bacillus DNA, indicating that they are not suitable for distinguishing SYJ15 from the other 5 Bacillus species: B. licheniformis, B. subtilis, B. pumilus, B. cereus and B. thuringiensis.

Agarose gel electrophoresis of PCR products using three different primer pairs to distinguish SYJ15 from other Bacillus species. M: DNA marker DL2000; lanes 1–6: PCR products using primers SYJ135F and SYJ135R with DNA template of B. pumilus, B. licheniformis, B. subtilis, B. thuringiensis, B. cereus and SYJ15, respectively; Lanes 7–12: PCR products using primers SYJ140F and SYJ140R with DNA template of B. pumilus, B. licheniformis, B. subtilis, B. thuringiensis, B. cereus and SYJ15, respectively; Lanes 13–18: PCR products using primers SYJ140F and SYJ140R with DNA template of B. pumilus, B. licheniformis, B. subtilis, B. thuringiensis, B. cereus and SYJ15, respectively
Discussion
SYJ15 is a highly pathogenic bacillus for Chinese soft shell turtle. The Blasting of 16S rDNA sequencing results demonstrated that SYJ15 shared 99% homology with B. cereus, B. thuringiensis, and B. anthracis. SYJ15 was identified as B. cereus using Phoenix 100 automatic microorganisms analyzer. SYJ15 infection leads to sickness in Chinese soft shell turtle with similar clinical symptoms caused by B. thuringiensis described by Chen et al. (2014). Tan et al. isolated a dominant bacterium strain from diseased Chinese soft shell turtle with some similar symptoms caused by SYJ15, and identified it as B. cereus based on its physiological and biochemical characteristics and 16S rRNA gene sequence (Tan et al. 2011). We anticipate that SYJ15, B. thuringiensis described by Chen and B. cereus described by Tan are the same pathogen for Chinese soft shell turtle. Based on the above, SYJ15 should belong to Bacillus cereus group. However, it needs further discussion on exact classification status.
In this study, we for the first time analyzed the whole-genome profile of SYJ15 using single molecular sequence technology. The group of B. cereus is composed of six closely related species, namely B. cereus, B. thuringiensis, B. anthracis, B. weihenstephanensis, B. mycoides and Bacillus pseudomycoides. The strains of B. cereus sensustricto, B. thuringiensis, and B. anthracis share highly conserved chromosomes but differ in the virulence encoding plasmids (Rasko et al. 2005). B. thuringiensis is an insect pathogen (Roh et al. 2007). B. cereus is a well-known food poisoning bacterium able to cause diarrhea, vomiting, and other more severe symptoms (Drobniewski 1993). B. anthracis is the etiological agent of anthrax worldwide and able to infect virtually all mammals. Whether these bacteria represent three distinct species or are subspecies of B. cereus sensulato is still controversy (Daffonchio et al 2000; Vilas-Bôas et al. 2007). The specific phenotype and pathogenicity of the species are often plasmid encoded (Jensen et al. 2003; Rasko et al. 2005), such as the toxins and capsule of B. anthracis (Dale et al. 2018), the insecticidal crystal proteins of B. thuringiensis (Berry et al. 2002), and the cereulide synthesis of emetic B. cereus strains (Ehling-Schulz et al. 2006), but other virulence-related characters including hemolysis, motility as well as resistance to antibiotics are encoded by the chromosome (Drobniewski 1993). Moreover, the pathogen is able to cause edema and cell death through a tripartite toxin consisting of a protective antigen, an edema factor, and a lethal factor (Puhar and Montecucco 2007). The production of polyglutamic acid capsule allows the organism to escape immune system (Missiakas and Schneewind 2017). All these virulence factors are encoded by the toxin plasmid pXO1 (Dale et al. 2018) and the capsule plasmid pXO2 (Koehler 2002). Although sequence of pXO1 and sequence of pXO2 to a lesser extent are widely distributed among strains of B. cereus group (Moayeri et al. 2015; Pannucci et al. 2002), the presence of plasmids with genes encoding toxin and capsule is rare. We identified 86 genes encoding virulence factors in the genome of SYJ15 based on database of Virulence Factors of Pathogenic Bacteria. Among them, 77 genes are located in the chromosome-encoding cytolysin, enterobactin, capsule and other proteins and 9 genes are located in the large plasmid encoding anthrax toxin, capsule, hyaluronic acid capsule, etc. Of the eight genes encoding capsule proteins, six genes are located in chromosome and two genes are located in the large plasmid. Moreover, three genes related to anthrax toxin are > 90% homologous to VFDB.
The complete genome of SYJ15 provides insights into the genes involved in its pathogenicity and further opens up the opportunities for potential exploitation of effective treatments against SYJ15. In addition, due to the similar morphology and high genomic homology between SYJ15 and other Bacillus species, we further selected a primer pair designed based on virulence gene sequence located on the large plasmid of SYJ15 and confirmed that the primer pair can be used in PCR to distinguish SYJ15 from other Bacillus species for molecular detection and accurate diagnosis of the disease caused by SYJ15.
References
Berry C, O’Neil S, Ben-Dov E, Jones AF, Murphy L, Quail MA, Holden MT, Harris D, Zaritsky A, Parkhill J (2002) Complete sequence and organization of pBtoxis, the toxin-coding plasmid of Bacillus thuringiensis subsp. israelensis. Appl Environ Microbiol 68:5082–5095
Chen ZY, Zheng JC, Jiang YL (1999) A new iridovirus isolated from soft-shelled turtle. Virus Res 63:147–151
Chen Q, Yang J, Yu F, Song T (2011) Isolation and identification of Bacillus mycoides in Trionyx sinensis. Fujian J Animal Husb Vet Med 33:4–7
Chen J, Zhu N, Kong L, Bei Y, Zheng T, Ding X, He Z (2013) First case of soft shell disease in Chinese soft-shelled turtle (Trionyx sinensis) associated with Aeromonassobria—A. veronii complex. Aquaculture 406:62–67
Chen J, Zhu N, Kong L, Bei Y, Zheng T, Ding X, He Z (2014) First reported fatal Bacillus thuringiensis infections in Chinese soft-shelled turtles (Trionyx sinensis). Aquaculture 428–429:16–20
Daffonchio D, Cherif A, Borin S (2000) Homoduplex and heteroduplex polymorphisms of the amplified ribosomal 16S–23S internal transcribed spacers describe genetic relationships in the ‘‘Bacillus cereus group’’. Appl Environ Microbiol 66:5460–5468
Dale JL, Raynor MJ, Ty MC, Hadjifrangiskou M, Koehler TM (2018) A dual role for the Bacillus anthracis master virulence regulator AtxA: control of sporulation and anthrax toxin production. Front Microbiol 9:482
Drobniewski FA (1993) Bacillus cereus and related species. Clin Microbiol Rev 6:324–338
Ehling-Schulz M, Fricker M, Grallert H, Rieck P, Wagner M, Scherer S (2006) Cereulide synthetase gene cluster from emetic Bacillus cereus: structure and location on a mega virulence plasmid related to Bacillus anthracis toxin plasmid pXO1. BMC Microbiol 6:20
Goodwin AE, Roy JS Jr, Grizzle JM, Goldsby MT Jr (1994) Bacillus mycoides: a bacterial pathogen of Channel catfish. Dis Aquat Org 18:173–179
Jensen GB, Hansen BM, Eilenberg J, Mahillon J (2003) The hidden lifestyles of Bacillus cereus and relatives. Environ Microbiol 5:631–640
Koehler TM (2002) Bacillus anthracis genetics and virulence gene regulation. Curr Top Microbiol Immunol 271:143–164
Li X, Zhang CL, Fang WH, Lin FC (2008) White-spot disease of Chinese soft-shelled turtles (Trionyx sinensis) caused by Paecilomyceslilacinus. J Zhejiang Univ Sci B 9:578–581
Lin Q, Zhu L, Li Z, Xu B, Xie N (2008) Isolation, determination and antimicrobial susceptibility test of the Citrobacterfreundii septicemia from soft-shelled turtle Trionyx sinensis. Fish Sci 27:42–43
Luo S, Zhou Y, Zhang W (1997) A case of corneal ulcer cause by Bacillus mycoides. Chin J Ophthalmol 33:91
Luo YW, Hao ZK, Wan YG, Qu JB (2009) A strain of bacillus cereus causing stigmatosis sympton complex in sea cucumber Apostichopusjaponicu. Fish Sci Technol Inf 36:60–63
Missiakas D, Schneewind O (2017) Assembly and function of the Bacillus anthracis S-Layer. Annu Rev Microbiol 71:79–98
Moayeri M, Leppla SH, Vrentas C, Pomerantsev AP, Liu S (2015) Anthrax pathogenesis. Annu Rev Microbiol 69:185–208
Pan XY, Hao GJ, Yao JY, Xu Y, Shen JY, Ying WL (2010) Identification and pathogenic facts studying for Edwardsiella tarda from Edwardsiellosis of Trionyx sinensis. Freshw Fish 40:40–45
Pannucci J, Okinaka RT, Williams E, Sabin R, Ticknor LO, Kuske CR (2002) DNA sequence conservation between the Bacillus anthracis pXO2 plasmid and genomic sequence from closely related bacteria. BMC Genomics 3:34
Puhar A, Montecucco C (2007) Where and how do anthrax toxins exit endosomes to intoxicate host cells? Trends Microbiol 15:477–482
Pychynski T, Malanowska T, Kozlowski M (1981) Bacterial flora in branchionecrosis of Carp (particularly Bacillus cereus and Bacillus subtilis). Medycyna Weterynaryjna 37:742–743
Rasko DA, Altherr MR, Han CS, Ravel J (2005) Genomics of the Bacillus cereus group of organisms. FEMS Microbiol Rev 29:303–329
Roh JY, Choi JY, Li MS, Jin BR, Je YH (2007) Bacillus thuringiensis as a specific, safe, and effective tool for insect pest control. J Microbiol Biotechnol 17:547–559
Savransky V, Sanford D, Syar E, Austin J, Tordoff K, Anderson M, Stark G, Barnewall R, Briscoe C, Lemiale-Biérinx L, Park S, Ionin B, Skiadopoulos M (2013) Pathology and pathophysiology of inhalational anthrax in a guinea pig model. Infect Immun 81(4):1152–1163
Shen J (2011) Animal toxicology, 2nd edn. Chinese Agricultural Press, Beijing
Tan A, Zhao F, Jiang L, Luo L, Wang WL, Peng H, Chen Y, Zou W (2011) Isolation and identification of Bacillus cereus from Trionyxsinensis. Guangdong Agric Sci 20:115–119
Vilas-Bôas GT, Peruca AP, Arantes OM (2007) Biology and taxonomy of Bacillus cereus, Bacillus anthracis, and Bacillus thuringiensis. Can J Microbiol 53:673–687
Xiao GH, Wang P, Liu MC, Jiang XH, Liu YS (2011) Isolation, identification and drug sensitive test of Aeromonas hydrophila from Chinese soft-shelled turtle. J Econ Anim 15:56–60
Yu M, Li Z, Wen G (2007) Progress on the application of Bacillus in aquaculture. Guangdong Agric Sci 11:78–81
Acknowledgements
This study was supported by grants of the Zhejiang Provincial Science and Technology Program (No. 2019YSZX002) and the Key Project of Zhejiang Science and Technology (No. 2016C02055-4).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Shuang-Jiang Liu.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yuan, X., Lyu, S., Zhang, H. et al. Complete genome sequence of novel isolate SYJ15 of Bacillus cereus group, a highly lethal pathogen isolated from Chinese soft shell turtle (Pelodiscus Sinensis). Arch Microbiol 202, 85–92 (2020). https://doi.org/10.1007/s00203-019-01723-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-019-01723-y