
Abstract
Wine industry needs a simple method for rapid diagnosis of the dominance of inoculated strains that could be performed routinely during the fermentation process. We present a suitable, high-throughput, and low-cost method to monitor rapidly the dominance of inoculated yeast strains in industrial fermentations of red and white wines using an activated carbon cleaning pretreatment, and a rapid DNA extraction method plus multiplex PCR-SSR analysis. We apply this technique directly to samples of fermenting wines without previously isolating yeast colonies. Results are obtained in a maximum time of 4.5 h.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Inoculation with selected yeasts as starter culture is often referred to establish a dominant population of a selected strain from the beginning of fermentation (Rodríguez et al. 2010; Suzzi et al. 2012). Risks associated with spontaneous or inoculated fermentations include both slow or arrested fermentations and the proliferation of contaminant yeasts (Fleet 2008; Rodríguez et al. 2010). To avoid these problems, commercially available wine yeasts exhibit great diversity in degrees of robustness, but unfortunately the most resilient strains do not necessarily deliver optimal sensory characteristics to the wine (Suzzi et al. 2012; Varela et al. 2009). In addition, the use of commercial starters could mask the distinctive properties of some local wines (Capello et al. 2004; Rodríguez et al. 2010; Varela et al. 2009). Some oenologists believe that wines with a geographical distinction can be obtained only with selected native yeast starters, originated from the micro-area where wines are produced and the use of native wine yeasts selected from each vine growing area is widespread (Cordero-Bueso et al. 2016; Capece et al. 2011; Esteve-Zarzoso et al. 2000). Nevertheless, the addition of the selected yeast is a key strategy to control alcoholic fermentation. If the starter culture and/or the inoculation management is not well controlled, the selected yeast will not gain dominance, and wild yeasts can manage the process due to a good adaptation to the wine cellar environment (Barrajón et al. 2009; Delteil, 1992; Fleet 2008). The molecular techniques commonly used in enology laboratories to differentiate between yeast strains of the same species are pulsed field gel electrophoresis (PFGE) and restriction analysis of the mitochondrial DNA (mtDNA RFLP) (González et al. 2007; López et al. 2001a; Rodríguez et al. 2010, 2011; Santamaría et al. 2005). The application of these techniques demonstrates that, in many cases, the population of a fermenter is “taken over” by wild yeasts, which relegate the inoculated strains to a minority presence (Esteve-Zarzoso et al. 2000; Lopes et al. 2007; Raspor et al. 2002). In order to minimize the impact of unwanted ecological evolutions, the industry needs a simple method for rapid diagnosis of the dominance of inoculated strains that could be performed routinely during the fermentation process (Ambrona et al. 2006; López et al. 2003). Despite the fact that some authors such as López et al. (2001a) and Rodríguez et al. (2011) enhanced yeast dominance and implantation detection protocols, these methods are still too time-consuming for handling higher numbers of samples and the material and reagent costs for extractions are high considering the price of commercial DNA extraction kits or the necessary volume of reagents. Moreover, the limitation of the majority of these methods is that they are culture-dependent techniques or cannot be applied directly to all type of wines (Rodríguez et al. 2011). DNA quality is critical because the efficiency of PCR amplification can be reduced by inhibitors from the matrix. In the case of red wines, it is not easy to obtain a clean yeast cell pellet from a centrifuged sample due to high levels of polysaccharides, anthocyanins and other colored polyphenols, and other organic constituents that interfere with DNA isolation and purification, or with high-throughput applications, such as mtDNA RFLP mapping or PCR-based analysis. DNA extraction of red wines has been highlighted as a limitation of culture-independent methods (Abriouel et al. 2006; Cankar et al. 2006). Some DNA extraction protocols have been adopted as pretreatments to have a clean centrifuged yeast pellet before the DNA extraction by using several compounds such as EDTA-PVP (0.15 M NaCl, 0.1M EDTA and 2 % (v/w) polyvinyl pyrrolidone (PVP)) or using NaCl (1 %) and frozen overnight; however, it requires elevated reagents costs or a long time to have results (Jara et al. 2008). Activated carbon is used to purify liquids and gases in a variety of applications, including municipal drinking water, food and beverage processing, odor removal, industrial pollution control, and point-of-use filters in the home. Activated carbon is also applied in some enological practices such as wine clarification (Salazar et al. 2007; López et al. 2001b), ochratoxin A and geosmin removal (Lisanti et al. 2014, Olivares-Marín et al. 2009), or winery wastewaters color removal (Devesa-Rey et al. 2011). Thus, the use of activated carbon filters could be an economic and suitable alternative to have a clean pellet before the DNA extraction.
In previous works, we analyzed the fermentation time courses of a winery during more than ten consecutive years, to evaluate the dominance of the inoculated yeasts by monitoring the proportions of inoculated and wild yeast strains during the fermentation process using mtDNA-RFLP, obtaining the results in about 7 h in the case of white wines and in 20 h in red wine samples (Rodríguez et al. 2011). We herein present a suitable, high-throughput, and rapid method, reducing the time to a maximum of 4.5 h, to monitor rapidly the dominance of inoculated yeast strains during industrial fermentations of white and red wines. We have used activated carbon as wine samples cleaning agent. We have optimized a quick DNA extraction method and applied multiplex PCR-SSR analysis as strategy directly to samples of fermenting wines, avoiding previous isolation of yeast colonies. Simultaneously, we tested different red and white grape varieties at laboratory and comparing the results using RFLP of the mitochondrial DNA to ensure the validity of the results obtained from the multiplex PCR-SSR analysis.
Materials and methods
Yeast strains and cultures
Saccharomyces cerevisiae strains UCAY-1001, UCAY-1009, UCAY-1013, UCAY-1032, and UCAY-2001 were obtained from the University of Cádiz Yeast Collection (Puerto Real, Cádiz, Spain) which are maintained at −80 °C in a 40 % glycerol solution. UCAY-1001 was isolated from a commercial winery of the D.O. Jerez-Xèrés-Sherry (Rodríguez et al. 2010). Strain UCAY-1009 was isolated from the commercial active dry yeast preparation Lalvin Rhône 2056 (Lallemand, Ontario, Canada). UCAY-1032 is the commercial yeast Saccharomyces cerevisiae var. bayanus Lalvin EC1118 (Lallemand, Canada). UCAY-1013 was obtained from the commercial yeast AWRI 796 (AB Mauri Yeast, Australia). UCAY-2001 was isolated from a Georgian wild grapevine (Vitis vinifera ssp. sylvestris (Gmelin) Hegi). All yeast strains are killer positive (K+). Killer behavior was assayed using the seeded agar plate technique as described by Sangorrín et al. (2002). Yeasts were inoculated into 5 mL of YPD liquid media (1 % yeast extract, 2 % peptone, 2 % dextrose) and incubated with shaking (150 rpm) at 28 °C overnight. After, cultures were washed twice with sterile distilled water (SDW) and cultured in 5 mL of natural Chardonnay and Cabernet Sauvignon musts at 25 °C with agitation. Cells growth was monitored by Thoma camera counts (Brand GMBH, Wertheim, Germany). When the culture reached 2 × 10−7 cells/mL, yeast cells were inoculated into fermentors to give an initial density of 1 × 10−6 cells/mL. Mixed cultures were inoculated with a concentration about 2.5 × 10−5 cell/mL for each yeast strain.
Laboratory fermentation assays
Two varieties (Chardonnay and Cabernet Sauvignon) were tested as well as five different yeast strains at laboratory scale to ensure the validity of the method. Monoculture ferments of each of the five yeast strains were also carried out in triplicate as control. Mixed culture micro-vinifications (coded as C1, C2 and C3) were conducted in triplicate at the laboratory using different proportions of S. cerevisiae yeast strains in order to determine from which proportion the genetic fingerprint obtained becomes that of a specific strain within the mixture (Table 1). Each combination of yeast strains was inoculated into three sterilized 1-L wool-plugged conical flasks containing 500 mL of natural Chardonnay must (225 g/L sugar concentration, pH 3.2) and three with 500 mL of Cabernet Sauvignon red must (230 g/L sugar content, pH 3.3). The total amount of fermentors was 18. Musts were not filter-sterilized. Fermentation conditions were the same as those previously described by Cordero-Bueso et al. (2011). Glucose and fructose were measured using an enzymatic kit (Roche, Mannheim, Germany). One hundred milliliters of fermenting musts were taken after 24 h of the inoculation and 100 mL at the midpoint of fermentation. The midpoint of fermentation was considered when 50 % of the initial sugar had been consumed. The remaining 300 mL were also filtered and centrifuged when sugar concentration was under 2 g/L. All samples were used for DNA extraction by using the protocol proposed in this work.
Industrial fermentations and sampling plan procedures
Industrial fermentations were performed in a commercial winery of Southwest of Spain. White wine fermentations were carried out in 48 stainless steel vessels of 400,000 L containing Palomino Fino white must, and the inoculations were carried out using pure cultures of the strain UCAY-1001 preparing a starter as described in Rodríguez et al. (2010). Scaling-up rounds were performed as stated Rodríguez et al. (2011), when the °Bé reached a value between 1 and 2. In each round, the fermentation volume was increased tenfold to give high initial levels of inoculum (>60 × 106 viable cells/mL) and ensuring the correct development of the inoculated strain. Samples were taken 24 h after refills with fresh must, which were done at either the initial, middle, or final fermentation stages in 48 vessels with wines at different °Bé. Three 500-mL sterile plastic bottles were filled with the Palomino Fino white wine from different parts of the each industrial vessel, during the scaling-up process, before each refill, and after partial refills, at the initial (11–6 °Bé), middle (6–3 °Bé) and final stage (3–0 °Bé) of the fermentation progress. Samples were kept under refrigeration (4 °C) and transported to the laboratory. A volume of 450 mL of each sample was used for DNA extraction and analysis of the multiplex PCR-SSR and mtDNA RFLP studies as below. The remaining 50 mL of each sample volume was kept in 40 % glycerol in the original media for conservation at −80 °C if subsequent analysis was needed.
Red wines industrial fermentations were performed in stainless steel vessels of 27,000 L, and no refills of must were carried out. Grape varieties used were Cabernet Sauvignon, Tempranillo, Syrah, Tintilla de Rota and Merlot. Fermentors were inoculated with the S. cerevisiae yeast strains UCAY 1009 and UCAY 1013 by preparing an inoculum in the same red must, after the hydration of yeast, and allowing it to ferment for 24 h to adapt the yeast to the final conditions of the fermentation (Rodríguez et al. 2011). Samples were taken at the initial, middle and final phases of the alcoholic fermentation. For the molecular analysis, samples of 250 mL of red wine were taken from the central part of the vessels. Of this sample volume, 225 mL was used for analysis as described below. The remaining 25 mL of each sample volume was suspended in 20 % glycerol for conservation at −80 °C for subsequent analysis.
Activated carbon cleaning pretreatment
Commercial activated carbon cartridge Maxtra (BRITA GmbH, Taunusstein, Germany) was installed in a BRITA® Slim™ pitcher and was soaked with SDW prior to pouring wine samples through it in the pitcher. Each wine sample was added to the upper reservoir for filtering and was allowed to flow through the filter into the bottom reservoir. When the wine sample was filtered, 200 mL of SDW was added into the top reservoir in order to clean the filter and to collect the maximum of possible yeast trapped in the filter. Filtered wine sample was transferred to a clean plastic bottle. The activated carbon filter and the pitcher were cleaned adding 1 L of SDW at 65 °C before to filter each wine sample. Optical density (OD) at 600 nm was measured in all samples with a Jenway 7315 spectrophotometer (Bibby Scientific, Staffordshire, UK) before and after filtering to test if using this filtration method implies biomass losses.
Rapid assay based on a microextraction of DNA and multiplex PCR-SSR analysis
Recently, Drumonde-Neves et al. (2013) have been reported a method to obtain yeast DNA from 96-well microplates in a concentration of 20 to 100 ng/μL, for direct PCR amplifications without further concentration adjustments. Thus, we modified and adapted this method to obtain a quickly and a reliable concentration of DNA directly from each collected red or white wine sample in less than 75 min and without a previous yeast growth needed. Several steps of the original procedure (Drumonde-Neves et al. 2013) were simplified or modified. The new 96-well microplate DNA extraction protocol was defined after several approaches for optimized PCR-quality DNA recovery. The procedure is as follows:
Yeast recovery
Each carbon-activated filtered wine sample was centrifuged at 1971×g for 5 min at 4 °C to collect the cells, which were washed twice with SDW to eliminate the wine debris and obtaining a clean biomass. Biomass was resuspended in 15 mL (between 107 and 108 cells/mL) of SDW and 100 µL were transferred to 96-well microplates, with lids with condensation rings, with a multichannel pipette for DNA extraction.
DNA extraction
Cells were spun down by centrifugation for 2 min at 1971×g, at 4 °C with a centrifuge with a microplate rotor (Serie Consul 21R, Orto-Alresa, Madrid, Spain), pouring off the supernatants with the help of the multichannel pipette. Then, the pellets were resuspended with 30 μL of buffer I (0.1 M EDTA, 0.9 M sorbitol, pH 7.5) containing 50 mg/mL lysing enzymes from Trichoderma harzianum (Sigma) and 14 mM of β-mercaptoethanol (1 µL/mL), noticing that lytic enzyme and β-mercaptoethanol should be added to the buffer just before distribution and incubated at 37 °C for 25 min with shaking (150 rpm). At this point, Master Mix for SSR PCR could be prepared (see PCR procedure below). After incubation, cells were centrifuged at 1971×g for 5 min at 4 °C, extracting the supernatants with the help of a multichannel micropipette. The resulting pellets were newly resuspended with 30 μL of buffer II (Tris–HCl 50 mM, EDTA 20 mM, SDS 0.35 M, pH 7.4). Multiwell plates were sealed in plastic bags and incubated at 65 °C for 5 min. Ten microliters of cold 5 M potassium acetate was immediately added and briefly mixed, incubating the plates at −20 °C for 5–8 min. After this step, plates were centrifuged at 4 °C for 12 min at 1971×g and supernatants were transferred to a clean 96-well plate containing 25 μL of isopropanol and incubated at room temperature for 5 min. Then, cells were centrifuged for 2 min at 1971×g, removing the supernatants by aspiration, and washing the nucleic acid pellets with 50 μL ethanol 70 % (v/v). Microplate lids were removed and pellets were air-dried for 6 min with a Speed-Vac apparatus (Eppendorf, Germany). Finally, the pellets were rehydrated in 50 μL of free DNA and RNA sterile water. The efficiency of the DNA extraction was checked by quantification of the DNA concentration with a NanoDrop 2000c (Thermo Scientific, USA).
Multiplex PCR-SSR analysis
For this technique, the primers SC8132X (Fw: CTGCTCAACTTGTGATGGGTTTTGG; Rv: CCTCGTTACTATCGTCTTCATCTTGC), YOR267C (Fw: GGTGACTCTAACGGCAGAGTGG; Rv: GGATCTACTTGCAGTATACGGG) and SCPTSY7 (Fw: AAAAGCGTAAGCAATGG TGTAGAT; Rv: AAATGATGCCAATATTGAAAAGGT), proposed by Vaudano and Garcia-Moruno (2008), were used (MWG Biotech AG, Ebersberg, Germany). PCR amplifications were performed in a Applied Biosystems SimplyAmp Thermal Cycler (Waltham, Massachussetts, USA) in 25 μL volumes constituted as follows: 1.5 μL of extracted DNA solution (20–80 ng of DNA), 2.5 mM MgCl2, 200 μM of each dNTP, 5 μL of 5X PCR buffer without magnesium, 2 U of GoTaq® G2 Flexi DNA polymerase (Promega, Madison, Wisconsin, USA), 10 pmol of each primer for locus SCYOR267C, 15 pmol of primers for locus SC8132X and 40 pmol of primers for locus SCPTSY7. The protocol, optimized for multiplex amplification of the three loci was: 4 min at 94 °C, 28 cycles of 30 s at 94 °C, 45 s at 56 °C, 30 s at 72 °C and, finally, 10 min at 72 °C. Amplified products were separated on an agarose gel (2.5 % w/v) with a final concentration of 5 µg/mL of ethidium bromide, in 1X TBE buffer at 115 V for 45 min. DNA fragment sizes were determined by comparison with a molecular ladder marker of 50 bp (BioLab, London, UK). The image of the gel was digitalized in a Molecular Imager apparatus (Gel-Doc XR) and analyzed using Quantity One 1-D software (Bio-Rad).
Validation of the multiplex PCR-SSR analysis
Simultaneously, to validate the results obtained by multiplex PCR-SSR assays, samples were also analyzed by RFLP of mtDNA following our published protocol (Rodríguez et al. 2011). Five microliters of the DNA sample was digested with 10 units of the endonuclease HinfI (Promega, Wisconsin, Madison, USA) by incubation at 37 °C for 2 h. The restriction fragments were separated by electrophoresis in 1 % agarose gels made with 1X TBE buffer, 0.5 μg/mL of ethidium bromide. The image of the gel was digitalized in a Molecular Imager apparatus (Gel-Doc XR) and analyzed using Quantity One 1-D software (Bio-Rad). Additionally, digestions were also carried out in a microwave oven as proposed by López et al. (2001a), which incubation conditions consist in three heating times at maximum power of the microwave 1250 W with duration of 20 s each, giving a spin between each time.
Results and discussion
Optimization of DNA extraction procedure directly from wine samples and multiplex PCR-SSR assays
At least 24–48 h is the time required to obtain DNA from yeast colonies growth in liquid or YPD agar medium and it varies depending on the species used and on the volume of medium employed. For industrial purposes, this time is too long, especially when a rapid identification of the microorganisms is required. With this protocol, it is possible to obtain enough yeast biomass directly from the wine and used for a subsequent DNA extraction, without a previous growth in a synthetic medium. All samples were subjected to activated carbon filtration. Mean OD600 was not significantly different among filtered and non-filtered samples, and thus all yeast strains were equally released from the filters. When higher cell concentrations are required, the volume of sterile water for cells resuspension can be decreased. In addition, as reported by Drumonde-Neves et al. (2013), significant time savings during DNA extraction can be achieved by using various multiwell plates at the same time. DNA from directly filtered and centrifuged wines can be obtained in <75 min. Moreover, a great quantity of samples can be analyzed at the same time and significantly reducing material and reaction volumes and costs. This is an advantage if there are many industrial vessels to control. Sometimes, depending on the yeast strain used, lysing enzymes is not effective, thus adding β-mercaptoethanol ensures disruption of protein structures avoiding an over degradation of the DNA. Using the proposed extraction protocol, values obtained ranged between 20 and 70 ng/μL of DNA, suitable for PCR amplification and enzymatic restriction without the need for further dilution. When higher DNA concentrations are required, the protocol can be modified by decreasing the volume of water for DNA rehydration.
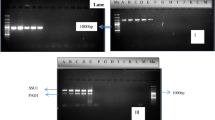
We chose the method of multiplex PCR of microsatellite loci proposed by Vaudano and Garcia-Moruno (2008) because of the high degree of polymorphism of the primers used and its discriminatory power, combined with its robustness and applicability to raw DNA templates. Although the authors defined the method as culture-dependent, following the proposed DNA extraction protocol, a culture-independent application of microsatellite multiplex PCR have been developed avoiding the colony step and having a rapid tool for the control of implantation enhancing the mtDNA technique. Our criteria for considering the result positive was obtaining identical patterns for the total cells recovered from must/wine from the different vessels and the inoculated yeast strains. In the case of a monoculture, the starter yeast culture should be taken to be dominant in the fermentation. The results should be considered negative when different patterns to the inoculated yeast, or additional or absences of bands were observed, in that case neither the presence for the dominance of the inoculated yeast strain can be assured. For example, Fig. 1a shows the patterns obtained by multiplex PCR-SSR analysis from mixed fermentation C2 of Cabernet sauvignon red wine. The proportion of patterns obtained in the mixed culture C2 after 24 h ranged from 33 to 66 % for each inoculated strain, although an optimal proportion of 33 % for each strain was desired. The proportion of the profiles of the yeast strains changed at the midpoint of fermentation, being UCAY 1013 the larger proportion (Fig. 1a). Although UCAY 1013 fits the 100 % of the profiles of the yeast population at the midpoint, it had decreased to 66 % at the end of fermentation. Similar results were obtained in the mixed cultures C1 and C3 (data not shown). For monocultures, the dominance of each inoculated yeast strain was evident at the end of the fermentation (data not shown).

Comparison of multiplex PCR-SSR sequence amplification patterns (a) and mtDNA RFLP with the endonuclease HinfI profiles (b) of S cerevisiae UCAY 1009, UCAY 1013 and UCAY 2001 obtained after activated carbon filtration and centrifugation of 1 L of Cabernet Sauvignon red wine after 24 h, at midpoint and at the end of the fermentation DNA was isolated from multiwell plates without previously adjusted cell and DNA concentrations
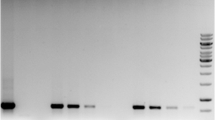
During the vintage of 2015, all industrial inoculated vessels (red and white wines) were analyzed in triplicate at the beginning, middle and end of the fermentation process. In all cases, multiplex PCR-SSR assays bring positive results (Figs. 2a, 3a). Noise represented by proteins and other cellular components ran very rapidly and did not interfere with DNA migration.

Comparison of multiplex PCR-SSR sequence amplification patterns of S cerevisiae UCAY 1001 obtained directly from non-filtered and centrifuged Palomino Fino white wine at the beginning (a) and at the end (c) of the fermentations in the industrial vessels A, G, D, E, L and P, and mtDNA RFLP with the endonuclease HinfI profiles of S cerevisiae UCAY 1001 obtained from the same deposits at the beginning (b) and at the end (d) of the fermentations following the previously established protocol in our laboratory

Comparison of multiplex PCR-SSR sequence amplification patterns of S cerevisiae UCAY 1013 obtained directly from filtered and centrifuged red wine at the beginning (a) and at the end (c) of the fermentations in the industrial vessels R1, R4, R10, R11, R23 and R25, and mtDNA RFLP with the endonuclease HinfI profiles of S cerevisiae UCAY 1013 obtained from the same deposits at the beginning (c) and at the final (d) of the fermentations following the previously established protocol in our laboratory
Validation of the multiplex PCR-SSR analysis
To validate the results obtained by SSR-multiplex PCR, all amplified samples were simultaneously analyzed by mtDNA restriction analysis with the endonuclease HinfI and following the protocol of Rodríguez et al. (2011) as we have been doing for many years. The comparison of profiles obtained after agarose electrophoresis of PCR products with those obtained by mtDNA RFLP confirmed that the patterns obtained were identical, corresponding to the inoculated yeast strains (Figs. 1b, 2b, 3b). On the contrary, applying the protocol proposed by López et al. (2001a), which could to reduce the analysis time by using a microwave oven during digestion process, we were unable to obtain successful results in some of the endonuclease digestions of DNA.
In conclusion, this pretreatment with activated carbon and the adaptation of DNA extraction and multiplex PCR-SSR protocols is presented as a new, reliable, low-cost, saving time and high-throughput method. The method has been applied in a real commercial winery to quality assurance in the yeast industry to detect wild yeast contamination and to control inoculums purity in white and red wines, allowing the winemakers to intervene rapidly during fermentation.
References
Abriouel H, Omar NB, López RL, Martínez-Cañamero M, Keleke S, Gálvez A (2006) Culture-independent analysis of the microbial composition of the African traditional fermented foods poto poto and dégué by using three different DNA extraction methods. Int J Food Microbiol 111:228–233
Ambrona J, Vinagre A, Maqueda M, Álvarez ML, Ramírez M (2006) Rhodaminepinkas a genetic marker for yeast populations in wine fermentation. J Agric Food Chem 54:2977–2984
Barrajón N, Arévalo-Villena M, Rodríguez-Aragón LJ, Briones A (2009) Ecological study of wine yeast in inoculated vats from La Mancha Region. Food Control 20:778–783
Cankar K, Stebih D, Dreo T, Zel J, Gruden K (2006) Critical points of DNA quantification by real-time PCR-effects of DNA extraction method and sample matrix on quantification of genetically modified organisms. BMC Biotechnol 6:37–52
Capece A, Pietrafesa R, Romano P (2011) Experimental approach for target selection of wild wine yeasts from spontaneous fermentation of “Inzolia” grapes. World J Microbiol Biotechnol 27:2775–2783
Capello MS, Bleve G, Grieco F, Dellagio F, Zacheo G (2004) Characterization of Saccharomyces cerevisiae isolated from must of grape grown in an experimental vineyard. J Appl Microbiol 97:1274–1280
Cordero-Bueso G, Arroyo T, Serrano A, Tello J, Aporta I, Vélez MD, Valero E (2011) Influence of the farming system and vine variety on yeast communities associated with grape-berries. Int J Food Microbiol 45:132–139
Cordero-Bueso G, Esteve-Zarzoso B, Gil-Díaz M, García M, Cabellos JM, Arroyo T (2016) Improvement of Malvar wine quality by use of locally-selected Saccharomyces cerevisiae strains. Fermentation 2:7
Delteil D (1992) Gestione della fermentazione con i lieviti enologici selezionati. Vignevini 9:35–38
Devesa-Rey R, Bustos G, Cruz JM, Moldes AB (2011) Optimisation of entrapped activated carbon conditions to remove coloured compounds from winery wastewaters. Biores Technol 102:6437–6442
Drumonde-Neves J, Vieira E, Lima MT, Araujo I, Casal M, Schuller D (2013) An easy, quick and cheap high-throughput method for yeast DNA extraction from microwell plates. J Microbiol Methods 93:206–208
Esteve-Zarzoso B, Gostincar A, Bobet R, Uruburu F, Querol A (2000) Selection and molecular characterization of wine yeasts isolated from the “El Penedes” area Spain. Food Microbiol 17:553–562
Fleet GH (2008) Wine yeast for the future. FEMS Yeast Res 8:979–995
González SS, Barrio E, Querol A (2007) Molecular identification and characterization of wine yeasts isolated from Tenerife (Canary Islands, Spain). J Appl Microbiol 102:1018–1025
Jara C, Mateo E, Guillamón JM, Torija MJ, Mas A (2008) Analysis of several methods for the extraction of high quality DNA from acetic acid bacteria in wine and winegar for characterization by PCR-based methods. Int J Food Microbiol 128:336–341
Lisanti MT, Gambuti A, Genovese A, Piombino P, Moio L (2014) Earthy off-flavour in wine: evaluation of remedial treatments for geosmin contamination. Food Chem 154:171–178
Lopes CA, Rodríguez ME, Sangorrín M, Querol A, Caballero AC (2007) Patagonian wines: implantation of an indigenous strain of Saccharomyces cerevisiae in fermentations conducted in traditional and modern cellars. J Ind Microbiol Biotechnol 34:139–149
López V, Querol A, Ramón D, Fernández-Espinar MT (2001a) A simplified procedure to analyse mitocondrial DNA from industrial yeasts. Int J Food Microbiol 81:63–71
López S, Castro R, García E, Pazo JA, Barroso C (2001b) The use of activated charcoal in combination with other fining agents and its influence on the organoleptic properties of sherry wine. Eur Food Res Technol 212:671–675
López V, Fernández-Espinar MT, Barrio E, Ramón D, Querol A (2003) A new PCRbased method for monitoring inoculated wine fermentations. Int J Food Microbiol 81:63–71
Olivares-Marín M, Del Prete V, García-Moruno E, Fernández-González C, Macías-García A, Gómez-Serrano V (2009) The development of an activated carbon from cherry stones and its use in the removal of ochratoxin A from red wine. Food Control 20:298–303
Raspor P, Cus F, Povhe Jemec K, Zagorc T, Cadez N, Nemanic J (2002) Yeast population dynamics in spontaneous and inoculated alcoholic fermentations of Zametovka must. Food Technol Biotechnol 40:95–102
Rodríguez ME, Infante JJ, Molina M, Domínguez M, Rebordinos L, Cantoral JM (2010) Genomic characterization and selection of wine yeast to conduct industrial fermentations of White wine produced in a SW Spain winery. J Appl Microbiol 108:292–1302
Rodríguez ME, Infante JJ, Molina M, Rebordinos L, Cantoral JM (2011) Using RFLP-mtDNA for the rapid monitoring of the dominant inoculated yeast strain in industrial wine fermentations. Int J Food Microbiol 145:331–335
Salazar FN, De Brujin JPF, Seminario L, Güell C, López F (2007) Improvement of wine crossflow microfiltration by a new hybrid process. J Food Eng 79:1329–1336
Sangorrín M, Zajonskovsky I, van Broock M, Caballero A (2002) The use of killer biotyping in an ecological survey of yeast in an old Patagonian winery. World J Microbiol Biotechnol 18:115–120
Santamaría P, Garijo P, López R, Tenorio C, Gutiérrez AR (2005) Analysis of yeast population during spontaneous alcoholic fermentation: effect of the age of the cellar and practice of inoculation. Int J Food Microbiol 103:49–56
Suzzi G, Arfelli G, Schirone M, Corsetti A, Perpetuini G, Tofalo R (2012) Effect of grape indigenous Saccharomyces cerevisiae strains on Montepulciano d’Abruzzo red wine quality. Food Res Int 46:22–29
Varela C, Siebert T, Cozzolino D, Rose L, McLean H, Henschke P (2009) Discovering a chemical basis for differentiating wines made by fermentation with “wild” indigenous and inoculated yeasts: role of yeast volatile compounds. Aus J Grape Wine Res 15:238–248
Vaudano E, Garcia-Moruno E (2008) Discrimination of Saccharomyces cerevisiae wine strains using microsatellite multiplex PCR and band pattern analysis. Food Microbiol 25:56–64
Acknowledgments
We thank Bodegas Barbadillo S.L. (Sanlúcar de Barrameda, Cádiz, Spain) by the support provided to perform this investigation in their industrial wineries and José Herrera Jiménez for their kindly help during the sampling plan and in the lab. This project has received funding from the “Consejo de Administración del Centro de Desarrollo Tecnológico Industrial (CDTI)”, reference; IDI20141202, UCA18DGUEII02 and from the European Union’s Seventh Framework Programme via the Marie Curie Action, “Co-funding of Regional, National and International Programs” to stimulate research activities without mobility restrictions, co-financed by the ‘Junta de Andalucía’ and the European Commission under Grant Agreement No. 291780.
Conflict of interest
Authors declare no financial or personal interests that might inappropriately influence the content of this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Erko Stackebrandt.
Rights and permissions
About this article

Cite this article
Cordero-Bueso, G., Rodríguez, M.E., Garrido, C. et al. Rapid and not culture-dependent assay based on multiplex PCR-SSR analysis for monitoring inoculated yeast strains in industrial wine fermentations. Arch Microbiol 199, 135–143 (2017). https://doi.org/10.1007/s00203-016-1287-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-016-1287-4