
Abstract
Summary
To develop a population pharmacokinetic model that describes the absorption and low plasma levels of risedronate in the body. The impact of patients’ characteristics on risedronate kinetics is investigated. Simulations revealed the high variability in the concentration levels after different dosage schemes. No dosage adjustment is required in renal impairment.
Introduction
Risedronate exhibits very low plasma levels and high residence time in the body. The aim of this study is to describe and explain the risedronate transit through the body. The impact of volunteers’ characteristics on the kinetics of risedronate is also investigated. Simulations are used to compare the risedronate plasma levels after different dosage schemes and assess the need for dose adjustment in patients with impaired kidney functionality.
Methods
Plasma concentration—time data were obtained from a four-period, two sequence, single-dose, crossover bioequivalence study. The effects of several covariates (e.g., weight, albumin, creatinine, alkaline phosphatase, and calcium) on model parameters were tested. Non-linear mixed-effect modeling was applied and a variety of models were evaluated placing emphasis on absorption and disposition properties. The modeling and simulation work was implemented in MonolixTM 2020R1.
Results
Following oral administration, the kinetics of risedronate was best described by a two-compartment model with lag time, first-order absorption, and elimination. The extent of peripheral distribution (i.e., bones) was found to be remarkably high. No volunteer characteristics were identified to affect significantly the disposition of risedronate. Using simulations, risedronate plasma profiles were obtained for different doses and frequencies of administration.
Conclusion
The absorption and disposition kinetics of risedronate were successfully characterized. Simulations revealed the high discrepancy in the concentration levels observed after different dosage regimens, implying the safety profile of risedronate. In virtual patients with renal impairment, the blood levels of risedronate are increased, but not in an extent requiring dose adaptation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Prevention and management of bone diseases, such as osteoporosis, have become more important as the aging population increases [1]. Osteoporosis affects millions of people worldwide over the age of 50, which is associated with significant morbidity, mortality, and costs for the health systems [2]. Paget’s disease is the second bone metabolic disease, with a prevalence around 2% among adults older than 55 years and 10% among people over 80 years [3]. Those skeletal disorders are caused because of an imbalance in the bone remodeling process, characterized by an excessive and anarchic remodeling ending up in a disorganization of the bone morphology.
For the treatment of both conditions, several pharmacotherapy strategies exist, where bisphosphonates (BPs) exert a predominant role [4,5,6,7]. BPs can be classified in two general groups: (a) the non-nitrogen-containing, also called first-generation, such as clodronate and etidronate and (b) the nitrogen-containing BPs which include the second generation (pamidronate, ibandronate, and alendronate), and the third generation (risedronate and zoledronic acid) [7]. BPs diminish bone resorption through different mechanisms [8]. Non-nitrogen-containing compounds principally obstruct osteoclast activity by inhibiting ATP-dependent enzymes by forming non-hydrolyzable analogs of ATP. The nitrogen-containing bisphosphonates exert their action in two ways: first, in the extracellular space act as calcium chelators by binding to and stabilizing calcium phosphate within bone matrix and subsequently preventing bone mineral dissolution. Secondly, they lead to multiple intracellular effects within osteoclasts; most notably, they inhibit the mevalonate pathway [9]. Bisphosphonates should not be administered indefinitely due to the limited efficacy data beyond 5 years of use and the increased risk of serious adverse events such as atypical femur fractures and osteonecrosis of the jaw. However, the optimal duration of treatment and the parameters for restarting remains unclear [9].
Risedronate, along with other BPs (like alendronate, ibandronate, and zoledronic acid), represents a popular choice for the treatment of osteoporosis. Risedronate is a potent antiresorptive agent, possessing high affinity for hydroxyapatite, which was found to reduce the risk of fracture in women with postmenopausal osteoporosis in randomized, controlled clinical trials [10, 11]. Currently, risedronate is approved by the EMA and FDA to prevent or manage osteoporosis and to treat Paget’s disease with doses ranging from 5mg daily to 150mg once a month [12,13,14]. Alike to other BPs, risedronate is not recommended for use in patients with reduced creatinine clearance.
Common characteristics of the second and third generation BPs refer to their very low bioavailability and long residence inside the body [12, 13, 15]. BPs are absorbed to low extent after oral administration and around half of the absorbed drug is taken up by the bones and the remaining is eliminated unchanged by the kidneys [16]. Similarly, risedronate has a bioavailability of 0.63% which can be further reduced in fed conditions, especially if diet is rich in calcium or divalent cations that can bind with the negative groups of phosphate formed at physiological pH. Once in general circulation, risedronate distributes and remains in bones for months [12, 13]. As all nitrogen-containing bisphosphonates, risedronate is widely distributed throughout the body, primarily in the bones, but also in soft tissues such as the liver, kidney, and spleen [7]. Risedronate is primarily cleared by the kidneys and it stays inside the body for a long time. In postmenopausal women, the terminal exponential half-life was found to be close to 24 days, implying that more than 4 months are required for its complete removal from the body [12, 13, 15, 17, 18].
Thus, it is intriguing to try to describe mathematically the two common characteristics of risedronate (and subsequently of all BPs) kinetics, namely, the long residence in the body and the low plasma levels. In case of risedronate, some typical pharmacokinetic analyses have appeared in the literature; however, a study explaining its kinetics has not been published so far. A valuable tool towards this direction is the application of non-linear mixed effect modeling approaches [19]. According to these methodologies, a structural mathematical model can be used to explain mechanistically/physiologically the transit of medicines through the body and concomitantly to account for the between-subject and within-subject variabilities of the people. In addition, the role of several subjects’ characteristics (like weight, sex, calcium, alkaline phosphatase levels) can be explored for their impact on drug transit inside the body.
The aim of this study was to describe and explain the risedronate kinetics after oral administration, by applying non-linear mixed effect modeling approaches. The potential impact of volunteers’ characteristics on the kinetics of risedronate was also investigated. Emphasis is placed on the absorption properties of risedronate and on the models to describe it. Simulations of different dosage regimens and several levels of renal function were also done.
Materials and methods
Data
The data used in this analysis were obtained from an open-label, four-period, two-sequence, single-dose, crossover bioequivalence study comparing two immediate-release tablets containing 30mg of risedronate in 40 healthy volunteers (Study # 1522/08: Risedronate sodium 30mg fc tabs/Verisfield Pharmaceuticals SA vs. Actonel® (Risedronate sodium 30mg tabs)/Procter & Gamble Pharmaceuticals). Risedronate was administered in each period after an overnight fast with 240 ml of water. Each subject received four treatments within the study according to a 4-sequence randomization schedule. There was a 16-day washout period between each treatment. All subjects were adults, non-smokers between 18 and 55 years of age, having a body mass index (BMI) between 18.5 and 24.9 Kg/m2 and weight between 50.1 and 77.0 Kg. None of them had any evidence of underlying diseases or clinically significant abnormal laboratory values. All subjects voluntarily consented to participate in the study.
Assay methodology
Risedronate concentrations were measured one hour before the single administration and during 24h after dosing. A total of 17 blood samples were collected at times 0.167, 0.333, 0.5, 0.667, 0.833, 1.0, 1.333, 1.667, 2.0, 3.0, 4.0, 6.0, 8.0, 12.0, and 24 h in vacutainers containing K2-EDTA.
The analyte risedronic acid and its internal standard risedronic acid-d4 were extracted from a 250μl aliquot of human EDTA plasma using DEA, 100mg, LCR solid-phase extraction cartridges according to method SOP. The extracted samples were injected into a liquid chromatograph equipped with a Chirobiotic T, Astec 4.6 × 50mm, 5μm column. The mobile phase A was a mixture of acetonitrile/Milli-Q type water (90/10), ammonium acetate 0.5mM, and acetic acid 1%, and the mobile phase B was a mixture of acetonitrile / Milli-Q type water (80/20), ammonium acetate 1mM, and acetic acid 1%.
The chromatographic separation was isocratically performed at room temperature. The detection was made with a tandem mass spectrometry detector API 3000 (MDS Sciex). The validated calibration range employed during samples analysis was from 399.800 to 31984.000 pg/ml. The percentages of bias were from −3.81 to −0.05 and the coefficients of variation ranges lied between 3.62 and 6.09%.
Non-linear mixed effect modeling
Data were analyzed by a population approach, using MonolixTM 2020R1 (Simulation Plus) [20]. The Stochastic Approximation Expectation Maximization algorithm implemented in MonolixTM was used to estimate the parameters.
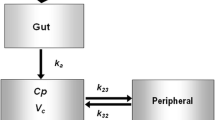
A variety of structural models were explored based on the literature information and the characteristics of risedronate (Fig. 1). In the case of absorption, models with lag-time, transit absorption compartments, and typical first-order absorption were investigated. In this context, three sets of models were tested: (a) models with simple first-order absorption (Fig. 1a and 1b), (b) models with a transit absorption compartment (Fig. 1c and 1d), and (c) models with lag time in absorption (Fig. 1e–h). Regarding distribution, the analyzed models included either one or up to four compartments, while for elimination, one or two parallel pathways were investigated. The models with two elimination pathways were explored since they take into consideration the reported disposition characteristics of bisphosphonates [7]. The latter comprises renal elimination (namely, the most important clearance mechanism) of risedronate and a secondary removal called “skeleton elimination” or long-term elimination. Since such kind of models, with two elimination pathways, were not included in the MonolixTM library, they were coded in the MlxTran language of MonolixTM.

Structural models investigated for the description of risedronate kinetics. For the characterization of absorption, three main approaches were used relying on the concept of simple first-order kinetics (a, b), the use of a deposit pre-absorption compartment (c, d), and the inclusion of delay (i.e., lag-time) (e–h). One-, two-, and three-compartment models were evaluated assuming one or two parallel elimination pathways. Key: Tlag, lag-time; F, bioavailability fraction; Ka, first-order absorption rate constant; Cl/F, apparent clearance; Vc/F, apparent volume of drug distribution of the central compartment; Vp/F: apparent volume of drug distribution of the peripheral compartment (tissues); Vp1/F: apparent volume of drug distribution of the first peripheral compartment; Vp2/F: apparent volume of drug distribution of the second peripheral compartment; Q/F: apparent inter-compartmental clearance; b, residual error; RSE%, relative standard error; SE, standard error; Omega, between subject variability
Between-subject variability was modeled assuming log-normal distribution of the model parameters, whereas the residual error models investigated included the combined, proportional, and additive. The impact of inter-occasion variability and correlation among parameters were further explored. In addition, the impact of several volunteers’ characteristics (age, weight, height, BMI, albumin, creatinine, creatinine clearance, alkaline phosphatase, and calcium) on structural model parameters was evaluated. In a first step, these covariates were added one-by-one on each parameter, starting with the strongest correlation observed. Covariates showing a significant association with structural parameters (p value <0.05) were kept and further tested in the final full model. In a subsequent step, models with covariates found significant in the univariate analysis were further simultaneously investigated using a backward elimination method. Only covariates remaining significant after this procedure would be kept in the final model.
To find the best model, different models were compared relying on their physiological plausibility and goodness of fit criteria like the Bayesian Information Criterion (BIC), the Akaike Information Criterion (AIC), -2⋅log-likelihood (-2LL) [19, 21]. The model with the lowest values in these statistical criteria, the best performance in the graphs, and having physiological rationale was finally selected.
Simulations
Using the estimates of the average and individual parameters from the final model, risedronate plasma concentration-time profiles were simulated for the official dosage schemes (Table 1) approved by the US FDA and the EMA [12, 13]. Simulations were also performed to explore the impact of renal impairment on risedronate pharmacokinetics. In this case, the concentration-time profiles after oral administration in patient with normal creatinine clearance were compared with the profiles obtained considering reduced renal elimination ability by 25%, 50%, and 75%. All simulation tasks were implemented in the Simulx tool of MonolixTM 2020R1.
Results
Forty volunteers were assessed in the four periods of the bioequivalence study. None of the identified adverse events was serious and all patients who showed unexpected reactions were treated appropriately and followed up until their health issue was resolved. Some selected demographic and laboratory data of the study population are presented in Table 2.
Non-linear mixed effect modeling
Depending on the absorption, distribution, and elimination characteristics, eight sets of structural models were explored (Fig. 1). As mentioned in the “Methods” section, these structural models were evaluated considering the plausibility and the goodness-of-fit criteria. In general, models with simple absorption (Fig. 1a, b) led to the worst performance, followed by the models assuming an initial absorption deposit site (Fig. 1c, d). The best results were obtained for models considering a lag-time in absorption of risedronate (Fig. 1e–h). Finally, the model that best described the data included lag time in absorption, two compartments, and elimination from the central compartment (Fig. 1e). In this structural model, it is considered that the central compartment represents blood and the peripheral compartment represents mainly the bones.
None of the covariates examined, namely, age, weight, height, BMI, albumin, creatinine, creatinine clearance, alkaline phosphatase, and calcium, was found to affect significatively the estimated pharmacokinetic model parameters. Similarly, treatment effect (i.e., the generic or the originator’s medicine) was not found to exert an impact on any part of risedronate kinetics. Inter-occasion variability, added to the volumes of distribution of the central, was found to be significant and improving all goodness of fit criteria. The residual error model with the best results was the proportional. The average model parameters, their between-subject variabilities, along with the estimation standard errors and residual standard errors (%) are listed in Table 3. It can be observed that for all parameters and their inter-individual variabilities, the RSE% values are less than 18.5%.
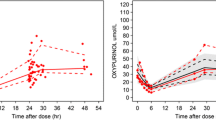
The validation plots are also in line with the statistical criteria indicating the appropriateness of the finally selected model. Figure 2 illustrates the virtual predictive check (VPC) plot and the predicted vs. observed (experimental) concentration values. The VPC plot (Fig. 2a) confirmed that the developed model can describe the data appropriately, since the observed plasma concentrations lie within the 90% prediction confidence interval at the 10th and 90th percentiles of the simulated drug concentrations. Similarly, the predicted vs. observed plot (Fig. 2b) shows that the individual predicted concentrations are close to the experimental values (namely, close to the ideal line of unity), underlining the good predictive performance of the developed model.

Diagnostic plots for the goodness-of-fit of the final best model (see Fig. 1e). a Visual predictive check plot showing the 90% prediction intervals at the 10th and 90th percentiles of the simulated risedronate concentrations. Dots and lines refer to the 10th, 50th, and 90th percentile of the experimental data. b Observed plasma concentration values versus the individual predicted concentrations from the final model. The diagonal line represents the identity
Simulations
The dosage regimens described in Table 1 were simulated for patients with normal kidney functionality (Fig. 3a) and patients with impaired renal function (Fig. 3b). In order to make the graphs easier to compare, all simulation graphs were performed until 12h, regardless of the fact that some dosing schemes refer to once-daily administration and others to once a month intake (see Table 1).

Simulated concentration vs. time profiles of risedronate for four different doses. a The average model parameters estimated in this analysis are used to simulate doses of 5, 30, 75, and 150mg. b For each dose (solid lines refer to the subjects with normal renal function), three levels of renal insufficiency are simulated (from top to bottom: 75%, 50%, and 25%)
Figure 3a clearly reveals that as the administered dose increases, the risedronate plasma levels also rise. Besides, the increase of renal impairment leads to increases in risedronate levels and this effect is more pronounced at the higher doses (Fig. 3b).
Discussion
The structural model found to best describe the risedronate data refers to a two-compartment model with lag time and proportional error model (Fig. 1e). Even though, in the risedronate summary of product characteristics (SmPC), it is stated that after an oral administration, the concentration-time profile shows three elimination phases; in this study, a two-compartment model (i.e., two elimination phases) led to the best performance [13]. The model quoted in the SmPC was also investigated in this study and resulted in worse statistical properties (compared to our final model), due to the fact that it included more parameters than the one finally selected.
From a semi-mechanistic point of view, a second elimination route referring to skeleton elimination was initially anticipated. However, in the investigated models, the inclusion of a second parallel elimination pathway led to a negligible contribution (maximum value estimated of 1.14 l/h) of this skeleton elimination compared to the total clearance (i.e., 108.55 l/h). Thus, only one clearance was finally utilized in the final model encompassing all routes of elimination. Alternatively, the physiologically anticipated skeleton elimination can also be considered part of the relatively high value of inter-compartmental clearance (i.e., Q/F=516.54 l/h in Table 3) leading to the very high peripheral distribution of risedronate. In this context, even though risedronate is highly hydrosoluble, it was found a very large apparent volume of distribution in the peripheral compartment (Vp/F =41,446.61 l), which is considered to reflect the high binding capacity of bones. This fact agrees with the extended distribution of risedronate, because it binds to hydroxyapatite in bone throughout the body [7].
In the early 1990s, etidronate studies in humans and other mammals revealed its very long residence inside the body [22]. Multi-compartmental approaches were used to explain etidronate bone disposition and re-absorption, while the inclusion of power-law decay models successfully described etidronate kinetics for up to 1000 days after administration [22]. In general, urine measurements were able to detect BPs years (which can be as high as 10 years) after treatment discontinuation [7, 23]. In this vein, it was quoted that power functions, rather than typical exponential decreases, are preferable for the description of long-term kinetics of BPs [7, 23]. The inclusion of power-law models is indicative of the necessity to explain the long-term kinetics of BPs, as in the case of other drugs with similar properties like amiodarone [24]. The very long residence (months, years) of BPs is also highlighted for alendronate, where the half-life was reported to be at least 10 years [25]. In this case, a three-compartment model was proposed with the central compartment referring to highly perfused tissues, while the two peripheral compartments ascribing to bones and other “non-calcified” tissues. To this point, it should be highlighted that in our study, C-t data were available for up to 24h, which is a short time period comparing to the long residence of risedronate inside the body. Therefore, it was not possible to capture the multi-compartmental character of risedronate disposition, and for this reason, the simplest two-compartment model was found to be better than the three- or four-compartmental models. Plausibly, no power functions were necessary for the description of kinetics, but a simple bi-exponential model sufficed.
The data utilized in this population pharmacokinetic analysis came from a bioequivalence study. Due to the nature of the bioequivalence trials, which focus mainly on differences in absorption between two pharmaceutical products, this analysis had the advantage of utilizing a dense blood sampling during the absorption phase of risedronate. This allowed us to study absorption phenomena and elucidate on the absorption properties of risedronate. In this context, several absorption models were explored and the one with the best ability to describe the concentration-time (C-t) data led to a model with lag-time. It was found a delay in absorption equal to 0.14h or equivalently 8.4min, which is in line with the delay mentioned in the clinical pharmacology and biopharmaceutics review of risedronate published by the US FDA [26]. This lag time can be attributed to the low permeability of risedronate [27].
Nine covariates were tested however none of them showed to significantly explain the inter-individual variability. In order to avoid the shrinkage phenomenon, the conditional distribution was used to calculate the random effects employed in the plots of correlation between random effects and individual parameters vs. covariates. The latter was done because it considers the uncertainty of the individual parameters, and in that way, it increases the reliability of diagnostic plots [28].
To the best of our knowledge, no other population pharmacokinetic model has been reported for risedronate, so it is not possible to directly compare our results with other risedronate models. Nevertheless, Pillai et al. (2004) proposed a pharmacokinetic-pharmacodynamic model for ibandronate, which is a N-containing bisphosphonate as in the case of risedronate. In their study, again, a model with one elimination pathway was found [29].
Another worth mentioning issue relies on the high diversity of approved doses for the treatment of osteoporosis. Simulation plots reveal the resulted high discrepancy of concentrations originating from these doses (Fig. 3a). For example, the maximum concentration levels range from 3ng/ml (after 5mg oral administration) up to 78ng/ml (after a 150mg oral dose). Even though creatinine clearance was not found to be a significant covariate in the model, simulations were performed to quantify the impact of renal impairment on risedronate levels. These simulations were performed relying on the literature evidence that risedronate is eliminated by the kidneys [12,13,14]. It should be clarified that in this study, creatinine clearance was not found to exert a significant role in the model, since the utilized data came from volunteers with normal kidney function; thus, it was not possible to identify renal function as a significant covariate. These simulations for risedronate showed increase of risedronate levels as renal impairment assumed to be more severe (Fig. 3b), which is expected since risedronate is eliminated by the kidneys. Currently, the clinical impact of increased risedronate levels is unclear in the literature, since the ideal concentration of BPs on bone mechanical properties is not completely clear [30]. In the same vein, the simulations performed in this study do not suggest that dose-adjustment with renal function is necessary, but these simulations were only performed to quantify the extent of increase in the risedronate levels in several levels of renal insufficiency. The simulations showed that the increase, due to renal insufficiency, is much less pronounced compared to the differences observed from the different dosage regimens. Taking into consideration that no adverse have been reported relevant to the plasma levels, this finding further underlines the fact that no dosage adjustment is required in patients with mild or moderate renal impairment [12, 13, 31].
A restriction of this study relies on the homogeneity of the tested population, since the volunteers were healthy subjects not older than 55 years; thus, no pathophysiological-related covariates could have been assessed. Nevertheless, the effect of kidney functionality, even though not found as significant covariate, was explored in the simulations in order to explore the impact of reduced renal clearance on risedronate plasma levels. Also, since risedronate pharmacokinetics depends on binding sites and bone turnover, then its pharmacokinetics might differ in different pathological conditions [16]. However, it is still questionable whether typical pharmacokinetic studies can identify these differences [16]. Another limitation is the short duration of blood sampling after risedronate administration. In order to capture the long-residence of risedronate inside the body, due to the slow dissociation and release from bones, a rather extended blood sampling regimen would be needed (e.g., for months or even years). However, in this study, data were available only up to 24h post-dose. Finally, in the simulations performed in this study, several doses were utilized. In all these cases, it was assumed that model parameters remain the same. In particular, this is true for doses up to 75mg, since it has been reported that absorption is independent of dose over the range 2.5 to 50mg or up to 75mg on two consecutive days a month [13, 32].
Conclusions
Risedronate exhibits very low plasma levels and long residence time in the body. Through the development of a population pharmacokinetic model, the absorption and disposition kinetics of risedronate were successfully characterized. Due to the dense blood sampling scheme, the C-t data used for model building offered the ability to study absorption kinetics. The extent of distribution, due to binding in bones, was found to be remarkably high. No volunteer characteristics were identified to affect significantly the disposition of risedronate. Simulations revealed the high discrepancy in the concentration levels observed after different dosage administrations, implying the safety profile of risedronate. In simulated patients with renal impairment, the blood levels of risedronate are increased, but not in an extent requiring dose adaptation.
References
Clynes MA, Harvey NC, Curtis EM, Fuggle NR, Dennison EM, Cooper C (2020) The epidemiology of osteoporosis. Br Med Bull 133:105–117. https://doi.org/10.1093/bmb/ldaa005
Ruiz-Adame M, Correa M (2020) A systematic review of the indirect and social costs studies in fragility fractures. Osteoporos Int 31:1205–1216. https://doi.org/10.1007/s00198-020-05319-x
Reid IR (2020) Management of Paget's disease of bone. Osteoporos Int 31:827–837. https://doi.org/10.1007/s00198-019-05259-1
Khosla S, Hofbauer LC (2017) Osteoporosis treatment: recent developments and ongoing challenges. Lancet Diabetes Endocrinol 5:898–907. https://doi.org/10.1016/S2213-8587(17)30188-2
Anastasilakis AD, Polyzos SA, Makras P (2018) Therapy of endocrine disease: denosumab vs bisphosphonates for the treatment of postmenopausal osteoporosis. Eur J Endocrinol 179:R31–R45. https://doi.org/10.1530/EJE-18-0056
Eastell R, Rosen CJ, Black DM, Cheung AM, Murad MH, Shoback D (2019) Pharmacological management of osteoporosis in postmenopausal women: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 104:1595–1622. https://doi.org/10.1210/jc.2019-00221
Cremers S, Drake MT, Ebetino FH, Bilezikian JP, Russell RGG (2019) Pharmacology of bisphosphonates. Br J Clin Pharmacol 85:1052–1062. https://doi.org/10.1111/bcp.13867
Russell RG, Watts NB, Ebetino FH, Rogers MJ (2008 Jun) Mechanisms of action of bisphosphonates: similarities and differences and their potential influence on clinical efficacy. Osteoporos Int 19(6):733–759. https://doi.org/10.1007/s00198-007-0540-8
Nayak S, Greenspan SL (2019) A systematic review and meta-analysis of the effect of bisphosphonate drug holidays on bone mineral density and osteoporotic fracture risk. Osteoporos Int 30:705–720. https://doi.org/10.1007/s00198-018-4791-3
Eastell R, Barton I, Hannon RA, Chines A, Garnero P, Delmas PD (2003) Relationship of early changes in bone resorption to the reduction in fracture risk with risedronate. J Bone Miner Res 18:1051–1056. https://doi.org/10.1359/jbmr.2003.18.6.1051
Ogura Y, Gonsho A, Cyong JC, Orimo H (2004) Clinical trial of risedronate in Japanese volunteers: single and multiple oral dose studies. J Bone Miner Metab 22:111–119. https://doi.org/10.1007/s00774-003-0458-y
Actonel® Full prescribing information. 2008 Procter & Gamble Pharmaceuticals, Inc. https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/020835s035lbl.pdf. Accessed 31 December 2020
Risedronate sodium 35mg. Summary of product characteristics. Sandoz limited. Date of revision of the text 27/11/2020. https://www.medicines.org.uk/emc/medicine/25017#gref. Accessed 31 December 2020
Risedronate sodium 75mg. Summary of product characteristics. Sandoz limited. https://mri.cts-mrp.eu/human/downloads/ES_H_0358_001_FinalPI_2of2.pdf. Accessed 11 February 2021
Soares AP, do Espírito Santo RF, Line SR, Md P, Santos Pde M, Toralles MB, do Espírito Santo AR (2016) Bisphosphonates: pharmacokinetics, bioavailability, mechanisms of action, clinical applications in children, and effects on tooth development. Environ Toxicol Pharmacol 42:212–217. https://doi.org/10.1016/j.etap.2016.01.015
Cremers SC, Pillai G, Papapoulos SE (2005) Pharmacokinetics/pharmacodynamics of bisphosphonates: use for optimisation of intermittent therapy for osteoporosis. Clin Pharmacokinet 44:551–570. https://doi.org/10.2165/00003088-200544060-00001
Perazella MA, Markowitz GS (2008) Bisphosphonate nephrotoxicity. Kidney Int 74:1385–1393. https://doi.org/10.1038/ki.2008.356
Mitchell DY, Barr WH, Eusebio RA, Stevens KA, Duke FP, Russell DA, Nesbitt JD, Powell JH, Thompson GA (2001) Risedronate pharmacokinetics and intra- and inter-subject variability upon single-dose intravenous and oral administration. Pharm Res 18:166–170. https://doi.org/10.1023/a:1011024200280
Bonate PL (2011) Pharmacokinetic-Pharmacodynamic Modeling and Simulation, 2nd edn. Springer US, New York
MonolixTM, Simulation Plus. URL: https://www.simulations-plus.com/software/monolix. Accessed 31 December 2020
Dziak JJ, Coffman DL, Lanza ST, Li R, Jermiin LS (2020) Sensitivity and specificity of information criteria. Brief Bioinform 21:553–565. https://doi.org/10.1093/bib/bbz016
Kasting GB, Francis MD (1992) Retention of etidronate in human, dog, and rat. J Bone Miner Res 7:513–522. https://doi.org/10.1002/jbmr.5650070507
Cremers S, Ebetino FH, Phipps R (2020) On the pharmacological evaluation of bisphosphonates in humans. Bone 139:115501. https://doi.org/10.1016/j.bone.2020.115501
Weiss M (1999) The anomalous pharmacokinetics of amiodarone explained by nonexponential tissue trapping. J Pharmacokinet Biopharm 27:383–396. https://doi.org/10.1023/a:1020965005254
Lin JH (1996) Bisphosphonates: a review of their pharmacokinetic properties. Bone 18:75–85. https://doi.org/10.1016/8756-3282(95)00445-9
US Food and Drug Administration. Center for Drug Evaluation and Research. Clinical pharmacology and biopharmaceutics review(s). Application number: 022560ORIG1S000", 2010. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/022560orig1s000clinpharmr.pdf. Accessed 31 December 2020
Hens B, Corsetti M, Spiller R, Marciani L, Vanuytsel T, Tack J, Talattof A, Amidon GL, Koziolek M, Weitschies W, Wilson CG, Bennink RJ, Brouwers J, Augustijns P (2017) Exploring gastrointestinal variables affecting drug and formulation behavior: Methodologies, challenges and opportunities. Int J Pharm 519:79–97. https://doi.org/10.1016/j.ijpharm.2016.11.063
Traynard P, Ayral G, Twarogowska M, Chauvin J (2020) Efficient pharmacokinetic modeling workflow with the monolixsuite: a case study of remifentanil. CPT Pharmacometrics Syst Pharmacol 9:198–210. https://doi.org/10.1002/psp4.12500
Pillai G, Gieschke R, Goggin T, Jacqmin P, Schimmer RC, Steimer JL (2004) A semimechanistic and mechanistic population PK-PD model for biomarker response to ibandronate, a new bisphosphonate for the treatment of osteoporosis. Br J Clin Pharmacol 58:618–631. https://doi.org/10.1111/j.1365-2125.2004.02224.x
Allen MR (2018) Recent advances in understanding bisphosphonate effects on bone mechanical properties. Curr Osteoporos Rep 16:198–204. https://doi.org/10.1007/s11914-018-0430-3
Mitchell DY, St Peter JV, Eusebio RA, Pallone KA, Kelly SC, Russell DA, Nesbitt JD, Thompson GA, Powell JH (2000) Effect of renal function on risedronate pharmacokinetics after a single oral dose. Br J Clin Pharmacol 49:215–222. https://doi.org/10.1046/j.1365-2125.2000.00135.x
Mitchell DY, Eusebio RA, Sacco-Gibson NA, Pallone KA, Kelly SC, Nesbitt JD, Brezovic CP, Thompson GA, Powell JH (2000) Dose-proportional pharmacokinetics of risedronate on single-dose oral administration to healthy volunteers. J Clin Pharmacol 40:258–265. https://doi.org/10.1177/00912700022008928
Acknowledgements
The authors wish to thank Verisfield Pharmaceuticals SA for providing the concentration-time data to perform the current modeling analysis
Code availability
A software (MonolixTM) was used for the modeling and simulation task of this study
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Conflicts of interest
None.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Cardozo, B., Karatza, E. & Karalis, V. Osteoporosis treatment with risedronate: a population pharmacokinetic model for the description of its absorption and low plasma levels. Osteoporos Int 32, 2313–2321 (2021). https://doi.org/10.1007/s00198-021-05944-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-021-05944-0