
Abstract
Summary
We sought to characterize the phenotypes and identify the SEC24D gene mutations associated with Chinese families of osteogenesis imperfecta (OI). Using whole-exome sequencing, we discovered two novel compound SEC24D mutations of OI patients. Our study extended both the phenotypic and the genotype of the OI patients with SEC24D mutations.
Introduction
To date, only three compound heterozygous mutations in the SEC24D gene have been found to cause recessively inherited forms of OI. We sought to characterize the phenotypes and to identify the SEC24D gene mutations associated with Chinese families with OI.
Methods
Using whole-exome sequencing in two probands, we identified two novel compound heterozygous mutations in SEC24D. In family 1, the proband was a 23-year-old male; he had recurrent fractures and dentinogenesis imperfecta. His anterior fontanel was not closed, and he showed facial dysmorphism. A computed tomography three-dimensional imaging of the cranium showed skull deformities associated with a broad ossification defect in the frontoapical area, a widened sagittal suture, and Wormian bones. In family 2, the proband was a 7-year-old boy, who also had recurrent fractures and dentinogenesis imperfecta. His anterior fontanel was not closed, and he did not have obvious facial dysmorphism.
Results
We identified one novel compound heterozygous missense substitution in the proband in family 1, including c.2723G>A (p. Cys908Tyr) and c.2842T>C (p. Ser948Pro). In the proband in family 2, we identified another novel compound heterozygous missense substitution, including c.938G>A (p. Arg313His) and c.875C>T (p. Pro292Leu).
Conclusions
We discovered two novel compound SEC24D mutations of autosomal recessive OI patients. Our study extended both the phenotypic and the genotypic spectrum of the autosomal recessive OI patients with SEC24D mutations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Osteogenesis imperfecta (OI) is a congenital connective tissue disorder characterized by low bone mass, fragile bones, and growth deficiency. Approximately 90% of cases of dominantly inherited OI are caused by mutations of the COL1A1 (MIM 120150) or the COL1A2 (MIM 120160) gene, which encode type I collagen mutations [1]. Recently, recessive mutations in genes have been identified in OI patients. In 2006, the first disease-associated gene of recessive OI, CRTAP (MIM 605497), was discovered [2, 3]. Over the past decade, another 16 genes have been identified that cause dominantly or recessively inherited forms of OI, such as (BMP1 [MIM 112264], CREB3L1 [HGNC ID 18856], FKBP10 [MIM 607063], IFITM5 [MIM 614757], LEPRE1 [MIM 610339], P4HB [MIM 176790], PLOD2 [MIM 601865], PLS3 [MIM 300131], PPIB [MIM 123841], SEC24D [MIM 607186], SERPINF1 [MIM 172860], SERPINH1 [MIM 600943], SP7 [MIM 606633], SPARC (MIM 182120), TMEM38B [MIM 611236], and WNT1 [MIM 164820]) (http://www.le.ac.uk/genetics/collagen/index.html). The pathogenic mechanism in OI arises from gene mutations that affect the synthesis, structure, folding, secretion, and matrix organization of type I collagen [4]. The majority of pathogenic mutations found in recessive OI genes are homozygous or compound heterozygous loss-of-function mutations that result in two null alleles, which are responsible for either no production or seriously decreased production of normal proteins [5]. In 2015, compound heterozygous mutations in the SEC24D gene were discovered to cause a novel severe form of recessively inherited OI in two Caucasian families. This type of OI was characterized by craniofacial malformations and skull ossification defects [6]. By contrast, Moosa et al. [7] reported a case involving a patient from a Chinese family with a more classical OI phenotype, who was carrying two novel compound heterozygous mutations in SEC24D but who lacked craniofacial malformations. Until now, these were the only reports of cases of OI that were caused by the SEC24D gene. China is a country with a large population; in previous studies, the pathogenic genes for OI reported in cases involving Chinese families have included COL1A1, COL1A2, LEPRE1, IFITM5, FKBP10, PLOD2, and TMEM38B [8–11]. In this study, we report two Chinese families with a moderate form of OI caused by compound heterozygous mutations in SEC24D.
Methods
Subjects
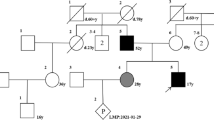
Two unrelated Chinese families with OI and 250 healthy control donors were included in this study. None of the patients belonged to a consanguineous family. All the subjects belonged to the Han ethnic group. The study was approved by the Ethics Committee of the Shanghai Jiao Tong University Affiliated Sixth People’s Hospital, and informed consent was obtained from every proband, family member, and volunteer before blood sampling and DNA analysis commenced.
Bone densitometry
The bone mineral density (BMD, g/cm2) of the lumbar spine (L1-L4) was measured using dual-energy X-ray absorptiometry (DXA). The probands of both families were assessed using Lunar Prodigy equipment (GE Lunar Corp., Madison, WI, USA). The Lunar device was calibrated daily, and the coefficient of variability (CV) value of the DXA measurements at L1-L4 was 1.39% [12]. Lumbar spine BMD results were converted to an age-specific and sex-specific Z-score according to the reference data [13].
Laboratory tests
Fasting blood samples were obtained in the morning between 8:00 and 10:00 AM, following both the manufacturer’s protocol and specialized laboratory assay quality control procedures. The following markers of calcium metabolism and bone turnover were measured: serum calcium (Ca), phosphorus (P), alkaline phosphatase (ALP), intact parathyroid hormone (PTH), 25(OH)D, β-CrossLaps of type I collagen containing cross-linked C-telopeptide (β-CTX), and osteocalcin (OC). Ca, P, and ALP were measured using a HITACHI 7600-020 automatic biochemistry analyzer, Tokyo, Japan. Other compounds were measured using the following kits (all from Roche Diagnostics, Mannheim, Switzerland): an intact PTH kit for PTH, a 25 hydroxy vitamin D3 kit for 25(OH)D, a β-CrossLaps kit for β-CTX, and an osteocalcin kit for OC. The intra- and inter-assay CVs, respectively, were 5.7 and 7.3% for 25(OH)D, 1.4 and 2.9% for PTH, 2.5 and 3.5% for β-CTX, and 2.9 and 4.0% for OC; these results were reported in a previous study [14]. The intra- and inter-assay CVs, respectively, were 1.5 and 2.0% for Ca, 2.1 and 2.3% for P, and 2.5 and 4.5% for ALP [15].
Whole-exome capture and massively parallel DNA sequencing
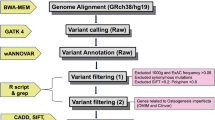
The exome of the probands of both families were sequenced to identify the pathogenic gene. Exon-enriched DNA was sequenced by the Illumina Genome Analyzer II platform following the manufacturer’s instructions (Illumina). The raw image files were processed using the Illumina Base Calling Software v. 1.7 with the default parameters, and the sequence of each individual DNA fragment was reported as 101-bp paired-end reads. The sequencing reads were aligned to the NCBI human reference genome (NCBI36.3) using the SOAPaligner [16, 17]. The SOAPsnp results were filtered using the following standards: the base quality was equal to or exceeded 20, the sequencing depth was between 4 and 200, the estimated copy number was less than two, and the distance between two SNPs was more than 5 bp [18–20]. Approximately 114 million reads were quantified and mapped to the hs37d5 human reference genome DNA sequence, leading to an average read depth of 78.8 for the exome of each proband. We collected reads that were among the target regions for SNP identification and subsequent analysis. The consensus sequence and quality of each allele was calculated by SOAPsnp.
SEC24D mutation confirmation
To confirm the mutation, fragments covering the mutation site in SEC24D which were identified by whole-exome sequencing were amplified by polymerase chain reaction (PCR). Primers for PCR amplification were designed using Primer3 software (http://bioinfo.ut.ee/primer3-0.4.0/). The primer of the family 1 were 5′-TGACTGGAATAAAGTCTGGCACAT-3′ for the forward primer and 5′- AAAGTCACATATTTTAGTTGTTTGAAAGT-3′ for the reverse primer. The primer of the family 2 were 5′-CCATTGCATCTAAATGCCTCTCA-3′ for the forward primer and 5′-CAGGATGCCAAAAACCATAGC-3′ for the reverse primer. The patients’ sequences were compared with the Ensembl reference gene sequence ENSG00000150961 (SEC24D). Direct sequencing was performed using the BigDye Terminator Cycle Sequencing Ready Reaction Kit, v. 3.1 (Applied Biosystems, Foster, CA), and the sequencing was analyzed with an ABI Prism 3130 automated sequencer.
Results
Clinical features
Family 1
In family 1, the proband was a 23-year-old male, the only son of nonconsanguineous parents. He was the product of a full-term pregnancy with a normal delivery. His birth weight and length were within normal limits. His height and weight at the time of the study were 143.0 cm and 75.0 kg, respectively. He experienced his first fracture, in the right humerus, at the age of only 1 month. He experienced six additional fractures, including both femurs, between the ages of 1 and 15 years. He fractured his elbow when he was 20 years old but had not experienced another fracture since then. The proband had evident deformities in both lower limbs. He had dentinogenesis imperfecta but did not exhibit blue sclera or hearing loss. His anterior fontanel was not closed and showed facial dysmorphism with down-slanting palpebral fissures, frontal protrusion, left ear dysplasia, and micrognathia (Fig. 1a, b). No family history of bone fragility was identified. A computed tomography three-dimensional imaging (CT 3D imaging) of the cranium showed skull deformities associated with a broad frontoapical ossification defect, a widened sagittal suture, and Wormian bones (Fig. 1c, d). Radiographs showed multiple fractures of both femurs and more severe bowing of the left femur (Fig. 1e). However, his lumbar spine bone mass was normal. Biological test results, including serum calcium, phosphate, alkaline phosphatase, and parathyroid hormone, were within normal ranges, both 25-hydroxyvitamin D and OC were deficient, and β-CTX was slightly above the normal range (Table 1). His parents did not show any symptoms of OI.

Photographs, brain CT scans, and X-ray of the proband of family 1. a The proband showed facial dysmorphism with down-slanting palpebral fissures and still not closed anterior fontanel. b Lateral view. Frontal protruded, mid-face hypoplasia, left ear dysplasia and micrognathia. c and d Three-dimensional CT scan of the cranium showed skull deformities associated with a broad ossification defect fronto apical, the sagittal suture widened, and Wormian bones. e Radiographs of the both legs of the proband of family 1 showed severe bowing of left femur, multiple old and new fractures of both femurs
Family 2
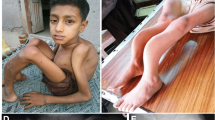
In family 2, the proband was a 7-year-old boy, the only son of non-consanguineous parents. He was the product of a full-term pregnancy with a normal delivery. His birth weight and length were within normal limits. His height and weight at the time of the study were 122.7 cm and 28.0 kg, respectively. He experienced his first fracture, in the right upper femur, at the age of 1.5 years old. He experienced a total of five fractures, including both femurs, between 1.5 and 4.5 years of age. He experienced a lumbar spine 3 fracture at the age of 7 years. He had dentinogenesis imperfecta but did not exhibit blue sclera or hearing loss. His anterior fontanel was not closed, and he did not have obvious facial dysmorphism (Fig. 2a, b). Radiographs showed fractures in both femurs and severe bowing of the left femur (Fig. 2c, d). The proband was treated with pamidronate (0.5 mg/kg for 3 days) when he was 3 years old. Then, he was treated with intramedullary nails that could be extended with a fixation operation. After the operation, he could walk independently and had not experienced further fractures in either femur (Fig. 2e). He exhibited low back pain when he was 7 years old, and the X-ray image showed a mild compression fracture of the third lumbar vertebra (Fig. 2f). The proband showed lower lumbar spine bone mass than that of healthy boys of the same age. Biological test results, including serum phosphate, alkaline phosphatase, parathyroid hormone, and 25-hydroxyvitamin D, were within normal ranges, whereas serum calcium and β-CTX were slightly above normal ranges (Table 1). His parents did not show any symptoms of OI.

Photographs and X-ray of the proband of family 2. a The proband showed facial dysmorphism with still not closed bregmatic. b Lateral view: without obvious micrognathia. c, d Radiographs of the both legs of the proband of family 2 showed severe bowing of left femur, fractures of both femurs. e Both femurs were operated with intramedullary nail can be extended fixation. f Mild compression fracture of lumbar 3
Whole-exome capture and massively parallel DNA sequencing
We performed exome sequencing on the probands of both families to search for the pathogenic gene. We filtered out all 1000G records and dbSNP records, and the filter region included exonic or splicing and overlapping noncoding RNA regions. Approximately 12.7 and 14.5 gigabases (Gb) of high-quality data were aligned to the target regions of the probands of family 1 and 2 with a per-base mismatch rate of 0.43 and 0.45%, respectively. As a result, the mean coverage sequencing depth of the official target regions of the probands of family 1 and family 2 was 85 and 101, respectively. The number of high-confidence SNPs was 208,147 and 217,868 SNPs, including 94,920 and 98,353 heterozygotes. We identified 18,869 and 19,972 high-confidence indels, of which 9352 and 10,299 were heterozygotes of the probands of family 1 and family 2, respectively. By filtering the data using public SNP databases, including dbSNP129, the UCSC Genome Browser Hg 18, and the 1000 Genome Browser, we identified the SEC24D gene mutations of the two probands. In the proband of family 1, we identified one novel compound heterozygous missense substitution, including one missense mutation, c.2723G>A (p. Cys908Tyr) and another mutation c.2842T>C (p. Ser948Pro); both mutations were in exon 21. In the proband of family 2, we identified one novel compound heterozygous missense substitution, including one missense mutation c.938G>A (p. Arg313His) in exon 8 and another mutation c.875C>T (p. Pro292Leu) in exon 7.
Validation of the SEC24D germ line mutation
The Sanger sequencing results for the entire SEC24D gene were consistent with the whole-exome sequencing results. The compound heterozygous missense mutations C908Y (c.2723G>A) (NM_014822.2) and S948P (c.2842T>C) (NM_014822.2) were confirmed in the proband of family 1 (Fig. 3a, b). The heterozygous C908Y (c.2723G>A) mutation was found in the father of the proband, and the heterozygous S948P (c.2842T>C) mutation was found in the mother. The compound heterozygous missense mutations R313H (c.938G>A) (NM_014822.2) and P292L (c.875C>T) (NM_014822.2) were confirmed in the proband of family 2 (Fig. 4a, b). The heterozygous R313H (c.938G > A) mutation was found in the father of the proband, and the heterozygous P292L (c.875C > T) mutation was found in the mother. A search of the data held in the OI variant database (https://oi.gene.le.ac.uk/variants.php?select_db=SEC24D&action=view_all) revealed that both mutations are novel. In addition, we compared the four mutated sequences with the 10 species, including human, zebra fish, mouse, rat, marmoset, king cobra, bovin, African clawed frog, sheep, and guinea pig. All four mutated sequences were at highly conserved positions (supplementary Fig. 1). We used the provean (Protein Variation Effect Analyzer) software tool to predict whether an amino acid substitution has an impact on the biological function of a protein, and the results showed that all four missense mutations were deleterious, and the provean scores of C908Y, S948P, R313H, and P292L were −7.442, −3.922, −4.792, and −9.185, respectively. The results were similar when the sequencing data were analyzed using polyphen (Polymorphism Phenotyping) tools.

Genetic analysis of the SEC24D gene mutation in 2 families. Direct DNA sequencing of the both probands. The proband 1 showed a compound heterozygote for SEC24D. a c.2723G > A (p. Cys908Tyr) the missense mutation in exon 21. b c.2842T>C (p. Ser948Pro) the other missense mutation in exon 21

Genetic analysis of the SEC24D gene mutation in 2 families. Direct DNA sequencing of the both probands. The proband 2 showed a compound heterozygote for SEC24D. a c.938G>A (p. Arg313His) the missense mutation in exon 8. b c.875C > T (p. Pro292Leu) the other missense mutation in exon 7
Discussion
Garbes et al. first reported mutations in SEC24D, which were revealed by whole-exome sequencing of OI patients in 2015, and described a 7-year-old Caucasian boy with a phenotype similar to the phenotype of patients with Cole-Carpenter syndrome [6]. Cole-Carpenter syndrome was first described in 1987 by Cole and Carpenter, who depicted two infants with OI who also had distinctive facial features (MIM 112240) [21]. Interestingly, in 2015, Rauch et al. [22] performed whole-exome sequencing on the genomic DNA of the two individuals described by Cole and Carpenter in 1987, both of whom are now adults, and discovered that Cole-Carpenter syndrome was caused by a heterozygous missense mutation in P4HB. When the symptoms of OI patients with SEC24D mutations were compared with those of OI patients with Cole-Carpenter syndrome, some differences were found. For example, the boy with SEC24D mutations reported by Garbes et al. showed macrocephaly but not hydrocephalus (as shown in patients with Cole-Carpenter syndrome), down-slanting palpebral fissures without ocular proptosis, and a wide sagittal suture with a broad ossification defect, in contrast to craniosynostosis. In our study, the proband of family 1 also showed facial dysmorphism with down-slanting palpebral fissures, frontal protrusion, left ear dysplasia, and micrognathia, which was similar to the 7-year-old boy reported by Garbes et al. [6]. In our study, a CT 3D imaging of the cranium of the proband of family 1 also showed skull deformities associated with a broad frontoapical ossification defect and a widened sagittal suture. Therefore, the specific facial dysmorphisms, including down-slanting palpebral fissures, ear dysplasia, and micrognathia, may be characteristics of OI patients with SEC24D mutations.
In 2015, Moosa et al. [7] also reported a case involving an OI patient carrying two novel compound heterozygous mutations in SEC24D. Compared with the probands reported in our study, this Chinese OI patient showed a more classical OI phenotype, including blue-gray sclerae, osteopenia, and Wormian bones. The discrepancy between our report and the case reported by Moosa et al. [7] relates to whether the craniofacial malformations were the simultaneous symptoms of OI patients with SEC24D mutations. In our study, the proband of family 2 was more similar to the patient reported by Moosa et al. [7]. Both patients showed an enlarged anterior fontanel without other craniofacial malformations. In fact, regardless of whether a patient showed evidence of facial dysmorphism, all the patients with the SEC24D mutations showed an unclosed or enlarged anterior fontanel. Therefore, the disturbed ossification of the skull might be a characteristic of OI patients with SEC24D mutations. Both of the patients were surgically treated, and their recoveries were satisfactory. Therefore, it can be concluded that the OI patients with SEC24D mutations had a good response to the orthopedic operations.
SEC24D is an element of the COPII complex that was exported from the endoplasmic reticulum (ER) [6]. The COPII complex is a set of cytoplasmic coat proteins that generate membrane-bound vesicles or carriers to export procollagen from the ER [23]. The medaka fish mutant vbi, an animal model with a SEC24D nonsense mutation, shows craniofacial malformations, including impaired ossification of the neurocranium, and OI [24]. To date, only three compound SEC24D mutations of OI patients have been reported worldwide [6, 7]. In this study, we reported two novel compound SEC24D mutations of OI patients, and all were compound missense mutations. Those four missense mutations were all found at highly conserved positions and were predicted to be deleterious by provean and polyphen-2 software. The parents of both families were confirmed to be heterozygous for one of the missense mutations. Therefore, an autosomal recessive inheritance pattern was confirmed. In our study, the proband of family 1 showed both compound mutations on exon 21, and he showed more obvious facial dysmorphism than the boy described by Garbes et al., whose mutation sites were on exons 5 and 23 [6]. The proband of family 2 in our study showed compound mutations on exons 7 and 8, and his phenotype was a more classical OI phenotype without obvious facial dysmorphism, as shown in the girl described by Moosa et al., whose mutations were on exons 2 and 19 [7]. We thus assumed that the closer the mutation was to the carboxyl end, the more obvious the facial dysmorphism would be. More OI patients with SEC24D mutations should be analyzed to verify our assumption.
In conclusion, we discovered two novel compound SEC24D mutations of autosomal recessive OI patients, including c.2723G>A (p. Cys908Tyr) and c.2842T>C (p. Ser948Pro) in one proband and c.938G>A (p. Arg313His) and c.875C>T (p. Pro292Leu) in the other proband. Our study extended both the phenotypic and the genotypic spectrum of autosomal recessive OI patients with SEC24D mutations.
References
Cheung MS, Glorieux FH (2008) Osteogenesis imperfecta: update on presentation and management. Rev Endocr Metab Disord 9:153–160
Morello R, Bertin TK, Chen Y, Hicks J, Tonachini L, Monticone M, Castagnola P, Rauch F, Glorieux FH, Vranka J, Bachinger HP, Pace JM, Schwarze U, Byers PH, Weis M, Fernandes RJ, Eyre DR, Yao Z, Boyce BF, Lee B (2006) CRTAP is required for prolyl 3-hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell 127:291–304
Barnes AM, Chang W, Morello R, Cabral WA, Weis M, Eyre DR, Leikin S, Makareeva E, Kuznetsova N, Uveges TE, Ashok A, Flor AW, Mulvihill JJ, Wilson PL, Sundaram UT, Lee B, Marini JC (2006) Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N Engl J Med 355:2757–2764
Forlino A, Cabral WA, Barnes AM, Marini JC (2011) New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol 7:540–557
Van Dijk FS, Sillence DO (2014) Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A 164a:1470–1481
Garbes L, Kim K, Riess A, Hoyer-Kuhn H, Beleggia F, Bevot A, Kim MJ, Huh YH, Kweon HS, Savarirayan R, Amor D, Kakadia PM, Lindig T, Kagan KO, Becker J, Boyadjiev SA, Wollnik B, Semler O, Bohlander SK, Kim J, Netzer C (2015) Mutations in SEC24D, encoding a component of the COPII machinery, cause a syndromic form of osteogenesis imperfecta. Am J Hum Genet 96:432–439
Moosa S, Chung BH, Tung JY, Altmuller J, Thiele H, Nurnberg P, Netzer C, Nishimura G, Wollnik B (2015) Mutations in SEC24D cause autosomal recessive osteogenesis imperfecta. Clin Genet. doi:10.1111/cge.12678 [Epub ahead of print]
Zhang ZL, Zhang H, Ke YH, Yue H, Xiao WJ, Yu JB, Gu JM, Hu WW, Wang C, He JW, Fu WZ (2012) The identification of novel mutations in COL1A1, COL1A2, and LEPRE1 genes in Chinese patients with osteogenesis imperfecta. J Bone Miner Metab 30:69–77
Zhang Z, Li M, He JW, Fu WZ, Zhang CQ, Zhang ZL (2013) Phenotype and genotype analysis of Chinese patients with osteogenesis imperfecta type V. PLoS One 8:e72337
Zhou P, Liu Y, Lv F, Nie M, Jiang Y, Wang O, Xia W, Xing X, Li M (2014) Novel mutations in FKBP10 and PLOD2 cause rare Bruck syndrome in Chinese patients. PLoS One 9:e107594
Lv F, Xu XJ, Wang JY, Liu Y, Asan WJW, Song LJ, Song YW, Jiang Y, Wang O, Xia WB, Xing XP, Li M (2016) Two novel mutations in TMEM38B result in rare autosomal recessive osteogenesis imperfecta. J Hum Genet 61:539–545
Zhang H, He JW, Gao G, Yue H, Yu JB, Hu WW, Gu JM, Hu YQ, Li M, Fu WZ, Liu YJ, Zhang ZL (2010) Polymorphisms in the HOXD4 gene are not associated with peak bone mineral density in Chinese nuclear families. Acta Pharmacol Sin 31:977–983
Maynard LM, Guo SS, Chumlea WC, Roche AF, Wisemandle WA, Zeller CM, Towne B, Siervogel RM (1998) Total-body and regional bone mineral content and areal bone mineral density in children aged 8–18 y: the Fels Longitudinal Study. Am J Clin Nutr 68:1111–1117
Lu HK, Zhang Z, Ke YH, He JW, Fu WZ, Zhang CQ, Zhang ZL (2012) High prevalence of vitamin D insufficiency in China: relationship with the levels of parathyroid hormone and markers of bone turnover. PLoS One 7:e47264
He J, Zhang H, Wang C, Zhang Z, Yue H, Hu W, Gu J, Fu W, Hu Y, Li M, Liu Y, Zheng H, Zhang Z (2014) Associations of serum sclerostin and polymorphisms in the SOST gene with bone mineral density and markers of bone metabolism in postmenopausal Chinese women. J Clin Endocrinol Metab 99:E665–E673
Li Y, Vinckenbosch N, Tian G, Huerta-Sanchez E, Jiang T, Jiang H, Albrechtsen A, Andersen G, Cao H, Korneliussen T, Grarup N, Guo Y, Hellman I, Jin X, Li Q, Liu J, Liu X, Sparso T, Tang M, Wu H, Wu R, Yu C, Zheng H, Astrup A, Bolund L, Holmkvist J, Jorgensen T, Kristiansen K, Schmitz O, Schwartz TW, Zhang X, Li R, Yang H, Wang J, Hansen T, Pedersen O, Nielsen R, Wang J (2010) Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat Genet 42:969–972
Zhang Z, Xia W, He J, Zhang Z, Ke Y, Yue H, Wang C, Zhang H, Gu J, Hu W, Fu W, Hu Y, Li M, Liu Y (2012) Exome sequencing identifies SLCO2A1 mutations as a cause of primary hypertrophic osteoarthropathy. Am J Hum Genet 90:125–132
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43:491–498
Shi Y, Li Y, Zhang D, Zhang H, Li Y, Lu F, Liu X, He F, Gong B, Cai L, Li R, Liao S, Ma S, Lin H, Cheng J, Zheng H, Shan Y, Chen B, Hu J, Jin X, Zhao P, Chen Y, Zhang Y, Lin Y, Li X, Fan Y, Yang H, Wang J, Yang Z (2011) Exome sequencing identifies ZNF644 mutations in high myopia. PLoS Genet 7:e1002084
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:e164
Cole DE, Carpenter TO (1987) Bone fragility, craniosynostosis, ocular proptosis, hydrocephalus, and distinctive facial features: a newly recognized type of osteogenesis imperfecta. J Pediatr 110:76–80
Rauch F, Fahiminiya S, Majewski J, Carrot-Zhang J, Boudko S, Glorieux F, Mort JS, Bachinger HP, Moffatt P (2015) Cole-carpenter syndrome is caused by a heterozygous missense mutation in P4HB. Am J Hum Genet 96:425–431
Miller EA, Schekman R (2013) COPII—a flexible vesicle formation system. Curr Opin Cell Biol 25:420–427
Ohisa S, Inohaya K, Takano Y, Kudo A (2010) sec24d encoding a component of COPII is essential for vertebra formation, revealed by the analysis of the medaka mutant, vbi. Dev Biol 342:85–95
Acknowledgements
We thank the patients and their family members for their invaluable cooperation. This study was supported by the National Basic Research Program of China (2014CB942903), National Natural Science Foundation of China (NSFC) (No. 81370978, 30800387, and 81200646), Science and Technology Commission of Shanghai municipality (14JC1405000 and 14ZR1431900), and Chongqing City Fundamental and Advanced Research Projects (CSTC2013jcyjC00009).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
The study was approved by the Ethics Committee of the Shanghai Jiao Tong University Affiliated Sixth People’s Hospital, and informed consent was obtained from every proband, family member, and volunteer before blood sampling and DNA analysis commenced.
Electronic supplementary material
ESM 1
(DOC 235 kb)
Rights and permissions
About this article

Cite this article
Zhang, H., Yue, H., Wang, C. et al. Novel mutations in the SEC24D gene in Chinese families with autosomal recessive osteogenesis imperfecta. Osteoporos Int 28, 1473–1480 (2017). https://doi.org/10.1007/s00198-016-3866-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-016-3866-2