
Abstract
Summary
Bone loss after spinal cord injury (SCI) is rapid, severe, and refractory to interventions studied to date. Mice with sclerostin gene deletion are resistant to the severe sublesional bone loss induced by SCI, further indicating pharmacological inhibition of sclerostin may represent a promising novel approach to this challenging medical problem.
Introduction
The bone loss secondary to spinal cord injury (SCI) is associated with several unique pathological features, including the permanent immobilization, neurological dysfunction, and systemic hormonal alternations. It remains unclear how these complex pathophysiological changes are linked to molecular alterations that influence bone metabolism in SCI. Sclerostin is a key negative regulator of bone formation and bone mass. We hypothesized that sclerostin could function as a major mediator of bone loss following SCI.
Methods
To test this hypothesis, 10-week-old female sclerostin knockout (SOST KO) and wild type (WT) mice underwent complete spinal cord transection or laminectomy (Sham).
Results
At 8 weeks after SCI, substantial loss of bone mineral density was observed at the distal femur and proximal tibia in WT mice but not in SOST KO mice. By μCT, trabecular bone volume of the distal femur was markedly decreased by 64 % in WT mice after SCI. In striking contrast, there was no significant reduction of bone volume in SOST KO/SCI mice compared with SOST KO/sham. Histomorphometric analysis of trabecular bone revealed that the significant reduction in bone formation rate following SCI was observed in WT mice but not in SOST KO mice. Moreover, SCI did not alter osteoblastogenesis of marrow stromal cells in SOST KO mice.
Conclusion
Our findings demonstrate that SOST KO mice were protected from the major sublesional bone loss that invariably follows SCI. The evidence indicates that sclerostin is an important mediator of the marked sublesional bone loss after SCI, and that pharmacological inhibition of sclerostin may represent a promising novel approach to this challenging clinical problem.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Spinal cord injury (SCI) is a devastating neurological disorder that causes rapid and severe osteoporosis with increased risk of fracture below the level of injury [1]. In patients with neurologically motor-complete SCI, bone mass may be reduced by more than 50 % at the distal femur and proximal tibia [1, 2]. Sublesional bone loss is most rapid during the first 6 to 12 months after SCI [3, 4] and continues beyond the initial rapid phase of bone loss in both the cortical and trabecular compartments [5]. The mechanisms responsible for SCI-induced bone loss remain largely unknown, and, as such, there is no targeted approach that has been identified to adequately preserve the mass and structural integrity of bone after motor-complete SCI [6]. A better understanding of the molecular mechanism and risk factors for SCI-related bone loss is critical in identifying therapeutic targets for the prevention or treatment of osteoporosis, fracture, and associated comorbidity.
The Wnt/β-catenin pathway has been reported to have an anabolic action on bone by regulating osteoblast function [7, 8]. Regulation of Wnt signaling in bone by Wnt inhibitors including sclerostin, DKK1, and sFRPs is crucial in the pathogenesis of disuse osteopenia [9]. Sclerostin, a product of the SOST gene, is produced predominantly by osteocytes and is a potent inhibitor of bone formation. Sclerostin and DKK1 act by binding to the Wnt coreceptor LRP5/6 thereby inhibiting Wnt signaling, whereas sFRPs bind Wnts to reduce their biological activity [9]. In humans and mice, loss of function mutations in SOST increases bone mass [10, 11]. Following in vivo mechanical loading, SOST/sclerostin is downregulated [12]. Conversely, expression of sclerostin is increased in osteocytes after hindlimb unloading [12, 13]. Importantly, targeted deletion of the sclerostin gene in mice increased bone formation and bone strength [13, 14] and conferred resistance to mechanical unloading-induced bone loss [13]. Sclerostin antibody restored bone mass after ovariectomy [15] and increased bone mass in aged male rats [16] and, furthermore, it mitigated bone loss in animal models of hindlimb-immobilization [17]. In recent phase I and II clinical trials in postmenopausal women, sclerostin antibody increased bone mineral density (BMD) and bone formation markers while it decreased a marker of bone resorption [18, 19]. Recent clinical studies suggest elevation of serum sclerostin levels after acute SCI [20]. Most recently, the application of sclerostin antibody increased bone formation and prevented bone loss and osteocyte morphology/structure changes in rat models of acute SCI [21, 22], suggesting that sclerostin is probably responsible for modulating SCI-related bone loss.
Based on these consideration, we hypothesize that sclerostin could function as a major mediator of bone loss following SCI. To test this hypothesis, it would require investigating the response of mice to SCI-induced extreme mechanical unloading in the absence of sclerostin gene. We sought to test the effects of a complete spinal cord transection (T10) on bone loss in sclerostin knockout (SOST KO) mice and compare findings to those of wild-type (WT) mice with SCI. If sclerostin is important for SCI-induced bone loss, then the mice with targeted deletion of sclerostin gene should be resistant to such bone loss.
Materials and methods
Animals, surgery, and tissue collection
Ten-week-old female SOST KO mice (20–25 g) and WT control mice were obtained from Amgen as previously described [14]. All animals were maintained on a 12:12-h light/dark cycle with lights on at 07:00 h in a temperature-controlled (20 ± 2 °C) vivarium, and all procedures were approved by the James J Peters VA Medical Center IACUC. Animals were provided water and standard chow ad libitum.
Four groups of animals were studied: WT/Sham, WT/SCI, SOST KO/Sham, and SOST KO/SCI (n = 11–15 per group). Spinal cord transection surgery was performed, as previously described, with some modifications [23, 24]. In brief, animals were anesthetized by inhalation of isoflurane and the spinal cord was transected at the interspace between the ninth and tenth vertebral bodies. Urine was expressed three times daily until automaticity of bladder developed, then as needed. Baytril was administered for the first 3 days postoperatively then as indicated for cloudy or bloody urine or for overt wound infection. Sham-transected animals received an identical surgery, including a laminectomy, except that the spinal cord was not manipulated.
For dynamic histomorphometric analysis, newly formed bones were labeled with fluorochromes by subcutaneous injection of calcein (10 mg/kg body weight) and xylenol orange (90 mg/kg) on day −6 and −2 before euthanasia, respectively. Eight weeks after SCI, animals were euthanized by inhalation of isoflurane prior to harvesting of tissue for study. The leg was removed using sterile technique; careful dissection was performed to free the head of the femur from the pelvis. The gastrocnemius muscle was removed by careful dissection, weighed, and normalized relative to body weight prior to spinal cord transection (pre-operative body weight). Blood was collected by cardiac puncture. To preserve bone for micro-CT and histomorphometry studies (n = 6–8 per group), the left leg was removed and placed into tubes containing 4 % PFA overnight, after which the PFA was replaced with 70 % ethanol for storage. The right femur and tibia (n = 4–5 per group) were placed in ice-cold Minimum Essential Alpha Medium (α-MEM) and then immediately processed for bone marrow cell cultures.
Dual-energy X-ray absorptiometry
Areal BMD measurements were performed by using a small animal dual-energy X-ray absorptiometer (DXA) (Lunar Piximus, WI, USA) as described [22, 25, 26]. Hindlimbs were positioned on the DXA platform with the knee flexed at an angle of 135 degrees, and DXA images were acquired with Lunar Pixmus software. The instrument was calibrated with a phantom following the procedures recommended by the manufacturer on each day of use prior to analysis of experimental samples. The metaphysis of the distal femur and proximal tibia was selected as regions of interest (ROI). The coefficient of variation for the repeated measurements for the ROI was approximately 1.5 %.
Volumetric BMD and bone architecture of the distal femur were assessed by a Scanco μCT scanner (μCT-40; Scanco Medical AG, Switzerland) at 16 mm isotropic voxel size, as previously described [22]. Image reconstruction and 3D quantitative analysis were performed using software provided by Scanco. Scans were initiated at the growth plate and moved proximally for a total of approximately 300 slices. A region of interest consisting of 100 slices beginning 0.5 mm proximal to the growth plate and continuing in a proximal direction was included in the analysis. Standard nomenclature and methods for bone morphometric analysis were employed [22].
Bone histomorphometric analysis
For fluorochrome-based determination of rates of bone formation by dynamic histomorphometry, the distal femurs were embedded in methyl methacrylate plastic and were then cut in 6 μm thicknesses of sections using a Reichert-Jung sledge microtome. Xylenol orange and calcein were visualized by fluorescent microscopy (OsteoMeasure™ system, OLYMPUS, Japan), and the distance between labeled layers was used as a measure of the rate of bone formation as determined by morphometry software [22, 27].
Ex vivo osteoblastogenesis assay
Procedures for osteoblast formation from bone marrow stem cells were performed, as previously described [22, 24, 25]. Briefly, cells were flushed from the marrow cavity with α-MEM and seeded into tissue culture wells in this medium, and the harvested bone marrow cells were then cultured in α-MEM supplemented with 15 % preselected FCS (Hyclone, Logan, UT, USA) and ascorbic acid-2-phosphate (1 mM). Recruitment of marrow stromal cells to the osteoblast lineage was assessed at 10 days of culture by counting the number of bone marrow stromal cells that were positively stained for alkaline phosphatase for colony-forming unit-fibroblastic (CFU-F) (Sigma). In separate plates, the cell culture was continued until day 28, when the number of colonies producing mineralized bone matrix (CFU-osteoblastic (OB) staining, CFU-OB) was determined by von Kossa staining (Sigma) [22, 27–33]. Colonies were counted according to their diameters: 1.0–2 mm for one colony, 2–4 mm for two colonies, 4–6 mm for three colonies, and 6 to 8 mm for four colonies, as previously described [34].
Isolation of total RNA and quantitative PCR
Total RNA from ex vivo cultured osteoblasts was isolated and used for real-time PCR determination of mRNA levels as described previously [22, 24, 25]. qPCR was performed with an Applied Biosystems (ABI) Via 7 thermal cycler using ABI Taqman 2X PCR mix and ABI Assay on Demand qPCR primers. Changes in expression were calculated using the 2-∆∆Ct method using 18S RNA as the internal control [22, 24, 25].
Statistics
Data are expressed as mean ± SEM; the number of independent samples (n) is provided in the legend of each figure. The statistical significance of differences among means was tested using one-way analysis of variance and a Newman–Keuls post hoc test to determine the significance of differences between individual pairs of means using a p value of 0.05 as the cutoff for significance. Statistical calculations were performed using Prism 4.0c (GraphPad Software, La Jolla, CA, USA).
Results
SOST deletion in mice prevented SCI-induced bone loss
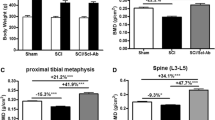
Body weight and the mass of gastrocnemius were significantly and similarly reduced after SCI in both WT and SOST KO mice (Fig. 1a, b).

Deletion of SOST gene prevents loss of BMD after SCI (a and b). Masses of body and gastrocnemius muscle in each group are shown. Gastrocnemius muscle weights at sacrifice were normalized relative to body weight prior to spinal cord transection (pre-operative body weight). Areal bone mineral density (aBMD) measurements in each group are shown at the distal femur (c), proximal tibia (d), and spine (L3–L5) (e), respectively. Data are expressed as mean ± SEM. WT/Sham group, n = 11; WT/SCI group, n = 12; KO/Sham group, n = 13; and KO/SCI group, n = 11. Significance of differences was determined using one-way analysis of variance with a Newman–Keuls test post hoc. *p < 0.05, **p < 0.01, and ***p < 0.001 versus the indicated group; NS no significant difference
Using a small animal dual-energy X-ray absorptiometer, BMD was determined. In the WT mice with SCI, significant loss of BMD was observed at the distal femur (−18 %) and at the proximal tibia (−28 %), as compared to WT/Sham control animals; a small, non-significant decrease in BMD of lumbar vertebra L3–L5 was also noted after SCI (Fig. 1c–e). Compared to WT/Sham mice, bone mass in SOST KO/Sham mice was higher at each of these sites (+55 % at the distal femur, +27 % at the proximal tibia, and +67 % at the L3–L5, respectively). Importantly, in SOST KO/SCI mice, no loss of BMD was observed at the distal femur or the proximal tibia, and bone loss of the vertebra was non-significant, when compared with the SOST KO/Sham animals (Fig. 1c–e).
Trabecular architecture was examined by high-resolution μCT (Fig. 2 (a)). SCI resulted in a markedly decrease of trabecular bone volume (BV/TV, −64 %) at the distal femur of WT mice. In striking contrast, there was no significant reduction of trabecular BV/TV in SOST KO/SCI mice compared with SOST KO/Sham mice, whose trabecular BV/TV was about 8-fold higher than that in WT/Sham mice (Fig. 2 (B—a)). Similarly, significant changes of the trabecular bone number (−52 %, Fig. 2 (B—b)), thickness (−30 %, Fig. 2 (B—c)), separation (+190 %, Fig. 2 (B—d)), connectivity density (−81 %, Fig. 2 (B—e)), and structure model index (+46 %, Fig. 2 (B—f)) were found in WT/SCI animals when compared to WT/sham animals, but not in SOST KO/SCI animals compared to the SOST KO/Sham animals. A statistically non-significant reduction (−34 %) of trabecular BV/TV was noted in SOST KO/SCI mice, compared to SOST KO/Sham mice; however, the trabecular BV/TV in SOST KO/SCI mice remained five times higher than that in WT/Sham mice.

Effects of SCI on trabecular architecture of the distal femur in WT and SOST KO mice. A Representative micro-CT 3D images of trabecular microarchitecture are displayed. B Measurements are shown for (a) trabecular bone volume per total tissue volume (BV/TV); (b) trabecular number (Tb.N, mm−1); (c) trabecular thickness (Tb.Th (mm)); (d) trabecular separation (Tb.Sp (mm)); (e) connectivity density (conn.D (mm−3)), and (f) structure model index (SMI). Data are expressed as mean ± SEM. WT/Sham group, n = 8; WT/SCI group, n = 7; KO/Sham group, n = 7; and KO/SCI group, n = 6. Significance of differences was determined by using one-way analysis of variance with a Newman–Keuls test post hoc. *p < 0.05, **p < 0.01, and ***p < 0.001 versus the indicated group
High-resolution μCT was also used to assess the effects of sclerostin deficiency on the cortical architecture of the femur midshaft (Fig. 3a). SCI significantly decreased the femur midshaft BV/TV (∼15 %) in WT animals, but not in SOST KO/SCI animals compared with SOST KO/Sham mice, whose BV/TV was about +27 % higher than that in WT/Sham mice (Fig. 3b).

Effects of SCI on cortical architecture of the femur midshaft in WT and SOST KO mice. a Representative micro-CT 3D-images of cortical microarchitecture are displayed. b Cortical bone volume over total tissue volume (BV/TV). Data are expressed as mean ± SEM. WT/Sham group, n = 8; WT/SCI group, n = 7; KO/Sham group, n = 7; and KO/SCI group, n = 6. Significance of differences was determined by using one-way analysis of variance with a Newman–Keuls test post hoc. *p < 0.05 versus the indicated group
SCI did not alter bone formation in SOST KO animals
Dynamic histomorphometric analysis (Fig. 4a) was conducted to measure the mineral apposition rate (MAR) and mineralizing surface/bone surface (MS/BS) for trabecular bone of the distal femur metaphysis. SCI significantly reduced MAR (Fig. 4 (B—a)) and bone formation rate (BFR/BS, Fig. 4 (B—c)) in WT mice but not in SOST KO mice. No significant difference in MS/BS was observed in WT mice after SCI. The increases in MS/BS and BFR/BS were observed in SOST KO mice when compared to WT mice as previously reported [14], although these changes did not reach statistic significance.

Effects of SCI on bone formation parameters in WT and SOST KO mice. A Representative dynamic histomorphometric analysis images are displayed. B Measurement of (a) mineral apposition rate (MAR); (b) mineralizing surface/bone surface (MS/BS); (c) bone formation rate over bone surface (BFR/BS) and (d) bone formation rate over total tissue volume (BFR/TV) for trabecular bone of the distal femur metaphysis. Data are expressed as mean ± SEM. WT/Sham group, n = 8; WT/SCI group, n = 7; KO/Sham group, n = 7; and KO/SCI group, n = 6. Significance of differences was determined using one-way analysis of variance with a Newman–Keuls test post hoc. *p < 0.05 and **p < 0.01 versus the indicated group; NS no significant difference
SCI did not affect osteoblastogenesis in SOST KO animals
The potential of bone marrow stromal cells to undergo osteoblastogenic differentiation was determined by counting the number of bone marrow stromal cells that were positively stained for CFU-F. The commitment of osteoprogenitors was assessed by determining the number of colonies producing mineralized bone matrix (CFU-OB) by von Kossa staining. Consistent to the previous findings [22, 24], decreases in osteoblastogenesis (CFU-F and CFU- OB staining) and the levels for transcripts encoding the osteoblast differentiation markers Runx2 and osteocalcin were observed in WT animals following SCI (Fig. 5a, b). Notably, there were an +180 % or +168 % increase in the numbers of CFU-F positive staining or mineralized nodules (CFU-OB) cells, respectively, in SOST KO/Sham mice when compared to WT/Sham mice; of interest, there was no change in CFU-F positive staining or CFU-OB cells in SOST KO/SCI mice when compared to SOST KO/Sham mice (Fig. 5a, b). Consistently, in ex vivo cultures of osteoblasts derived from marrow stromal stem cells, mRNA levels of Runx2, osteocalcin, Tcf7, and Lef-1 were significantly increased by about 1.7- to 2.3-fold, in the SOST KO/Sham group compared to the WT/Sham group; a non-significant increase in mRNA levels for Bmp2 or Axin2 was also observed. Of note, there was no significant change in mRNA expression of Runx2, osteocalcin, Tcf7, Lef-1, Bmp2, or Axin2 in SOST KO/SCI mice when compared to SOST KO/Sham mice (Fig. 5c–h).

Differentiation potential of bone marrow progenitor cells generated from WT and SOST KO mice following SCI. a Representative images showing alkaline phosphatase staining (CFU-F) of cultures of marrow stromal cells and cell counts of alkaline phosphatase–positive cells and CFU-OB staining of colonies producing mineralized bone matrix after von Kossa staining. b Counts of alkaline phosphatase–positive cells and colonies by CFU-OBs staining. c–h Changes in gene expression in osteoblasts developed by primary culture of bone marrow stromal cells. mRNA levels were determined by real-time PCR. Data are expressed as mean ± SEM; WT/Sham group, n = 5; WT/SCI group, n = 4; KO/Sham group, n = 5; and KO/SCI group, n = 4. Significance of differences was determined using one-way analysis of variance with a Newman–Keuls test post hoc. *p < 0.05, **p < 0.01, and ***p < 0.001 versus the indicated group; NS no significant difference. CFU-F colony forming unit–fibroblastic; CFU-OBs CFU-osteoblasts
Discussion
The bone loss following SCI is associated with several unique pathological features that differentiate it different from other forms of osteoporosis. These features include permanent immobilization, neurological dysfunction, systemic hormonal alternations, and associated metabolic disorders [1]. It is unknown how these complex pathophysiological changes are linked to molecular alterations that could influence bone metabolism and adaptations in SCI. To address this fundamental question, our study compared the effects of a complete spinal cord transection on bone loss in WT animals and in SOST KO animals. The targeted deletion of sclerostin gene in mice was resistant to sublesional loss of trabecular and cortical bone mass due to SCI, as characterized by no change of BMD, bone volume or BFR in SOST KO mice after SCI. Furthermore, at the cellular level, SCI did not alter osteoblastogenesis in SOST KO mice. Sclerostin appears to plays an essential role in mediating the severe bone deterioration that results from SCI.
Recent clinical studies indicate elevation of serum sclerostin levels after short-term SCI, suggesting that rapid bone loss due to motor-complete SCI results, at least in part, from elevated sclerostin levels during the acute and sub-acute phases after SCI, and that blocking sclerostin activity could represent a therapeutic option for patients with short-term SCI [20, 35]. This notion is supported by the recent findings from ours and others in which sclerostin antibody can prevent sublesional bone loss in rat models of acute SCI [21, 22]. However, whether sclerostin plays a role in the bone loss after SCI at the chronic stage remains controversial. In one study, a decline in the levels of serum sclerostin during chronic SCI and a strong correlation between serum sclerostin levels and BMD values in SCI patients were reported, suggesting that sclerostin may not be an appropriate therapeutic marker to employ in the treatment of osteoporosis in those with chronic SCI [35]. In contrast, a recent study by Invernizzi et al. demonstrated significantly higher values of serum sclerostin in long-term SCI compared with healthy subjects [36].
Recent work in rats with motor-complete SCI has demonstrated that sublesional loss of trabecular BV/TV rapidly occurred as early as 1 week after injury [37], with additional loss of bone by −61 to 67 % within the first 2–3 weeks [37], −70 % at 33 days [38], and −67 % at 56 days [22, 39]. In the present study, trabecular BV/TV in WT mice at 56 days after SCI is lost by −64 %, a magnitude similar to the one typically observed in SCI rats at the same duration of post-injury that is almost equivalent to the maximal degree of bone loss in rats with chronic SCI [22, 38, 39]. From the prior discussion, it would follow that the bone adaptation in animals injured 56 after SCI would serve as an appropriate model for capturing the effects of both the acute and chronic phases of sublesional bone loss. In this regard, our findings that SOST KO mice are resistant to the severe bone loss at 56 days after SCI, clearly define the key role for sclerostin in the development of bone loss not only during the acute phase but also during the chronic phase. Because there is currently no clinically proven pharmacological approach to prevent osteoporosis in acute SCI, a majority of the 12,000 new cases of SCI each year in the USA progress to the chronic phase without any treatment to halt the development of osteoporosis [1]. Thus, our current findings have important therapeutic implications, strongly suggesting that sclerostin antibodies could be a valid option to prevent the bone loss in patients with acute and, possibly, mitigate additional bone loss in those with chronic SCI.
Mechanical loading is one of the primary factors controlling bone mass with the removal of stress and strain on the skeleton resulting in bone loss [40, 41]. Osteocytes that comprise 90 to 95 % of all bone cells and have been considered as an orchestrator of bone remodeling through regulation of osteoclast and osteoblast activity by sensing and transducing mechanical signals into anabolic chemical signals [42]. It has been reported that osteocyte expression of sclerostin is reduced by mechanical loading and increased by unloading [12, 14, 43], suggesting that sclerostin expression is responsive to mechanical stimuli. In neurologically motor-complete SCI, sublesional bone loss proceed at a rate of up to 1 % per week for the first 6–12 months at the sites most severely affected [1, 3], a rate that is substantially greater than that observed in other types of disuse osteoporosis such as microgravity (0.25 %/week) [44] and prolonged bed rest (0.1 %/week) [45], as well as that of post-menopausal women (3–5 %/year) [46]. Using a mechanical unloading model achieved by tail suspension, Lin et al. found that SOST KO mice are resistant to immobilization-induced bone loss [13]. Although such a mechanical unloading paradigm is not a condition as extreme as that of SCI, the finding in immobilization is similar to our finding in that SOST KO mice with SCI, with both mice models protected from bone loss, suggesting a common role of sclerostin in regulation of bone homeostasis in different conditions of skeleton unloading.
Recently, several mice strains with the targeted deletion of SOST full allele or SOST distal enhancer were generated and characterized [13, 14] [47]. In these animal models and in our current study, mice with SOST deficiency show a high bone mass phenotype as characterized by marked increase in BMD, bone volume, and bone formation, indicating that sclerostin antagonizes a powerful bone formation pathway. Of note, the present work reveals that genetic inactivation of sclerostin prevents SCI-related bone loss, which is tightly associated with the prevention of the SCI-induced reduction in bone formation, as reflected in SOST KO mice with SCI by the absence of change in BFR and MAR. Specifically, the normalization of MAR suggests that there is a rescue in the amount of bone matrix deposited per active osteoblast cluster. This evidence indicates that sclerostin serves as a key player of bone formation pathway in conditions of extreme immobilization after SCI. A statistically non-significant reduction (−34 %) of trabecular BV/TV was observed in SOST KO/SCI mice, compared to SOST KO/Sham mice. Thus, mice with the deletion of sclerostin were largely, but not fully, protected against bone loss after SCI. Although it may be tempting to suggest that the predominant role of sclerostin deficiency in our mouse model of SCI was on bone formation, with a lesser impact on bone resorption, because the degree of inhibition of bone resorption was not quantitated, the relative contribution on bone formation versus resorption remains merely conjecture.
The present work provides the first evidence that SOST gene deletion has a favorable impact on osteoblastic differentiation potential of bone marrow progenitors by our finding of enhanced osteoblastogenesis. The targeted deletion of SOST gene increased the number of in ex vivo-cultured osteoblasts and abrogated SCI-induced impairment in osteoblastogenesis. This data is consistent with recent reports that SCI or other conditions of unweighting reduced numbers of colonies of marrow stromal cells, alkaline phosphatase-positive osteoblasts, and bone nodules; inhibition of sclerostin activity by Scl-Ab reversed each of these adverse changes [22, 48]. Furthermore, SOST gene deletion significantly increased gene expression in cultured osteoblasts of gene expression for osteoblast differentiation genes (Runx2 and osteocalcin) and Wnt signaling (Tcf7 and Lef-1). The effects of these changes would be expected to lead to the activation of Wnt signaling within mesenchymal stem cells (MSCs) and, thereby, to promote the recruitment of MSCs into osteoblast lineage and enhance bone formation. Although quite preliminary, it is yet tempting to postulate with the advent of recent evidence that SCI causes a unfavorable shift from osteoblastogenesis to adipogenesis within bone marrow MSCs though adverse regulation of peroxisome proliferator-activated receptor-γ and Wnt signaling, resulting in bone loss [49]. Whether sclerostin-mediated Wnt signaling cascade is involved in the aforementioned coordinated molecular and cellular events in models of SCI is an area worthy of investigation in future studies.
Currently, there are no well-accepted interventions for the prevention or treatment of SCI-induced osteoporosis. Bisphosphonates are effective for osteoporosis in the elderly, but appear, at best, to transiently reduce SCI-related bone loss in individuals with SCI who are ambulatory [1, 50]. In those with motor-complete SCI, bisphosphonates have no effect at the knee [51, 52]. Development of a safe and effective therapy for SCI-induced bone loss is a high clinical priority. Our current findings strongly suggest that sclerostin appeared to be a therapeutic target to treat SCI-induced bone loss. Indeed, recent preclinical studies confirmed effects of sclerostin inhibition using sclerostin antibody in SCI-related bone loss [21, 22]. Further investigation on the upstream regulator, as well as downstream effectors, of sclerostin will be needed to dissect the molecular mechanisms of sclerostin in the regulation of SCI-related bone loss. Screening of sclerostin inhibitors could also be performed and further tested in SCI animal models for drug discovery studies because such an approach would offer easier administration, lower cost, and less side effects.
The current study reveals the resistance to SCI-induced bone loss in SOST KO mice 56 days following SCI, yet it is premature to speculate with any confidence as to whether the prevention of bone loss in rodents could be translated to chronic SCI in patients in an effort to reverse bone loss. Additional investigation will focus on effects of sclerostin inhibition on long-term SCI, when adverse pathological changes including neurological dysfunction, permanent immobility, systematic hormonal change, and metabolic disorders are more fully developed and when extensive trabecular bone loss has already occurred. Such a study is being actively pursued in our laboratory.
In summary, complete spinal cord transection induced significant loss of bone mass and bone formation below the level of lesions in WT mice but not in SOST KO mice. Mechanisms underlying sclerostin deficiency-mediated promotion of bone formation are linked to the prevention of the impaired osteoblastogenic potential of bone marrow progenitors after SCI; one can assume through the activation of Wnt pathway. Our findings establish that the targeted disruption of sclerostin will provide resistance to SCI-induced severe bone loss, strongly suggesting that blockade of sclerostin could be a promising new therapeutic approach in the preservation of bone mass and integrity after motor-complete SCI. Additionally, these findings provide relevant information with regard to therapeutic options and mechanistic insights for other conditions associated with disuse osteoporosis as a consequence of neurological disorders and immobilization, such as those of stroke, Parkinson’s disease, multiple sclerosis, poliomyelitis, bed rest, and space flight.
Abbreviations
- BFR:
-
Bone formation rate
- BMD:
-
Bone mineral density
- CFU-F:
-
Colony-forming unit-fibroblastic
- DXA:
-
Dual-energy x-ray absorptiometer
- KO:
-
Knockout
- MAR:
-
Mineral apposition rate
- MS/BS:
-
Mineralizing surface/bone surface
- MSCs:
-
Mesenchymal stem cells
- PFA:
-
Paraformyldahyde
- SCI:
-
Spinal cord injury
- SOST :
-
Sclerostin
References
Qin W, Bauman WA, Cardozo C (2010) Bone and muscle loss after spinal cord injury: organ interactions. Ann N Y Acad Sci 1211:66–84
Qin W, Bauman WA, Cardozo CP (2010) Evolving concepts in neurogenic osteoporosis. Curr Osteoporos Rep 8:212–218
Szollar SM, Martin EM, Sartoris DJ, Parthemore JG, Deftos LJ (1998) Bone mineral density and indexes of bone metabolism in spinal cord injury. Am J Phys Med Rehabil 77:28–35
Garland DE, Adkins RH, Scott M, Singh H, Massih M, Stewart C (2004) Bone loss at the os calcis compared with bone loss at the knee in individuals with spinal cord injury. J Spinal Cord Med 27:207–211
Modlesky CM, Majumdar S, Narasimhan A, Dudley GA (2004) Trabecular bone microarchitecture is deteriorated in men with spinal cord injury. J Bone Miner Res 19:48–55
Battaglino RA, Lazzari AA, Garshick E, Morse LR (2012) Spinal cord injury-induced osteoporosis: pathogenesis and emerging therapies. Curr Osteoporos Rep 10:278–285
Zaidi M (2007) Skeletal remodeling in health and disease. Nat Med 13:791–801
Glass DA 2nd, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, Karsenty G (2005) Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell 8:751–764
Bonewald LF, Johnson ML (2008) Osteocytes, mechanosensing and Wnt signaling. Bone 42:606–615
Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M, Lacza C, Wuyts W, Van Den Ende J, Willems P, Paes-Alves AF, Hill S, Bueno M, Ramos FJ, Tacconi P, Dikkers FG, Stratakis C, Lindpaintner K, Vickery B, Foernzler D, Van Hul W (2001) Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet 10:537–543
Morvan F, Boulukos K, Clement-Lacroix P, Roman Roman S, Suc-Royer I, Vayssiere B, Ammann P, Martin P, Pinho S, Pognonec P, Mollat P, Niehrs C, Baron R, Rawadi G (2006) Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J Bone Miner Res 21:934–945
Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, Mantila SM, Gluhak-Heinrich J, Bellido TM, Harris SE, Turner CH (2008) Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem 283:5866–5875
Lin C, Jiang X, Dai Z, Guo X, Weng T, Wang J, Li Y, Feng G, Gao X, He L (2009) Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/beta-catenin signaling. J Bone Miner Res 24:1651–1661
Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D’Agostin D, Kurahara C, Gao Y, Cao J, Gong J, Asuncion F, Barrero M, Warmington K, Dwyer D, Stolina M, Morony S, Sarosi I, Kostenuik PJ, Lacey DL, Simonet WS, Ke HZ, Paszty C (2008) Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res 23:860–869
Li X, Ominsky MS, Warmington KS, Morony S, Gong J, Cao J, Gao Y, Shalhoub V, Tipton B, Haldankar R, Chen Q, Winters A, Boone T, Geng Z, Niu QT, Ke HZ, Kostenuik PJ, Simonet WS, Lacey DL, Paszty C (2009) Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res 24:578–588
Li X, Warmington KS, Niu QT, Asuncion FJ, Barrero M, Grisanti M, Dwyer D, Stouch B, Thway TM, Stolina M, Ominsky MS, Kostenuik PJ, Simonet WS, Paszty C, Ke HZ (2010) Inhibition of sclerostin by monoclonal antibody increases bone formation, bone mass, and bone strength in aged male rats. J Bone Miner Res 25:2647–2656
Tian X, Jee WS, Li X, Paszty C, Ke HZ (2010) Sclerostin antibody increases bone mass by stimulating bone formation and inhibiting bone resorption in a hindlimb-immobilization rat model. Bone 48:197–201
Padhi D, Jang G, Stouch B, Fang L, Posvar E (2001) Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res 26:19–26
McClung MR, Grauer A (2014) Romosozumab in postmenopausal women with osteopenia. N Engl J Med 370:1664–1665
Battaglino RA, Sudhakar S, Lazzari AA, Garshick E, Zafonte R, Morse LR (2012) Circulating sclerostin is elevated in short-term and reduced in long-term SCI. Bone 51:600–605
Beggs LA, Ye F, Ghosh P, Beck DT, Conover CF, Balaez A, Miller JR, Phillips EG, Zheng N, Williams AA, Aguirre JI, Wronski TJ, Bose PK, Borst SE, Yarrow JF (2015) Sclerostin inhibition prevents spinal cord injury-induced cancellous bone loss. J Bone Miner Res 30:681–689
Qin W, Li X, Peng Y, Harlow LM, Ren Y, Wu Y, Li J, Qin Y, Sun J, Zheng S, Brown T, Feng JQ, Ke HZ, Bauman WA, Cardozo CC (2015) Sclerostin antibody preserves the morphology and structure of osteocytes and blocks the severe skeletal deterioration after motor-complete spinal cord injury in rats. J Bone Miner Res 30:1994–2004
Arima H, Hanada M, Hayasaka T, Masaki N, Omura T, Xu D, Hasegawa T, Togawa D, Yamato Y, Kobayashi S, Yasuda T, Matsuyama Y, Setou M (2014) Blockade of IL-6 signaling by MR16-1 inhibits reduction of docosahexaenoic acid-containing phosphatidylcholine levels in a mouse model of spinal cord injury. Neuroscience 269:1–10
Sun L, Pan J, Peng Y, Wu Y, Li J, Liu X, Qin Y, Bauman WA, Cardozo C, Zaidi M, Qin W (2013) Anabolic steroids reduce spinal cord injury-related bone loss in rats associated with increased Wnt signaling. J Spinal Cord Med 36:616–622
Bramlett HM, Dietrich WD, Marcillo A, Mawhinney LJ, Furones-Alonso O, Bregy A, Peng Y, Wu Y, Pan J, Wang J, Guo XE, Bauman WA, Cardozo C, Qin W (2014) Effects of low intensity vibration on bone and muscle in rats with spinal cord injury. Osteoporos Int 25:2209–2219
Cardozo CP, Qin W, Peng Y, Liu X, Wu Y, Pan J, Bauman WA, Zaidi M, Sun L (2010) Nandrolone slows hindlimb bone loss in a rat model of bone loss due to denervation. Ann N Y Acad Sci 1192:303–306
Qin W, Sun L, Cao J, Peng Y, Collier L, Wu Y, Creasey G, Li J, Qin Y, Jarvis J, Bauman WA, Zaidi M, Cardozo C (2013) The central nervous system (CNS)-independent anti-bone-resorptive activity of muscle contraction and the underlying molecular and cellular signatures. J Biol Chem 288:13511–13521
Di Gregorio GB, Yamamoto M, Ali AA, Abe E, Roberson P, Manolagas SC, Jilka RL (2001) Attenuation of the self-renewal of transit-amplifying osteoblast progenitors in the murine bone marrow by 17 beta-estradiol. J Clin Invest 107:803–812
Abe E, Marians RC, Yu W, Wu XB, Ando T, Li Y, Iqbal J, Eldeiry L, Rajendren G, Blair HC, Davies TF, Zaidi M (2003) TSH is a negative regulator of skeletal remodeling. Cell 115:151–162
Sun L, Peng Y, Sharrow AC, Iqbal J, Zhang Z, Papachristou DJ, Zaidi S, Zhu LL, Yaroslavskiy BB, Zhou H, Zallone A, Sairam MR, Kumar TR, Bo W, Braun J, Cardoso-Landa L, Schaffler MB, Moonga BS, Blair HC, Zaidi M (2006) FSH directly regulates bone mass. Cell 125:247–260
Zhang R, Supowit SC, Klein GL, Lu Z, Christensen MD, Lozano R, Simmons DJ (1995) Rat tail suspension reduces messenger RNA level for growth factors and osteopontin and decreases the osteoblastic differentiation of bone marrow stromal cells. J Bone Miner Res 10:415–423
Kondo H, Nifuji A, Takeda S, Ezura Y, Rittling SR, Denhardt DT, Nakashima K, Karsenty G, Noda M (2005) Unloading induces osteoblastic cell suppression and osteoclastic cell activation to lead to bone loss via sympathetic nervous system. J Biol Chem 280:30192–30200
Basso N, Jia Y, Bellows CG, Heersche JN (2005) The effect of reloading on bone volume, osteoblast number, and osteoprogenitor characteristics: studies in hind limb unloaded rats. Bone 37:370–378
Owen ME, Cave J, Joyner CJ (1987) Clonal analysis in vitro of osteogenic differentiation of marrow CFU-F. J Cell Sci 87(Pt 5):731–738
Morse LR, Sudhakar S, Danilack V, Tun C, Lazzari A, Gagnon DR, Garshick E, Battaglino RA (2012) Association between sclerostin and bone density in chronic spinal cord injury. J Bone Miner Res 27:352–359
Invernizzi M, Carda S, Rizzi M, Grana E, Squarzanti DF, Cisari C, Molinari C, Reno F (2015) Evaluation of serum myostatin and sclerostin levels in chronic spinal cord injured patients. Spinal Cord 53:615–620
Minematsu ANY, Imagita H, Sakata S (2014) Time course changes in trabecular bone microstructure in rats with spinal cord injury. J Life Sci 8:522–528
Zamarioli A, Battaglino RA, Morse LR, Sudhakar S, Maranho DA, Okubo R, Volpon JB, Shimano AC (2013) Standing frame and electrical stimulation therapies partially preserve bone strength in a rodent model of acute spinal cord injury. Am J Phys Med Rehabil 92:402–410
Voor MJ, Brown EH, Xu Q, Waddell SW, Burden RL, Burke DA, Magnuson DS (2012) Bone Loss Following Spinal Cord Injury in a Rat Model. J Neurotrauma 29(8):1676–168
Albright F BC, Cope O (1941) Acute atrophy of bone (osteoporosis) stimulating hypreparathyroidism. J Clin Endocrinol Metab 1:711–716
Bauman WA, Cardozo CP (2015) Osteoporosis in individuals with spinal cord injury. PM R 7(2):188–201
Bonewald LF (2011) The amazing osteocyte. J Bone Miner Res 26:229–238
Tu X, Rhee Y, Condon KW, Bivi N, Allen MR, Dwyer D, Stolina M, Turner CH, Robling AG, Plotkin LI, Bellido T (2012) Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone 50:209–217
Vico L, Collet P, Guignandon A, Lafage-Proust MH, Thomas T, Rehaillia M, Alexandre C (2000) Effects of long-term microgravity exposure on cancellous and cortical weight-bearing bones of cosmonauts. Lancet 355:1607–1611
Leblanc AD, Schneider VS, Evans HJ, Engelbretson DA, Krebs JM (1990) Bone mineral loss and recovery after 17 weeks of bed rest. J Bone Miner Res 5:843–850
Berarducci A (2009) Stopping the silent progression of osteoporosis. Am Nurse Today 3:18
Collette NM, Genetos DC, Economides AN, Xie L, Shahnazari M, Yao W, Lane NE, Harland RM, Loots GG (2012) Targeted deletion of Sost distal enhancer increases bone formation and bone mass. Proc Natl Acad Sci U S A 109:14092–14097
Shahnazari M, Wronski T, Chu V, Williams A, Leeper A, Stolina M, Ke HZ, Halloran B (2012) Early response of bone marrow osteoprogenitors to skeletal unloading and sclerostin antibody. Calcif Tissue Int 91:50–58
Yan J, Li B, Chen JW, Jiang SD, Jiang LS (2012) Spinal cord injury causes bone loss through peroxisome proliferator-activated receptor-gamma and Wnt signalling. J Cell Mol Med 16:2968–2977
Chang KV, Hung CY, Chen WS, Lai MS, Chien KL, Han DS (2013) Effectiveness of bisphosphonate analogues and functional electrical stimulation on attenuating post-injury osteoporosis in spinal cord injury patients- a systematic review and meta-analysis. PLoS One 8:e81124
Bauman WA, Wecht JM, Kirshblum S, Spungen AM, Morrison N, Cirnigliaro C, Schwartz E (2005) Effect of pamidronate administration on bone in patients with acute spinal cord injury. J Rehabil Res Dev 42:305–313
Bauman WA, Cirnigliaro CM, La Fountaine MF, Martinez L, Kirshblum SC, Spungen AM (2015) Zoledronic acid administration failed to prevent bone loss at the knee in persons with acute spinal cord injury: an observational cohort study. J Bone Miner Metab 33:410–421
Acknowledgments
This work was supported by the Veterans Health Administration, Rehabilitation Research, and Development Service (grants 5I01RX001313 and 5I01RX000687 to WQ; B9212-C and B2020-C to WAB). Ministry of Science and Technology PRC grant 2014DFA32120 and the Natural Science Foundation of China (NSFC) grant 81471000 to YW. Amgen Inc. provided SOST KO mice. Authors’ roles: CPC, HK, XL, WAB, and WQ were responsible for study design and data analysis. YP, LH, WZ, JL, YQ, YW, LR, and WQ conducted the bone biology study. Jay Cao performed microCT analysis. The manuscript was written by WZ and WQ and was revised and approved by all authors. WQ takes responsibility for the integrity of the data analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Yuanzhen Peng, Wei Zhao, Xiaodong Li, Lauren M Harlow, Jiliang Li, Yiwen Qin, Jianping Pan, Yingjie Wu, Liyuan Ran, Hua Zhu Ke, William A. Bauman, Christopher Cardozo, and Weiping Qin declare that they have no conflict of interest.
Disclosure
YP, WZ, LH, JL, YQ, JP, YW, LR, CPC, WAB, and WQ have nothing to disclose. XL is current employee and shareholder of Amgen Inc., and HZK is current employee and shareholder of UCB Pharma.
Rights and permissions
About this article

Cite this article
Qin, W., Zhao, W., Li, X. et al. Mice with sclerostin gene deletion are resistant to the severe sublesional bone loss induced by spinal cord injury. Osteoporos Int 27, 3627–3636 (2016). https://doi.org/10.1007/s00198-016-3700-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-016-3700-x