
Abstract
Summary
This study was to determine if antibody against sclerostin (Scl-Ab) could prevent glucocorticoid (GC)-induced osteoporosis in mice. We found that Scl-Ab prevented GC-induced reduction in bone mass and bone strength and that the anabolic effects of Scl-Ab might be partially achieved through the preservation of osteoblast activity through autophagy.
Introduction
Glucocorticoids (GCs) inhibit bone formation by altering osteoblast and osteocyte cell activity and lifespan. A monoclonal antibody against sclerostin, Scl-Ab, increased bone mass in both preclinical animal and clinical studies in subjects with low bone mass. The objectives of this study were to determine if treatment with the Scl-Ab could prevent loss of bone mass and strength in a mouse model of GC excess and to elucidate if Scl-Ab modulated bone cell activity through autophagy.
Methods
We generated reporter mice that globally expressed dsRed fused to LC3, a protein marker for autophagosomes, and evaluated the dose-dependent effects of GCs (0, 0.8, 2.8, and 4 mg/kg/day) and Scl-Ab on autophagic osteoblasts, bone mass, and bone strength.
Results
GC treatment at 2.8 and 4 mg/kg/day of methylprednisolone significantly lowered trabecular bone volume (Tb-BV/TV) at the lumbar vertebrae and distal femurs, cortical bone mass at the mid-shaft femur (FS), and cortical bone strength compared to placebo (PL). In mice treated with GC and Scl-Ab, Tb-BV/TV increased by 60–125 %, apparent bone strength of the lumbar vertebrae by 30–70 %, FS-BV by 10–18 %, and FS-apparent strength by 13–15 %, as compared to GC vehicle-treated mice. GC treatment at 4 mg/kg/day reduced the number of autophagic osteoblasts by 70 % on the vertebral trabecular bone surface compared to the placebo group (PL, GC 0 mg), and GC + Scl-Ab treatment.
Conclusions
Treatment with Scl-Ab prevented GC-induced reduction in both trabecular and cortical bone mass and strength and appeared to maintain osteoblast activity through autophagy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Patients treated chronically with glucocorticoids (GCs) have decreased bone formation and increased bone resorption and fragility [1, 2]. The mechanisms that account for GC-induced inhibition of bone formation include the reduction of lifespan and activity of osteoblasts [2]. GCs alter the function of bone-forming cells through multiple pathways that include the reduction of osteoblast proliferation through its suppression of growth factors BMP2 and TGFβ1 [3]; up-regulation of the Wnt antagonists (Dkk-1, Wif-1, and Sost) and down-regulation of the Wnt receptor complex (frizzled 4, 7, Dsh1, and Axin1) [3, 4], which can suppress osteoblast differentiation (alkaline phosphatase, akp2), maturation (osteocalcin), and activity [5, 6]. GCs may also directly affect osteocytes as genes primarily expressed in osteocytes, Dmp1, Phex, and Sost [7] and are associated with bone formation and mineralization, and were up-regulated following GC treatment.
GCs can also alter osteoblast and osteocyte cell viability through autophagy [8–11]. Autophagy is an intracellular digestive mechanism that removes or recycles the dysfunctional organelles and/or oxidized proteins, and functions as a mechanism for nutrient breakdown to maintain cell survival [12, 13]. The role for autophagy as a metabolic regulator for bone cells has been previously reported. Autophagic proteins, including Atg5, Atg7, and Atg4b, are important for generating the osteoclast-ruffled border, corresponding to the secretory function of osteoclasts, and thereby bone resorption activities in vitro and in vivo [14, 15]. Autophagy is activated during osteoblast differentiation, and the skeleton becomes osteogenic when focal adhesion kinase family interacting protein, a key regulator of autophagy induction, is ablated from osteoprogenitor cells [16]. Selective deletion of Atg 7 in terminally differentiated osteocytes led to an inhibition of bone turnover and an osteoporotic phenotype [17]. Also, autophagy is an active process in most cells to recycle and then remove waste. We observed that GC treatment of osteocytes in vitro induced autophagy in a dose-dependent manner. The autophagic pathway was still present in osteocytes at a GC dose of 0.7–2.1 mg/kg/day, but this protective mechanism was no longer effective when GC dose was increased (5.6 mg/kg/day) [10]. Changes in osteoblast and osteocyte viability may contribute to GC suppression of bone formation [18], and this may contribute to the observation of a the reduction in bone strength [5] that is independent of the bone mass in these experimental models and in patients treated with GCs [19]. Interventions that maintain the viability of osteoblasts and osteocytes in the presence of GCs may be a therapeutic approach for prevention and treatment of GC-induced osteoporosis.
Sclerostin (Sost) is a protein produced by osteocytes that can inhibit osteoblast maturation. [20–24]. A monoclonal antibody to sclerostin (Scl-Ab) has been developed that can inhibit the activity of sclerostin, and can stimulate bone formation [25, 26]. Our research group reported that GC treatment for 28 days increased Sost gene expression [3] and concurrent PTH treatment modestly reduced the Sost expression [27]. The effects of Scl-Ab on osteoblast viability through autophagy are not well known. Since chronic GC use can inhibit osteoblast activity and lifespan [3], the objectives of this study were to determine if treatment with a Scl-Ab could rescue the GC-induced suppression of bone formation, and to determine if the anabolic effects of the Scl-Ab were associated with autophagy in osteoblasts in a mouse model of GC-induced osteoporosis.
Methods
Generation of the dsRed-LC3 reporter mice
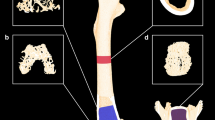
Transgenic mice were generated to globally express dsRed fused to LC3, a protein marker for autophagosomes. This mouse line allowed the examination of red fluorescent-LC3 (dsRed-LC3) in autophagic vacuoles in vitro and to identify the autophagic cells, including the osteoblast and osteocytes in vivo. A vector containing a dsRed–LC3 cassette was inserted between the CAG promoter (cytomegalovirus immediate-early (CMVie) enhancer and chicken b-actin promoter) [28] and the SV40 late polyadenylation signal. In this construct, dsRed was fused to the N terminus of rat LC3B (U05784) so as not to affect C-terminal PE conjugation. The 3.4-kbp CAG-dsRed-LC3-SV40 polyA fragment was microinjected into Swiss-Webster fertilized oocytes (Fig. 1a). This approach was derived from the GFP-LC3 transgenic technique that was previously published [29, 30]. The expression of dsRed-LC3 dots (autophagosomes) was confirmed in primary osteoblasts cultured from long bones (Fig. 1b, blue arrows). dsRed-LC3+ cells were observed within the bone marrow, and on bone surface approximate to the calcein-labeled mineralized bone surface (Fig. 1c, white arrows).

Generation of the dsRed-LC3 reporter mice. a Diagram of the dsRed-LC3 construct. b Primary osteoblasts were isolated from long bones of the dsRed-LC3 mice at 1 month of age. A portion of the osteoblasts demonstrated the classic autophagic dots (blue arrow head), a signature appearance for autophagosomes. c A representative distal femur frozen section from a dsRed-LC3 male mouse. Autophagic cells were observed within bone marrow or on the bone surface adjacent to the double calcein-labeled surface (white arrows). Calcein was given to the mice at 20 mg/kg 9 and 2 days prior to sacrifice. Ct cortical bone, Tb trabecular bone. Scale bar 100 μm
Animals and experimental procedures
Two-month-old male dsRed-LC3-reporter mice were maintained on commercial rodent chow (22/5 Rodent Diet; Teklad, Madison, WI) available ad libitum with 0.95 % calcium and 0.67 % phosphate. Mice were housed in a room that was maintained at 20 °C with a 12-h light/dark cycle. The dsRed-LC3-reporter mice were randomized into eight experimental groups. Slow-release pellets (Innovative Research of American, Sarasota, FL) of prednisolone (GC) or placebo were implanted respectively: Group 1, the control group, was implanted with a placebo pellet (PL, n = 10); groups 2–4 were implanted with slow-release prednisolone pellets, which were equivalent to 0.8, 2.8, and 4 mg/kg/day, respectively (n = 8–10/group); group 5 was implanted with PL pellet and received Scl-Ab treatment (25 mg/kg, SC, 2×/week., n = 7); and groups 6–8 received GC 0.8, 2.8, or 4 mg/kg/day and Scl-Ab treatment (25 mg/kg, SC, 2×/week., n = 8–10/group). The mice were sacrificed after 3 weeks of treatment. Calcein (20 mg/kg) was injected to all mice 9 and 2 days before sacrifice. All animals were treated according to the USDA animal care guidelines with the approval of the UC Davis Committee on Animal Research.
The mice were fasted for 12 h before sacrifice; serum samples were obtained during necropsy and were stored at −80 °C prior to the assessment of biochemical markers of bone turnover. At necropsy, the mice were exsanguinated by cardiac puncture. At the time of sacrifice, the fifth lumbar vertebral body (LVB) and right distal femurs (DF) were used for micro-CT scans. Cryosections were collected from the fourth LVB and the right femoral shafts and used for bone histomorphometry and autophagy evaluations. The sixth LVBs and the left femurs were used for biomechanical compression tests. The right tibiae were used protein extraction for Western blots.
Micro-CT
The fifth lumbar vertebra and right distal femurs were scanned with μCT (VivaCT 40, Scanco Medical AG, Bassersdorf, Switzerland) at 70 keV and 145 μA with an isotropic resolution of 10.5 μm in all three dimensions. The entire LVB5 was scanned. All trabeculae in the marrow cavity and within cortical shafts were evaluated. Scanning of the DF was initiated at the level of the growth cartilage-metaphyseal junction and extended proximally for 250 slices. Evaluations were performed on 150 slices beginning 0.2 mm proximal to the most proximal point along the boundary of the growth cartilage so as to exclude the growth plates. For each scan, the volume of interest was defined as ~0.25 mm internal to the boundary of the marrow cavity with the cortex. The methods used for calculating trabecular thickness (Tb.Th) and bone mineral density (BMD) have been described previously [5, 31]. For the right mid-femur, the scanning was performed at the middle femur and continued proximally 0.5 mm. All the slides were used to evaluate total volume (TV), cortical bone volume (BV), and cortical thickness (Ct.Th) [32–34].
Biochemical markers of bone turnover
Serum osteocalcin was measured using a Luminex Osteocalcin kit (EMD Millipore, Billerica, MA, USA), and serum CTX-1 was measured by ELISA (Immunodiagnostic Systems Inc., Gaithersburg, MD, USA) following the manufacturer’s instructions. The coefficient of variations for these tests in our laboratory was less than 10 %.
Bone histomorphometry
Eight micrometers thick cryosections of the fourth LVB were obtained with a Leica/Cryostat microtome. Bone histomorphometry was performed using a semi-automatic image analysis Bioquant system (Bioquant Image Analysis Corporation, Nashville, TN) linked to a microscope equipped with transmitted and fluorescence light [5]. A counting window, allowing for measurement of the entire trabecular bone and bone marrow within the growth plate and cortex, was created for the histomorphometric analysis. Static measurements included tissue area (T.Ar), bone area (B.Ar), and bone perimeter (B.Pm). Dynamic measurements included single- (sLS) and double-labeled surface (dLS), autophagic osteoblast surface, and inter-labeled thickness (Ir.L.Th). These indices were used to calculate mineralizing surface (MS/BS) and mineral apposition rate (MAR) and surface-based bone formation rate (BFR/BS) [35, 36]. One section from each mouse was used to stain with tartrate-resistant acid phosphatase for osteoclast measurement (OcS/BS). We used the terminology following the recommendation of the American Society for Bone and Mineral Research [36], and we have reported similar methodology in other experiments in our laboratory [5, 31, 37, 38]. Autophagic osteoblasts were defined as punctuated red-fluorescence staining cells laying on the trabecular surface that co-localized with the calcein labels. The results were presented as the percentage of autophagic osteoblast surface/bone surface [10].
The femoral diaphyses were dissected and fixed in 4 % paraformaldehyde, dehydrated in graded concentrations of ethanol and xylene, embedded un-decalcified in methyl methacrylate, and then cross-sectioned using a SP1600 microtome (Leica, Buffalo Grove, IL) into 40 μm sections. Total cross-sectional bone area (T.Ar), cortical area (Ct.Ar), and cortical width (Ct.Wi) were measured with the Bioquant Image analysis system. Single- and double-labeled surface and inter-labeled width were measured separately at the endocortical (Ec) and periosteal (Ps) bone surfaces. MAR and BFR/BS were calculated thereafter for both the endocortical and periosteal bone surfaces [32, 33, 39].
Real-time RT-PCR
Total RNA was obtained from the tibiae shaft using a modified two-step purification protocol employing homogenization (PRO250 Homogenizer, 10 mm × 105 mm generator, PRO Scientific IN, Oxford CT) in Trizol (Invitrogen, Carlsbad, CA) followed by purification over a QIAGEN RNeasy column (QIAGEN, Valencia, CA). RNA integrity was monitored by running RNA on 1 % Agarose gel in 120 V for 30 min. Intact RNA has both clear 18S and 28S ribosomal RNA bands with 28S rRNA approximately twice as intense as the 18S rRNA band. RNA was reverse-transcribed to single-stranded cDNA by using High-Capacity cDNA Archive Kit (ABI, California). Gene expressions were presented as fold changes from PL group [3].
Western blot
Tibial cortical bones were lysed in RIPA buffer with homogenization. The bone lysates were resolved on SDS-PAGE and electrophoretically transferred to polyvinylidene difluoride membranes. Membranes were incubated with primary antibodies that include β-actin (Santa Cruz Biotechnology, Santa Cruz, CA), anti-Atg 7, anti-Atg-16L, and anti-LC3 (Cell Signaling Technology, Danvers, MA) followed by species-specific horseradish peroxidase secondary antibody. Immunoreactive materials were detected by chemiluminescence (Pierce Laboratories, Thermo Fisher Scientific, Rockford, IL), then were imaged and quantitated by BIO-RAD ChemiDoc MP imaging system and analysis software [10, 40].
Biomechanical testing
For the vertebral bodies, the endplates of the lumbar vertebral body were polished using an 800-grit silicon carbide paper to create two parallel planar surfaces. Before testing, caudal and cranial diameter measurements were taken at the top, middle, and bottom of each vertebrae to obtain six measurements which were averaged as the diameter; the height along the long axis was recorded as well and the vertebrae were modeled as a cylinder. The lumbar vertebra were then loaded to failure under far-field compression along its long axis using an MTS 831 electro-servo-hydraulic testing system (MTS Systems Corp., Eden Prairie, MN) at a displacement rate of 0.01 mm/s with 90 N load cell; the tests were performed in 37 °C HBSS, and sample loads and displacements were continuously recorded throughout each test. Values for the maximum load and the apparent ultimate stress (bone strength) for compression were then determined, where the apparent ultimate stress was calculated using the expression σ = 4P/(πd 2), where P is the load and d is the average diameter [33, 39, 41].
To analyze the biomechanical properties of the cortical bone, the left femurs were subjected to four-point bending tests, with the bone loaded using an MTS 831 electro-servo-hydraulic testing system (MTS Systems Corp., Eden Prairie, MN) such that the posterior surface was in tension and the anterior surface was in compression; the major loading span was 14.5 mm. Each femur was loaded to failure in 37 °C HBSS at a displacement rate of 0.01 mm/s while its corresponding load and displacement were measured using a calibrated 90 N load cell. Two diameter measurements were taken at the fracture location, and averaged to model the femur as a cylinder. Values for the yield stress and load, maximum load and ultimate stress (bone strength) in the bending tests were then determined, with the apparent stress calculated from σ = PLy/4I; P is the load, L is the major loading span, y is the distance from the center of mass (d/2), and I is the moment of inertia (πd 4⁄64), where d is the average diameter [33, 39, 41]. A measure of apparent toughness was estimated in terms of the work of fracture, specifically the area under the load vs. displacement curve normalized by twice the fracture surface area.
Statistical analysis
The group means and standard deviations (SDs) were calculated for all outcome variables. The nonparametric Kruskal-Wallis test was used to determine the overall differences between the groups followed by Bonferroni post hoc test to compare the treatment groups with PL or GC groups (SPSS Version 14; SPSS Inc., Chicago, IL).
Results
Body weight
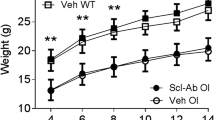
Vehicle- or Scl-Ab-treated mice with PL pellets gained body weight from 8 to 11 weeks of age. Mice in the groups treated with GC 2.8 and 4 mg/kg/day groups had nearly a 20 % weight loss from the baseline level during the experiment. Animals treated with Scl-Ab did not alter body weights as compared to the PL or GC vehicle-treated groups (Supplementary Table 1).
Bone volume and bone turnover changes in the trabecular bones
Measurements of trabecular bone at both the lumbar vertebral body (LVB) and the distal femurs (DF) were obtained, and in this manuscript, we report only on the LVB results. For the lumbar vertebra body (LVB), GC at 0.8 and 2.8 mg/kg/day levels did not affect trabecular bone volume/tissue volume (BV/TV), Tb.N, Tb.Th, Conn-Dens, and BMD significantly, while GC 4 mg/kg/day dose decreased BV/TV, Tb.Th, and Conn-Dens by 27, 11, and 19 %, respectively, as compared to the PL (vehicle group (p < 0.05) (Table 1). PL plus Scl-Ab treatment significantly increased the BV/TV compared to PL vehicle alone (p < 0.05 vs. PL). The increases in trabecular bone BV/TV were associated with greater Tb.Th and BMD in Scl-Ab-treated groups (Table 1). The changes in the BV/TV of the trabecular bone with GC alone or GC plus Scl-Ab combination treatments were overall more pronounced at the DFM site than the LVB. For example, PL plus Scl-Ab mice increased trabecular bone BV/TV by 139 % as compared to the PL alone. GC-induced reduction in the BV/TV or the increase in BV/TV with GC and Scl-Ab combination treatment were more pronounced in the DFM than in the LVB that GC + Scl-Ab completely prevented GC-induced bone loss in the DFM (supplementary Table 2).
Dynamic bone turnover was assessed by measuring serum biochemical markers of bone turnover and dynamic histomorphometry on the fourth LVB (Table 2). GC at 4 mg/kg/day decreased serum osteocalcin (OC) by 25 % and increased CTX-1 levels by 68 % compared to the PL (p < 0.05 vs. PL). GC at 4 mg/kg significantly decreased MS/MS by 50 % and BFR/BS by 60 % as compared to the PL group (p < 0.05 vs. PL). No significant differences in bone turnover markers and bone formation parameters were observed between the lower doses of GC and PL groups. However, Scl-Ab plus PL significantly increased bone formation parameters, MS/BS, MAR, and BFR/BS, by approximately 50 % compared to the PL group treated with vehicle (p < 0.05 vs. PL). The Scl-Ab treatments prevented the reduction of bone formation parameters in mice with GC at 4 mg/kg/day. GC at 4 mg/kg or GC + Scl-Ab had significantly higher OcS/BS as compared to the PL plus Scl-Ab group or PL treated with vehicle (p < 0.05 vs. PL) (Fig. 2a, Table 2).

GC reduced bone formation, which were preserved by Scl-Ab co-treatment. a Representative frozen section obtained from the fourth LVB in dsRed-LC3 reporter mice treated with vehicle or various doses of GC or GC plus Scl-Ab. Scale bar 100 μm. b Representative cross-sections were obtained from the middle-femoral shaft. Yellow arrows illustrate double-labeled endocortical surface and white arrow illustrate double-labeled periosteal surface. Original magnification ×4. All the mice received two calcein labels at days 2 and 9 prior to sacrifice
Bone volume and bone turnover changes in the cortical bone
Since the trends for total bone volume (TV) and cortical bone volume (BV) were similar for all the groups, we chose to report BV. Compared to the placebo-treated animals, the GC dose of 0.8 mg/kg did not change middle-femoral BV and BMD (Table 3). However, BV was 12 % lowered in GC 4 mg/kg group and BMD was 7 % lower than PL (p < 0.05 vs. PL). Also, endocortical bone mineralizing surface was reduced by about 70 % in GC 2.8 and 4 mg/kg/day dose groups (p < 0.05 vs. PL). Also, periosteal MS/BS was significantly lowered at GC 4 mg/kg/day dose level and BFR at the periosteal bone surface was reduced by approximately 80 % in both the GC 2.8 and 4 mg/kg/day groups as compared to the PL (p < 0.05 vs. PL). PL plus Scl-Ab increased cortical BV by 20 % over PL group alone (p < 0.05 vs. PL). Bone formation at the endocortical bone surface was increased by 160 % in PL plus Scl-Ab compared to PL alone (p < 0.05 vs. PL). No significant differences were observed in Ec. and Ps-MS and BFR between vehicle- and Scl-Ab-treated groups in mice treated with lowest dose of GC. Treatment with GC plus Scl-Ab prevented the reduction in both endocortical and periosteal bone formation observed with GC treatment alone (2.8 and 4 mg/kg/day) (Fig. 2b).
Bone strength
GCs dose-dependently decreased vertebral maximal load and ultimate stress (Table 4). Toughness was reduced by 40 % in the GC 4 mg/kg group compared to the PL group (p < 0.05 vs. PL). PL plus Scl-Ab treatment increased the apparent ultimate stress and toughness by 31 and 108 %, respectively, as compared to PL alone (p < 0.05 vs. PL) (Table 4). Maximum load, apparent ultimate stress, and toughness were also greater in Scl-Ab plus GC groups compared with respective controls with significant differences observed in mice with GC treatments of 2.8 and 4 mg/kg/day (Table 4).
Mice treated with GC 0.8 mg/kg/day tended to have higher maximum load and apparent ultimate stress of the femur diaphysis than PL but the overall cortical strength, apparent toughness, was decreased by 30 % compared to PL. Mice treated with GCs at 0.8–2.8 mg/kg/day did not have changes in cortical bone strength compared to the PL group; however, mice treated with the GC at the 4 mg/kg/day dose had 10–30 % decreased apparent ultimate stress and toughness as compared to PL (p < 0.05 vs. PL). Pl plus Scl-Ab-treated mice had statistically significant increased maximum load and apparent ultimate stress as compared to PL group treated with vehicle. No significant differences in femoral bone strength parameters were observed between vehicle- and Scl-Ab-treated mice with low dose of GC. GC plus Scl-Ab treatment prevented the reduction in apparent ultimate stress and toughness in mice with GC at 2.8 and 4 mg/kg/day alone (p < 0.05 vs. respective GC group) (Table 4).
Osteoblast autophagic in vivo
Exploratory studies to assess autophagic osteoblasts in vivo were performed in the high-dose GC (4 mg/kg/day) group, and the GC plus Scl-Ab combination treatment group. RNA was extracted from the distal tibia, and each sample included both the cortical shaft and the periosteum. We found the mice treated with GC at 4 mg/kg/day had reduced levels of autophagy–related genes, ATg7 and Atg-16L by 1.8- and 3.4-fold, respectively, in the tibial shafts. Concurrent treatment with GC plus Scl-Ab maintained autophagic gene expression at approximately twofold above PL level (Fig. 3a). We also evaluated the protein levels of autophagy–related proteins (Atg7, Atg-16L, and LC3-II) expressed in the tibial shafts, and found that the levels of the autophagy-related proteins were similar between WT and DsRed-LC3-reporter mice (data not shown). Western blot analyses performed from the tibial shafts found that GC exposure reduced the gene expression of Atg-7, Atg-16L, and the amount of LC3-II/LC3-I by more than 50 %, calculated as a semi-quantitative assessment of the intensity of the Western blots, suggesting reduced induction and formation of autophagosomes. Concurrent treatment with GC plus Scl-Ab maintained the expression of ATG-7 and ATG-16L, and LC3-II at the PL levels (Fig. 3b, c). At the lumber vertebral body, autophagic osteoblast surface/BS was reduced by nearly 70 % in GC 4 mg/kg group compared to the PL on the vertebral trabecular bone surface (p < 0.05 vs. PL). When compared to GC 4 mg/kg/day, high dose alone, GC plus Scl-Ab treatment significantly increased the autophagic osteoblast surface/BS by 133 % as compared to GC alone (p < 0.05 vs. GC) (Fig. 3d).

Activation of autophagy in vivo. a RNA was extracted from the tibial cortical bone including periosteum for autophagy quantitative RT-PCR. GC-4 or GC-4 plus Scl-Ab (n = 3) were used. Gene expression is shown by fold changes from PL group, which was set to “1”. b Protein was extracted from the tibial cortical bone. Samples were collected from PL, GC-4, or GC-4 plus Scl-Ab, (n = 3) respectively. Western blotting was performed for select proteins related to autophagy activation, Atg7 and Atg16L and autophagosome formation (LC3-II/I). c Quantitative density evaluation for the Western blots. d Quantitative measurement of the autophagic osteoblasts/total osteoblasts at the vertebral trabecular bone surface. * GC vs. PL p < 0.05; # PL/GC + Scl-Ab vs. PL/GC p < 0.05 at a same dose level
Discussion
Glucocorticoid-induced osteoporosis (GIOP) is the most common cause for secondary osteoporosis as 30–50 % of patients on chronic prednisolone treatment sustain a fracture [42]. Several clinical studies have demonstrated that bisphosphonates increase bone mineral density (BMD) at the spine and hip [43–48] and decrease the incidence of vertebral fractures, especially in post-menopausal women treated with GCs [49]. A new anti-resorptive agent, denosumab, which inhibits RANKL action, also prevented GC-induced loss of bone mass and strength in a hRANKL-knockin mouse model [50], and improved BMD of the lumbar spine and hip in rheumatoid arthritis patients treated with GCs [51]. A bone anabolic agent, rhPTH (1–34), is generally considered as a second-line treatment option for GIOP patients. In clinical studies, rhPTH significantly increased bone formation markers and BMD at the spine and hip in GIOP patients [52–54], and it is more effective than bisphosphonates for increasing spine BMD and reducing the incidence of new vertebral fractures in GIOP-treated populations [47, 55–57].
Sclerostin is a Wnt antagonist that is secreted primarily by osteocytes. Once it is released, it is thought to move through the canaliculi of the osteocytes and into the bone marrow where it will inhibit osteoblast maturation and activity. The efficacy of Scl-Ab on augmentation of bone formation and bone mass have been demonstrated in intact, estrogen-deficient, hind-limb immobilization rodent models [58–61], as well as in primates and humans [62]. The effect Scl-Ab treatment in glucocorticoid-induced bone loss was studied in 7-week-old BLAB/C mice. The mice were treated with 3 mg/kg/day dexamethasone by oral gavage for up to 9 weeks. GC treatment alone reduced trabecular bone volume and femoral areal BMD, and these changes were prevented by concurrent Scl-Ab treatment of 25 mg/kg, 2×/week [63]. In our study, we used 2-month-old male mice and evaluated if Scl-Ab would prevent the negative effects of GCs on bone formation. We found that Scl-Ab treatment increased bone formation primarily by increasing the osteoblast surface and activity (mineral apposition rate) such that surfaced-based (BFR/BS) and tissue-based (BFR/TV) bone formation were increased. These observations are consistent with other reports of Scl-Ab treatment in intact rats or mice [25, 63, 64], and confirm that Scl-Ab directly stimulates modeling-dependent gain in bone mass [65, 66] resulting in more than 100 % increase in both the vertebral trabecular bone both in mice with or without concurrent GC exposure. The increase in trabecular bone volume was associated with increased trabecular bone thickness and overall bone strength. Treatment with Scl-Ab also increased the mineral apposition at the endocortical bone surface and resulted in higher cortical bone volume and BMD that was associated with significantly higher cortical bone strength.
Interestingly, the lowest dose of GCs used in this study (0.8 mg/kg/day) modestly increased endosteal bone formation and increased trabecular and cortical bone volume compared to the placebo-treated mice. However, higher GC doses of 2.8 and 4 mg/kg/day resulted in a dose-dependent decrease in serum osteocalcin levels, bone formation indices (mineralized bone surface, mineral apposition rate, and bone formation rates), and trabecular bone volume, including trabecular thickness at both the distal femurs and the lumber vertebrae. Additionally, GCs decreased periosteal bone formation at 2.8 and 4 mg/kg/day doses. However, the combination of GCs plus Scl-Ab was effective in maintaining endocortical and periosteal bone formation, cortical bone volume, and bone strength. Since GCs at 2.8 and 4 mg/kg/day levels reduced the body weights of the study mice by more than 20 %, it was possible that the reduced body weights might have contributed to the GC-induced bone loss as well as the reduction in treatment efficacy with Scl-Ab as the GC dose was increased. Marenzana et al. treated mice with dexamethasone alone and with Scl-Ab [63] and reported a reduction in bone formation when Scl-Ab was combined with higher doses of GCs [3, 27]. Also, a retrospective post hoc analysis was performed on the effects of 18-month clinical study in which nearly 400 patients that were on low (≤5 mg/kg/day), medium (>5 and <15 mg/kg/day), and high (≥15 mg/kg/day) of GCs and treated with Teriparatide (20 μg/kg/day). It was found that the increase in lumbar spine BMD was greater in low GC group treated with teriparatide than in the high-dose GC group [67]. These findings suggest that high doses of GCs might impair bone active agents from preserving bone mass and bone strength.
Glucocorticoids have multiple effects on bone cells that alter bone remodeling and induce rapid bone loss [1, 2]. We have demonstrated that GC treatment of mice resulted in an early elevation in the expression of genes involved with osteoclast activities within the first 7 days of GC exposure [3], which was followed by a delayed but prolonged suppression of osteoblastogenesis [3, 68] with reduced bone formation measured by serum osteocalcin and surface-based histomorphometry [34]. Indeed, GC doses of 4 mg/kg significantly reduced the percentages of osteoblasts that underwent autophagy, suppressed bone formation at all bone surfaces, and reduced bone strength at both the trabecular and cortical bone. We also observed preservation of autophagic osteoblasts with Scl-Ab plus GC treatment that with GC alone suggesting that Scl-Ab treatment may sustain osteoblast viability which was otherwise depressed by GC excess. It is extremely challenging to monitor the level of autophagy in bone. Nevertheless, we used multiple approaches such as RT-PCR, Western blotting, and the dsRed-LC3 reporting mouse line to monitor osteoblast autophagy at the tissue level. We found that the higher GC dose level (4 mg/kg/day) suppressed autophagic gene expression and reduced autophagic osteoblast surface while GC (4 mg/kg/day) plus Scl-Ab maintained the autophagic osteoblasts at the placebo levels. These data support a mechanism for the Scl-Ab to override the suppressive effects of GC on bone formation and bone strength by partially maintaining the viability of bone forming cells through autophagy.
This study has a number of strengths including a validated mouse model for GIOP and the use of a number of doses of glucocorticoids to determine a dose response for the study endpoints. We explored the potential mechanism of Scl-Ab on autophagy, via a novel dsRed-LC3 reporter mouse line. However, there are also a number of limitations. We only studied male mice and at one time point (21 days). Also, we only addressed the effect of Scl-Ab on prevention of GC-induced bone loss, and we did not evaluate the therapeutic effects of Scl-Ab in this model. Lastly, we used young mice with skeletons that were still maturing and accruing bone mass. To confirm our results and allow for extrapolation to older age groups, our studies will need to be repeated in older animals. The shortcoming of this mouse line is that it is a global reporting mouse line, and all cells undergoing autophagy express dsRed fluorescent. Another interesting finding from this study is that we found Scl-Ab by itself tended to lower osteoclast number but increase serum CTX-1. We have carefully reviewed the CTX-1 data from all study groups, and we truly have no explanation as to why the CTX-1 level was higher in the placebo plus sclerostin-antibody group. While we tend to be confident that CTX-1 is a good reflection of bone turnover or osteoclast activity; in this case, we cannot be sure because osteoclast surface was not significantly different in the placebo plus Scl-Ab groups. One possible explanation is that the Scl-Ab could augment RANKL release from osteocytes and osteoblasts during the treatment, which could increase osteoclast activity and CTX-1 levels. While serum RANKL levels were not measured in this study, we did measure serum levels of OPG, and it was not different in the placebo plus Scl-Ab group as compared to the placebo. Additionally, Scl-Ab might presumably block endogenous sclerostin activity and if the high-dose GC treatment induced osteocyte apoptosis and reduced sclerostin synthesis and activity, Scl-Ab treatment may have been less efficacious. However, it is beyond the scope of this report to study apoptosis, and we do not have ex vivo data regarding the live/dead ratio of osteocytes that would have supported this potential mechanism.
In summary, treatment of young male mice with GCs at 2.8 or 4 mg/kg/day doses for 21 days reduced bone formation, bone mass, and bone strength of both the trabecular and cortical bone. Treatment with a Scl-Ab prevented the detrimental effects of GCs on bone formation, and improved both trabecular architecture and integral bone strength. These findings warrant further evaluation of Scl-Ab in patients on chronic glucocorticoids.
References
Dalle Carbonare L, Arlot ME, Chavassieux PM, Roux JP, Portero NR, Meunier PJ (2001) Comparison of trabecular bone microarchitecture and remodeling in glucocorticoid-induced and postmenopausal osteoporosis. J Bone Miner Res 16(1):97–103
Weinstein RS (2001) Glucocorticoid-induced osteoporosis. Rev Endocr Metab Disord 2(1):65–73
Yao W, Cheng Z, Busse C, Pham A, Nakamura MC, Lane NE (2008) Glucocorticoid excess in mice results in early activation of osteoclastogenesis and adipogenesis and prolonged suppression of osteogenesis: a longitudinal study of gene expression in bone tissue from glucocorticoid-treated mice. Arthritis Rheum 58(6):1674–1686
Ohnaka K, Taniguchi H, Kawate H, Nawata H, Takayanagi R (2004) Glucocorticoid enhances the expression of dickkopf-1 in human osteoblasts: novel mechanism of glucocorticoid-induced osteoporosis. Biochem Biophys Res Commun 318(1):259–264
Lane NE, Yao W, Balooch M, Nalla RK, Balooch G, Habelitz S, Kinney JH, Bonewald LF (2006) Glucocorticoid-treated mice have localized changes in trabecular bone material properties and osteocyte lacunar size that are not observed in placebo-treated or estrogen-deficient mice. J Bone Miner Res 21(3):466–476
Hurson CJ, Butler JS, Keating DT, Murray DW, Sadlier DM, O'Byrne JM, Doran PP (2007) Gene expression analysis in human osteoblasts exposed to dexamethasone identifies altered developmental pathways as putative drivers of osteoporosis. BMC Musculoskelet Disord 8:12
Rios HF, Ye L, Dusevich V, Eick D, Bonewald LF, Feng JQ (2005) DMP1 is essential for osteocyte formation and function. J Musculoskelet Nueronal Interact 5(4):325–327
Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC (1998) Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Investig 102(2):274–282
Xia X, Kar R, Gluhak-Heinrich J, Yao W, Lane NE, Bonewald LF, Biswas SK, Lo WK, Jiang JX (2010) Glucocorticoid-induced autophagy in osteocytes. J Bone Miner Res 25(11):2479–2488
Jia J, Yao W, Guan M, Dai W, Shahnazari M, Kar R, Bonewald L, Jiang JX, Lane NE (2011) Glucocorticoid dose determines osteocyte cell fate. FASEB J 25(10):3366–3376
Yao W, Dai W, Jiang JX, Lane NE (2013) Glucocorticoids and osteocyte autophagy. Bone 54(2):279–284
Todde V, Veenhuis M, van der Klei IJ (2009) Autophagy: principles and significance in health and disease. Biochim Biophys Acta 1792(1):3–13
White E, Lowe SW (2009) Eating to exit: autophagy-enabled senescence revealed. Genes Dev 23(7):784–787
DeSelm CJ, Miller BC, Zou W, Beatty WL, van Meel E, Takahata Y, Klumperman J, Tooze SA, Teitelbaum SL, Virgin HW (2011) Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev Cell 21(5):966–974
Chung YH, Yoon SY, Choi B, Sohn DH, Yoon KH, Kim WJ, Kim DH, Chang EJ (2012) Microtubule-associated protein light chain 3 regulates Cdc42-dependent actin ring formation in osteoclast. Int J Biochem Cell Biol 44(6):989–997
Liu F, Fang F, Yuan H, Yang D, Chen Y, Williams L, Goldstein SA, Krebsbach PH, Guan JL (2013) Suppression of autophagy by FIP200 deletion leads to osteopenia in mice through the inhibition of osteoblast terminal differentiation. J Bone Miner Res 28(11):2414–2430
Onal M, Piemontese M, Xiong J, Wang Y, Han L, Ye S, Komatsu M, Selig M, Weinstein RS, Zhao H et al (2013) Suppression of autophagy in osteocytes mimics skeletal aging. J Biol Chem 288(24):17432–17440
Weinstein RS, Jilka RL, Almeida M, Roberson PK, Manolagas SC (2010) Intermittent parathyroid hormone administration counteracts the adverse effects of glucocorticoids on osteoblast and osteocyte viability, bone formation, and strength in mice. Endocrinology 151(6):2641–2649
Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C (2003) Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 48(11):3224–3229
Paszty C, Turner CH, Robinson MK (2010) Sclerostin: a gem from the genome leads to bone-building antibodies. J Bone Miner Res 25(9):1897–1904
Poole KE, van Bezooijen RL, Loveridge N, Hamersma H, Papapoulos SE, Lowik CW, Reeve J (2005) Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J 19(13):1842–1844
van Bezooijen RL, Roelen BA, Visser A, van der Wee-Pals L, de Wilt E, Karperien M, Hamersma H, Papapoulos SE, ten Dijke P, Lowik CW (2004) Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med 199(6):805–814
Yang F, Tang W, So S, de Crombrugghe B, Zhang C (2010) Sclerostin is a direct target of osteoblast-specific transcription factor osterix. Biochem Biophys Res Commun 400(4):684–688
Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, Harris SE, Wu D (2005) Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem 280(20):19883–19887
Li X, Warmington KS, Niu QT, Asuncion FJ, Barrero M, Grisanti M, Dwyer D, Stouch B, Thway TM, Stolina M et al (2010) Inhibition of sclerostin by monoclonal antibody increases bone formation, bone mass and bone strength in aged male rats. J Bone Miner Res
McClung MR, Grauer A, Boonen S, Bolognese MA, Brown JP, Diez-Perez A, Langdahl BL, Reginster JY, Zanchetta JR, Wasserman SM et al (2014) Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med 370(5):412–420
Yao W, Cheng Z, Pham A, Busse C, Zimmermann EA, Ritchie RO, Lane NE (2008) Glucocorticoid-induced bone loss in mice can be reversed by the actions of parathyroid hormone and risedronate on different pathways for bone formation and mineralization. Arthritis Rheum 58(11):3485–3497
Niwa H, Yamamura K, Miyazaki J (1991) Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108(2):193–199
Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T (2004) LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 117(Pt 13):2805–2812
Mizushima N (2009) Methods for monitoring autophagy using GFP-LC3 transgenic mice. Methods Enzymol 452:13–23
Yao W, Cheng Z, Koester KJ, Ager JW, Balooch M, Pham A, Chefo S, Busse C, Ritchie RO, Lane NE (2007) The degree of bone mineralization is maintained with single intravenous bisphosphonates in aged estrogen-deficient rats and is a strong predictor of bone strength. Bone 41(5):804–812
Yao W, Guan M, Jia J, Dai W, Lay YA, Amugongo S, Liu R, Olivos D, Saunders M, Lam KS et al (2013) Reversing bone loss by directing mesenchymal stem cells to bone. Stem Cells 31(9):2003–2014
Guan M, Yao W, Liu R, Lam KS, Nolta J, Jia J, Panganiban B, Meng L, Zhou P, Shahnazari M et al (2012) Directing mesenchymal stem cells to bone to augment bone formation and increase bone mass. Nat Med 18(3):456–462
Dai W, Jiang L, Lay YA, Chen H, Jin G, Zhang H, Kot A, Ritchie RO, Lane NE, Yao W (2015) Prevention of glucocorticoid induced bone changes with beta-ecdysone. Bone 74C:48–57
Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR (1987) Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 2(6):595–610
Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR, Parfitt AM (2013) Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 28(1):2–17
Lane NE, Yao W, Nakamura MC, Humphrey MB, Kimmel D, Huang X, Sheppard D, Ross FP, Teitelbaum SL (2005) Mice lacking the integrin beta5 subunit have accelerated osteoclast maturation and increased activity in the estrogen-deficient state. J Bone Miner Res 20(1):58–66
Balooch G, Yao W, Ager JW, Balooch M, Nalla RK, Porter AE, Ritchie RO, Lane NE (2007) The aminobisphosphonate risedronate preserves localized mineral and material properties of bone in the presence of glucocorticoids. Arthritis Rheum 56(11):3726–3737
Yao W, Dai W, Shahnazari M, Pham A, Chen Z, Chen H, Guan M, Lane NE (2010) Inhibition of the progesterone nuclear receptor during the bone linear growth phase increases peak bone mass in female mice. PLoS One 5(7):e11410
Xia X, Kar R, Gluhak-Heinrich J, Yao W, Lane NE, Bonewald LF, Biswas SK, Lo WK, Jiang JX (2010) Glucocorticoid induced autophagy in osteocytes. J Bone Miner Res
Turner CH, Burr DB (1993) Basic biomechanical measurements of bone: a tutorial. Bone 14(4):595–608
Civitelli R, Ziambaras K (2008) Epidemiology of glucocorticoid-induced osteoporosis. J Endocrinol Investig 31(7 Suppl):2–6
Jacobs JW, de Nijs RN, Lems WF, Geusens PP, Laan RF, Huisman AM, Algra A, Buskens E, Hofbauer LC, Oostveen AC et al (2007) Prevention of glucocorticoid induced osteoporosis with alendronate or alfacalcidol: relations of change in bone mineral density, bone markers, and calcium homeostasis. J Rheumatol 34(5):1051–1057
Inoue Y, Shimojo N, Suzuki S, Arima T, Tomiita M, Minagawa M, Kohno Y (2008) Efficacy of intravenous alendronate for the treatment of glucocorticoid-induced osteoporosis in children with autoimmune diseases. Clin Rheumatol 27(7):909–912
Kaji H, Kuroki Y, Murakawa Y, Funakawa I, Funasaka Y, Kanda F, Sugimoto T (2010) Effect of alendronate on bone metabolic indices and bone mineral density in patients treated with high-dose glucocorticoid: a prospective study. Osteoporos Int 21(9):1565–1571
Sambrook PN, Roux C, Devogelaer JP, Saag K, Lau CS, Reginster JY, Bucci-Rechtweg C, Su G, Reid DM (2012) Bisphosphonates and glucocorticoid osteoporosis in men: results of a randomized controlled trial comparing zoledronic acid with risedronate. Bone 50(1):289–295
Devogelaer JP, Sambrook P, Reid DM, Goemaere S, Ish-Shalom S, Collette J, Su G, Bucci-Rechtweg C, Papanastasiou P, Reginster JY (2013) Effect on bone turnover markers of once-yearly intravenous infusion of zoledronic acid versus daily oral risedronate in patients treated with glucocorticoids. Rheumatology (Oxford) 52(6):1058–1069
Reid DM, Devogelaer JP, Saag K, Roux C, Lau CS, Reginster JY, Papanastasiou P, Ferreira A, Hartl F, Fashola T et al (2009) Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet 373(9671):1253–1263
Thomas T, Horlait S, Ringe JD, Abelson A, Gold DT, Atlan P, Lange JL (2013) Oral bisphosphonates reduce the risk of clinical fractures in glucocorticoid-induced osteoporosis in clinical practice. Osteoporos Int 24(1):263–269
Hofbauer LC, Zeitz U, Schoppet M, Skalicky M, Schuler C, Stolina M, Kostenuik PJ, Erben RG (2009) Prevention of glucocorticoid-induced bone loss in mice by inhibition of RANKL. Arthritis Rheum 60(5):1427–1437
Cohen SB, Dore RK, Lane NE, Ory PA, Peterfy CG, Sharp JT, van der Heijde D, Zhou L, Tsuji W, Newmark R (2008) Denosumab treatment effects on structural damage, bone mineral density, and bone turnover in rheumatoid arthritis: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, phase II clinical trial. Arthritis Rheum 58(5):1299–1309
Farahmand P, Marin F, Hawkins F, Moricke R, Ringe JD, Gluer CC, Papaioannou N, Minisola S, Martinez G, Nolla JM et al (2013) Early changes in biochemical markers of bone formation during teriparatide therapy correlate with improvements in vertebral strength in men with glucocorticoid-induced osteoporosis. Osteoporos Int 24(12):2971–2981
Eastell R, Chen P, Saag KG, Burshell AL, Wong M, Warner MR, Krege JH (2010) Bone formation markers in patients with glucocorticoid-induced osteoporosis treated with teriparatide or alendronate. Bone 46(4):929–934
Lane NE, Sanchez S, Modin GW, Genant HK, Pierini E, Arnaud CD (2000) Bone mass continues to increase at the hip after parathyroid hormone treatment is discontinued in glucocorticoid-induced osteoporosis: results of a randomized controlled clinical trial. J Bone Miner Res 15(5):944–951
Gluer CC, Marin F, Ringe JD, Hawkins F, Moricke R, Papaioannu N, Farahmand P, Minisola S, Martinez G, Nolla JM et al (2013) Comparative effects of teriparatide and risedronate in glucocorticoid-induced osteoporosis in men: 18-month results of the EuroGIOPs trial. J Bone Miner Res 28(6):1355–1368
Saag KG, Shane E, Boonen S, Marin F, Donley DW, Taylor KA, Dalsky GP, Marcus R (2007) Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med 357(20):2028–2039
Saag KG, Zanchetta JR, Devogelaer JP, Adler RA, Eastell R, See K, Krege JH, Krohn K, Warner MR (2009) Effects of teriparatide versus alendronate for treating glucocorticoid-induced osteoporosis: thirty-six-month results of a randomized, double-blind, controlled trial. Arthritis Rheum 60(11):3346–3355
Agholme F, Isaksson H, Li X, Ke HZ, Aspenberg P (2011) Anti-sclerostin antibody and mechanical loading appear to influence metaphyseal bone independently in rats. Acta Orthop 82(5):628–632
Li X, Ominsky MS, Warmington KS, Morony S, Gong J, Cao J, Gao Y, Shalhoub V, Tipton B, Haldankar R et al (2009) Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res 24(4):578–588
Li X, Warmington KS, Niu QT, Asuncion FJ, Barrero M, Grisanti M, Dwyer D, Stouch B, Thway TM, Stolina M et al (2010) Inhibition of sclerostin by monoclonal antibody increases bone formation, bone mass, and bone strength in aged male rats. J Bone Miner Res 25(12):2647–2656
Tian X, Jee WS, Li X, Paszty C, Ke HZ (2011) Sclerostin antibody increases bone mass by stimulating bone formation and inhibiting bone resorption in a hindlimb-immobilization rat model. Bone 48(2):197–201
Ominsky MS, Vlasseros F, Jolette J, Smith SY, Stouch B, Doellgast G, Gong J, Gao Y, Cao J, Graham K et al (2010) Two doses of sclerostin antibody in cynomolgus monkeys increases bone formation, bone mineral density, and bone strength. J Bone Miner Res 25(5):948–959
Marenzana M, Greenslade K, Eddleston A, Okoye R, Marshall D, Moore A, Robinson MK (2011) Sclerostin antibody treatment enhances bone strength but does not prevent growth retardation in young mice treated with dexamethasone. Arthritis Rheum 63(8):2385–2395
Li X, Ominsky MS, Warmington KS, Morony S, Gong J, Cao J, Gao Y, Shalhoub V, Tipton B, Haldankar R et al (2008) Sclerostin antibody treatment increases bone formation, bone mass and bone strength in a rat model of postmenopausal osteoporosis *. J Bone Miner Res
Ominsky MS, Niu QT, Li C, Li X, Ke HZ (2014) Tissue-level mechanisms responsible for the increase in bone formation and bone volume by sclerostin antibody. J Bone Miner Res 29(6):1424–1430
Li X, Ominsky MS, Warmington KS, Niu QT, Asuncion FJ, Barrero M, Dwyer D, Grisanti M, Stolina M, Kostenuik PJ et al (2011) Increased bone formation and bone mass induced by sclerostin antibody is not affected by pretreatment or cotreatment with alendronate in osteopenic, ovariectomized rats. Endocrinology 152(9):3312–3322
Devogelaer JP, Adler RA, Recknor C, See K, Warner MR, Wong M, Krohn K (2010) Baseline glucocorticoid dose and bone mineral density response with teriparatide or alendronate therapy in patients with glucocorticoid-induced osteoporosis. J Rheumatol 37(1):141–148
Shahnazari M, Yao W, Corr M, Lane NE (2008) Targeting the Wnt signaling pathway to augment bone formation. Curr Osteoporos Rep 6(4):142–148
Acknowledgments
This work was funded by a research grant to UC Davis by Amgen and National Institutes of Health grant R01 AR061366 (to WY) and K24 AR04884, P50 AR060752, P50 AR063043, and R01 AR043052 (to NEL).
Conflicts of interest
Wei Yao, Weiwei Dai, Li Jiang, Evan Yu-An Lay, Zhendong Zhong, Robert O. Ritchie and Nancy Lane declare that they have no conflict of interest. Xiaodong Li and Huazhu Ke were employees of Amgen at the time when the study was performed.
Author information
Authors and Affiliations
Corresponding author
Additional information
W. Yao and W. Dai contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
(PPTX 57 kb)
Supplementary Table 2
(PPTX 70 kb)
Rights and permissions
About this article

Cite this article
Yao, W., Dai, W., Jiang, L. et al. Sclerostin-antibody treatment of glucocorticoid-induced osteoporosis maintained bone mass and strength. Osteoporos Int 27, 283–294 (2016). https://doi.org/10.1007/s00198-015-3308-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-015-3308-6