
Abstract
Microbial arsenic transformations play essential roles in controlling pollution and ameliorating risk. This study combined high-throughput sequencing and PCR-based approaches targeting both the 16 S rRNA and arsenic functional genes to investigate the temporal and spatial dynamics of the soil microbiomes impacted by high arsenic contamination (9.13 to 911.88 mg/kg) and to investigate the diversity and abundance of arsenic functional genes in soils influenced by an arsenic gradient. The results showed that the soil microbiomes were relatively consistent and mainly composed of Actinobacteria (uncultured Gaiellales and an unknown_67 − 14 bacterium), Proteobacteria, Firmicutes (particularly, Bacillus), Chloroflexi, and Acidobacteria (unknown_Subgroup_6). Although a range of arsenic functional genes (e.g., arsM, arsC, arrA, and aioA) were identified by shotgun metagenomics, only the arsM gene was detected by the PCR-based method. The relative abundance of the arsM gene accounted for 0.20%–1.57% of the total microbial abundance. Combining all analyses, arsenic methylation mediated by the arsM gene was proposed to be a key process involved in the arsenic biogeochemical cycle and mitigation of arsenic toxicity. This study advances our knowledge about arsenic mechanisms over the long-term in highly contaminated soils.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Soil is an important sink of arsenic, in which the most common forms found are arsenite and arsenate. In addition to inorganic arsenic, organic arsenic is detected in soils and can be volatile, entering the atmosphere (Jia et al. 2013). The toxicity of organic arsenic is typically lower than that of inorganic arsenic. Thus, when inorganic arsenic is transformed into organic arsenic and subsequently released into the atmosphere, environmental risks and toxicity of arsenic in contaminated environments may decrease. Consequently, it is important to pay attention to arsenic transformations in soils in order to effectively control pollution and prevent risks. Microorganisms play important roles in arsenic transformations through arsenite oxidation, arsenate reduction, and arsenic methylation. Each arsenic-transforming mechanism is mediated by particular arsenic functional genes. Arsenite oxidation converts arsenite to arsenate, and this process is governed by the aioA gene (Muller et al. 2003). Detoxification arsenate reduction, transforming arsenate to arsenite, is controlled by the arsC gene, and this mechanism principally controls arsenic mobilization in oxic environments (Huang 2014). Another arsenate reduction mechanism is through the respiratory process catalyzed by the arrA gene (Saltikov and Newman 2003). Arsenic methylation, being catalyzed by the arsM gene, transforms inorganic arsenic to volatile methyl arsenic (Qin et al. 2006). Arsenic methylation is critical for decreasing the toxicity and mobility of arsenic within the environment. A global survey of arsenic functional genes in soil microbiomes from the USA, Canada, Brazil, Russia, and Malaysia suggested the importance of the local microbial community in arsenic transformation (Dunivin et al. 2019). Due to a large volume of uncultured microorganisms in the environment, microbial communities and the diversity of arsenic functional genes in soils impacted long-term by high arsenic contamination remains unclear. This study combined high-throughput sequencing and PCR-based approaches in order to advance our understanding of soil microorganisms and their arsenic functional genes in agricultural soils with chronically high arsenic levels. The objectives of this study were to investigate spatial and temporal variations of soil microbiomes impacted by high arsenic contamination and to analyze the diversity and abundance of arsenic functional genes in soils subject to different levels of arsenic contamination. Overall, this study will help identify the potential mechanisms involved in the biogeochemical cycling of arsenic, and the mitigation of arsenic toxicity.
Materials and Methods
Sampling Site Characteristics and Sample Collection
The study area was located in Dan Chang District, Suphan Buri Province, Thailand and it is mainly influenced by the presence of an abandoned tin mine and continuing agricultural activity. A previous study has shown that soils from this area are chronically contaminated by arsenic, with extremely high arsenic concentrations in soils (Tiankao and Chotpantarat 2018). Soil samples were collected from five locations (S1 to S5) (Supplemental Fig. 1). The soil samples from each location were collected at six-monthly intervals on three occasions (T1, T2, and T3). To make a composite sample, the soil samples from each sampling site were randomly collected from five locations and were subsequently pooled on-site. The samples were kept in plastic bags and stored on ice during transportation.
Analysis of Soil Properties
The concentrations of total arsenic in soils were analyzed at the Central Laboratory (Thailand) Co., Ltd., using an in-house method based on a handbook of soil analysis: chemical and physical methods, APSRDO, DOA: 1/2553 (Wright 1934). The concentrations of organic matter (OM), total nitrogen (TN), and total phosphorus (TP) in soils were measured by the Soil-Fertilizer-Environment Scientific Development Project, Department of Soil Science, Kasetsart University, Thailand, using the Walkley and Black Titration method, the Micro-Kjeldahl method, and the Vanado molybdophosphoric acid yellow method, respectively. Soil pH (1:1 soil to water ratio) was measured using a portable InLab® Expert Pro ISM-IP67 pH electrode (Mettler Toledo, USA).
DNA Extraction
DNA was extracted from soils using a DNeasy PowerSoil Pro Kit (Qiagen, USA) following the manufacturer’s protocol. DNA was extracted in triplicate and subsequently pooled before downstream molecular analyses. The extracted DNA was then qualified and quantified using agarose gel electrophoresis and a Nanodrop spectrophotometer ND-100 (Thermo Fisher Scientific, USA).
Analysis of 16S rRNA Gene Sequences
For 16 S rRNA library preparation, the V3-V4 hypervariable region of the 16 S rRNA gene was amplified using primers 341 F and 806R. The 16 S rRNA gene libraries of 15 soil samples were prepared by Novogene Co., Ltd. (Beijing, China) using a NEBNext® Ultra™ II DNA Library Prep Kit for Illumina®, following the manufacturer’s protocol. An Illumina NovaSeq 6000 platform was used to produce 250 bp paired-end reads. The raw sequencing data quality was assessed using reports from FastQC (Andrews 2010) and MultiQC (Ewels et al. 2016). Qualified reads of the 16 S rRNA amplicons were analyzed using mothur software version 1.44.1 (Schloss et al. 2009). Chimeric sequences were eliminated using de novo searching algorithm of UCHIME (Edgar et al. 2011). The processed sequences were clustered to operational taxonomic units (OTUs) based on de novo hierarchical clustering approach with 97% identity threshold of 16 S gene sequences. OTUs containing less than two sequences, referred to as singletons and doubletons, were removed as they could be considered to have occurred because of sequencing errors. OTUs abundance table was normalized by scaling to the minimum depth among all samples. Taxonomic classification of each OTU was performed using 16 S rRNA SILVA database version 132 (Quast et al. 2013). Rarefaction curves were calculated to observe a relation between community richness and sequencing depth of each sample. Alpha-diversity (e.g., Chao1, Ace, and Shannon indices) was measured via mothur. Relative taxa abundances were calculated based on the normalized OTUs abundance. Raw data of 16 S rRNA gene amplicon sequences were deposited in the Sequence Read Archive (SRA) under the Bioproject number PRJNA716501.
Analysis of Shotgun Metagenomic data
The metagenome sequencing libraries were prepared by Novogene Co., Ltd. (Beijing, China) using a NEBNext® Ultra™ TruSeq DNA Library Prep Kit V3 for Illumina®, following the manufacturer’s protocol. An Illumina NovaSeq 6000 platform was used to produce 150 bp paired-end reads. The quality of raw metagenomic sequencing reads was assessed using FastQC and MultiQC. The qualified reads were assembled using MetaSPAdes version 3.15.4 (Nurk et al. 2017). The open reading frames (ORFs) were predicted using Prokka version 1.14.6 (Seemann 2014). The sequences of arsenic functional genes were collected from the NCBI database and were blasted against the contigs to locate the genes. The qualified metagenomics reads were mapped to the contigs by BWA version 0.7.17 (Li and Durbin 2009). Raw data of shotgun metagenome were deposited in the SRA under the Bioproject number PRJNA824136.
Analysis of arsM Gene sequences
The arsM gene libraries of 5 soil samples (S1-3, S2-3, S3-3, S4-3, and S5-3) were constructed using the specific primer arsMF1 and arsMR2 (Jia et al. 2013). As with the 16 S rRNA gene libraries, the arsM gene libraries were prepared by Novogene Co., Ltd. (Beijing, China). Raw arsM sequences were initially cleaned by removing low quality sequences and trimming off barcodes and adapter sequences using FASTP (Chen et al. 2018). Cleaned sequences were subsequently constructed as the amplicon sequence variants (ASVs) using dada2 (Callahan et al. 2016). To construct the neighbor-joining tree with 1000 replicates of the bootstrap test, all representative ASVs were aligned with reference arsM sequences using MUSCLE through the MEGA package (version 7.0.21) (Kumar et al. 2016). Raw data of arsM sequences were deposited in the SRA under the Bioproject number PRJNA716505.
Quantification of arsM Gene
The abundance of arsM and 16 S rRNA genes was quantified using the primers arsMF1/arsMR2 (Jia et al. 2013) and 341 F/518R (Muyzer et al. 1993), respectively. The 16 S rRNA gene was quantified to represent the total microbial abundance. The qPCR mixture, in a total volume of 10 µl, contained 5 µl of SsoFast EvaGreen Supermix (Bio-Rad, USA), 0.03 µl of each primer (100 µM stocks), 0.02 µl of BSA (10 mg ml−1), 1 µl of genomic DNA template (10 ng µl−1), and 3.92 of nuclease-free water (Invitrogen, USA). A gradient thermal program of 51–60°C was conducted to verify an appropriate qPCR condition. The annealing temperature was set at 53 and 55°C for the quantifications of arsM and 16 S rRNA genes, respectively. The qPCR thermal program was conducted according to the manufacturer’s protocol. All qPCR amplifications were conducted in triplicate using a CFX96 real-time system (Bio-Rad, USA). Melt curve analysis and agarose gel electrophoresis were performed after each run to ensure the qPCR specificity.
Statistical Analysis
Correlations between soil properties and the microbial abundance were assessed using Spearman’s correlation coefficients and their corresponding p-values using MATLAB software (Math-Works, USA).
Results and Discussion
Soil Properties
The concentration of arsenic in the soil samples was categorized as extremely high (S2), high (S3), moderate (S4), and low (S1 and S5) levels, ranging from 583.69 to 911.88, 126.18-183.12, 30.05–71.09, and 9.13–29.41 mg/kg, respectively (Table 1). In all analyzed 15 samples, pH ranged from 6 to 8, indicating slightly acidic to slightly basic conditions. Although the concentration of OM was relatively high in S4-3, it was comparable across all analyzed samples. TN was detected in the range of 743.75-1,138.00 mg/kg across all analyzed soils. While the concentrations of OM and TN in soil samples were relatively consistent across the three sampling intervals, the concentrations of TP in soil samples varied according to the sampling period. The TP level was relatively high in the soil samples collected from T1 and T3, but it sharply dropped in those collected from T2 (Table 1). The loss of TP in T2 was potentially due to plant uptake and rainfall during the growing season. Previous studies suggested that factors contributing to TP losses from agricultural soils were surface runoff, soil characteristics, and plant consumption (King et al. 2015; Yi et al. 2022). To better understand the impact of arsenic on human health, the arsenic concentrations in agricultural products should also be analyzed.
Microbial Community in Arsenic-Contaminated Soils
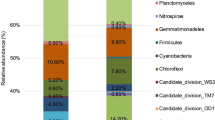
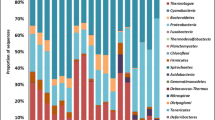
After removing chimera and low quality sequences, a total of 1,766,576 sequences were obtained, representing 286,183 unique sequences. The read depth was between 102,380 and 127,169 sequences per sample and the number of unique sequences was between 33,777 and 47,807 sequences per sample. Across all samples, a total of 19,637 OTUs were detected. Rarefaction curves indicated an optimal sequencing depth (Supplemental Fig. 2). The diversity indices of all 15 samples was comparable (Supplemental Table 1). The results suggested that core microbial phyla across 15 samples were relatively consistent (Fig. 1A). Major detected phyla were Actinobacteria (29%–43%), Proteobacteria (16%–26%), Firmicutes (11%–25%), Chloroflexi (5%–9%), and Acidobacteria (5%–14%) (Fig. 1A). Actinobacteria, Proteobacteria, and Firmicutes were also highly detected in soils highly contaminated with arsenic ranging from 34.11 to 821.23 mg/kg (Luo et al. 2014). ArsM protein associated with arsenic methylation was predominantly found in Proteobacteria, Bacteroidetes, Firmicutes, and Actinobacteria (Rahman et al. 2020). Other minor phyla detected in all samples were Gemmatimonadetes (2%–3%), Verrucomicrobia (1%–2%), and Bacteroidetes (1%–3%). Rokubacteria and Cyanobacteria were also detected at low abundance (< 1%–2%). All these phyla have been previously found in soil contaminated with arsenic (Bose et al. 2022). Spearman’s correlation coefficient indicated that the abundance Rokubacteria (r = -0.589, p = 0.021) showed significantly negative correlations with arsenic concentration. However, information on effects of arsenic concentration on the abundance of Rokubacteria is limited.

Relative abundance of soil microbiome at the phylum level in 15 soil samples (A). Heatmap based on the abundance of microbial taxa with more than 1% OTUs at least in one sample and the relative abundance of microbial taxa is indicated by the color intensity (B). Neighbor-joining phylogenetic tree of the arsM sequences and the heatmap indicates the abundance of each arsM sequence in the analyzed samples (C)
Dominant Taxa Impacted by Arsenic in Soils
A heatmap analysis revealed that dominant bacterial taxa across 15 soil samples with a broad range of arsenic concentrations were Bacillus, unknown_Subgroup_6, uncultured Gaiellales, and unknown_67 − 14 bacterium (Fig. 1B). Bacillus and uncultured Gaiellales have been previously found in soils contaminated with metals, including arsenic (Gong et al. 2023; Yang et al. 2023). Both are reported to tolerate high arsenic concentrations. Members of the genus Bacillus are able to transform arsenic through arsenate reduction, arsenite oxidation, and arsenic methylation, and they have been proposed for arsenic bioremediation (Bagade et al. 2020; Alotaibi et al. 2021). Spearman’s correlation coefficient indicated that the abundance of unclassified Bacillaceae (r = -0.804, p = 0.0003) and showed significantly negative correlation to arsenic concentrations. This implied that increased abundance of Bacillaceae members was related to a decrease in arsenic concentration. Although unknown_Subgroup_6, members of the phylum Acidobacteria, have been found in paddy and agricultural soils (Kandasamy et al. 2021; Bose et al. 2022), their association with arsenic is still unknown. The unknown_67 − 14 bacterium, a member of the phylum Actinobacteria, was commonly detected in arsenic-contaminated soils analyzed in this study; however, their association with arsenic is very limited. Spearman’s correlation coefficient showed that the abundance of uncultured Gaiellales (r = 0.586, p = 0.022), unknown IMCC26256 (r = 0.835, p = 0.0001) and unclassified Micromonosporaceae (r = 0.779, p = 0.001) were significantly positively correlated with arsenic concentrations. A previous study suggested that bacteria associated with Gaiellales and IMCC26256 were able to withstand high levels of arsenic and antimony in soil (Gong et al. 2023). Micromonosporaceae are known as polysaccharide-degrading bacteria (Yeager et al. 2017), but their association with arsenic is unclear.
Diversity and Abundance of the arsM Gene
Five soil samples from T3 were screened for the presence of aioA, arrA, arsC, and arsM genes using previously published primers (Sun et al. 2004; Quéméneur et al. 2010; Mirza et al. 2017). However, the amplifications of aioA, arrA, and arsC showed negative or unstable PCR signal. Only the arsM sequences were processed through high throughput sequencing. The phylogenetic tree showed that the majority of arsM sequences retrieved from this study were closely related to uncultured arsM sequences previously found in estuary sediment, paddy soil, arsenic-rich sediment, river water affected by acid mine drainage (AMD), and water-pit lake (Fig. 1C). In addition to uncultured arsM clones, some of the arsM sequences recovered from this study were phylogenetically related to those belonging to Rhodopseudomonas palustris, Nocardioides sp., Streptomyces griseorubiginosus, and Sphaerobacter thermophiles (Fig. 1C). The known arsM gene hosted by Rhodopseudomonas palustris are generally found in arsenic-contaminated paddy soils and composting manure (Zhao et al. 2013; Zhai et al. 2017). Metagenomics analysis revealed that Rhodopseudomonas palustris were highly detected in paddy soils with arsenic concentrations of less than 15 mg/kg (Xiao et al. 2016). A previous study also suggested that Rhodopseudomonas palustris could detoxify arsenic through the arsM gene when the arsenic concentrations were relatively high (Zhao et al. 2015). Streptomyces sp. and Sphaerobacter thermophiles harboring the arsM gene were previously found in paddy soils and composting pig manure (Zhao et al. 2013; Zhai et al. 2017). Streptomyces sp. and Nocardioides sp. were predicted to be resilient to soil heavily co-contaminated with arsenic and antimony (Li et al. 2021).
The resulting qPCR revealed that the relative abundance of the arsM gene accounted for 0.20%–1.57% of the total microbial abundance (Fig. 2A). A previous study revealed that the relative abundance of the arsM gene in paddy soils where the arsenic concentrations ranged from 3 to 81 mg/kg was approximately 0.01%–0.02% (Zhao et al. 2013). The abundance of arsenic functional genes in 13 Chinese paddy soils across arsenic concentrations of 11.7 to 25.4 mg/kg was quantified by qPCR, and the results showed that the relative abundance of the arsM gene accounted for 0.1 to 2.6% of the total microbial abundance (Zhang et al. 2015). The highest relative abundance of the arsM gene appeared in soils in which the arsenic concentrations were chronically the highest (Fig. 2A). This result implies that high concentrations of arsenic could enhance the abundance of the arsM gene in impacted soils. The arsM gene expression also indicated that the activity of arsM gene increased when arsenic concentration in impacted soils increased (Dong et al. 2020).

Relative abundance of arsM gene copy numbers and their corresponding arsenic concentrations (A). Abundance of arsenic functional genes analyzed by metagenomics (B)
Arsenic Functional Genes Analyzed by Metagenome Assembly-Based Methods
Two representative soil samples, S2-3 and S5-3, with extremely high and low arsenic concentrations were analyzed by metagenome assembly-based methods to compare the presence of arsenic functional genes. The results showed that the abundance of all detected arsenic functional genes, except the aioB gene, was higher in S2-3 than S5-3 (Fig. 2B). The concentration of arsenic in S2-3 was about 64 times higher than that in S5-3 (Table 1). The results likewise suggest that higher arsenic concentrations enhance the abundance of arsenic functional genes in soil. Zhang et al. (2021) has shown that the expression levels of ars and aioA genes in paddy soils increased according to the soil arsenic concentrations (2.5 to 104.0 mg/kg). The abundance of arsenic functional genes in soils with higher concentrations of arsenic (34.11 to 821.23 mg/kg) and antimony (226.67 to 3,923.07 mg/kg) was higher (Luo et al. 2014).
Among the detected arsenic functional genes, the arsM gene was present at the highest abundance in both S2-3 and S5-3 samples (Fig. 2B). The arsM gene was elsewhere found to be present in high levels in soil metagenomes, and its abundance was higher in the cultivation-independent than the cultivation-dependent soil samples (Dunivin et al. 2019). Overall, our results suggest that the arsM gene responsible for arsenic methylation likely plays a crucial role in driving the arsenic cycle in the long-term, and highly arsenic-contaminated, area analyzed in this study.
In addition to the arsM gene, metagenomics analysis can detect other arsenic functional genes, such as arsC, arrA, and aioA, which could not be recovered from our soils using the corresponding specific primers. Since a great many uncultured microorganisms with arsenic functional genes exist in the environment, the available primers for detecting these genes are insufficient. Shotgun metagenomics sequencing technology, however, is capable of capturing all functional genes, regardless of primer specificity and coverage.
Conclusions
The microbial communities and arsenic functional genes in soils from an area with chronic and severe arsenic contamination were explored using three omics analyses, including high-throughput sequencing techniques targeting both the 16 S rRNA and arsenic functional genes, as well as shotgun sequencing. This study revealed the spatial and temporal variations of soil microbiome impacted by agricultural activity and former tin-mining. Core microbial taxa remained relatively consistent throughout the year. While shotgun metagenomics identified a variety of arsenic functional genes (e.g., arsM, arsC, arrA, and aioA) of both known and unknown microorganisms, a PCR-based method detected only the arsM gene. Based on the analysis of arsenic functional genes and metagenomics, arsenic methylation mediated by the arsM gene could be considered a key arsenic-transforming process. High arsenic concentrations could enhance the numbers of arsM genes found in soils. Uncultured arsenic methylating bacteria and bacteria related to Rhodopseudomonas palustris, Nocardioides sp., Streptomyces griseorubiginosus and Sphaerobacter thermophiles likely played an important role in removing arsenic. Overall, this study shed light on arsenic methylation driven by uncultured soil microbiome in the chronically high arsenic contamination.
References
Alotaibi BS, Khan M, Shamim S (2021) Unraveling the underlying heavy metal detoxification mechanisms of Bacillus species. Microorganisms 9:1628. https://doi.org/10.3390/microorganisms9081628
Andrews S (2010) FastQC: a quality control tool for high throughput sequence data. Available online http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Bagade A, Nandre V, Paul D, Patil Y, Sharma N, Giri A, Kodam K (2020) Characterisation of hyper tolerant Bacillus firmus L-148 for arsenic oxidation. Environ Pollut 261:114124. https://doi.org/10.1016/j.envpol.2020.114124
Bose H, Sahu RP, Sar P (2022) Impact of arsenic on microbial community structure and their metabolic potential from rice soils of West Bengal, India. Sci Total Environ 841:156486. https://doi.org/10.1016/j.scitotenv.2022.156486
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP (2016) DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. https://doi.org/10.1038/nmeth.3869
Chen S, Zhou Y, Chen Y, Gu J (2018) Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34:i884–i890. https://doi.org/10.1093/bioinformatics/bty560
Dong Y, Gao M, Liu X, Qiu W, Song Z (2020) The mechanism of polystyrene microplastics to affect arsenic volatilization in arsenic-contaminated paddy soils. J Hazard Mater 398:122896. https://doi.org/10.1016/j.jhazmat.2020.122896
Dunivin TK, Yeh SY, Shade A (2019) A global survey of arsenic-related genes in soil microbiomes. BMC Biol 17:1–17. https://doi.org/10.1186/s12915-019-0661-5
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. https://doi.org/10.1093/bioinformatics/btr381
Ewels P, Magnusson M, Lundin S, Käller M (2016) MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32:3047–3048. https://doi.org/10.1093/bioinformatics/btw354
Gong Y, Yang S, Chen S, Zhao S, Ai Y, Huang D, Yang K, Cheng H (2023) Soil microbial responses to simultaneous contamination of antimony and arsenic in the surrounding area of an abandoned antimony smelter in Southwest China. Environ Int 174:107897. https://doi.org/10.1016/j.envint.2023.107897
Huang JH (2014) Impact of microorganisms on arsenic biogeochemistry: a review. Water Air Soil Pollut 225:1848. https://doi.org/10.1007/s11270-013-1848-y
Jia Y, Huang H, Zhong M, Wang FH, Zhang LM, Zhu YG (2013) Microbial arsenic methylation in soil and rice rhizosphere. Environ Sci Technol 47:3141–3148. https://doi.org/10.1021/es303649v
Kandasamy S, Weerasuriya N, White JF, Patterson G, Lazarovits G (2021) Soil’s physical and nutritional balance is essential for establishing a healthy microbiome. In: Dubey SK, Sharma DK, Sharma JD (eds) Microbiome stimulants for crops. Woodhead Publishing, Cambridge, pp 381–404. https://doi.org/10.1016/B978-0-12-822122-8.00004-2
King KW, Williams MR, Macrae ML, Fausey NR, Frankenberger J, Smith DR, Kleinman PJA, Brown LC (2015) Phosphorus transport in agricultural subsurface drainage: a review. J Environ Qual 44:467–485. https://doi.org/10.2134/jeq2014.04.0163
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. https://doi.org/10.1093/molbev/msw054
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25:1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Li Y, Zhang M, Xu R, Lin H, Sun X, Xu F, Gao P, Kong T, Xiao E, Yang N, Sun W (2021) Arsenic and antimony co-contamination influences on soil microbial community composition and functions: relevance to arsenic resistance and carbon, nitrogen, and sulfur cycling. Environ Int 153:106522. https://doi.org/10.1016/j.envint.2021.106522
Luo J, Bai Y, Liang J, Qu J (2014) Metagenomic approach reveals variation of microbes with arsenic and antimony metabolism genes from highly contaminated soil. PLOS ONE 9:e108185. https://doi.org/10.1371/journal.pone.0108185
Mirza BS, Sorensen DL, Dupont RR, McLean JE (2017) New arsenate reductase gene (arrA) PCR primers for diversity assessment and quantification in environmental samples. Appl Environ Microbiol 83:e02725–e02716. https://doi.org/10.1128/AEM.02725-16
Muller D, Lievremont D, Simeonova DD, Hubert JC, Lett MC (2003) Arsenite oxidase aox genes from a metal-resistant β-proteobacterium. J Bacteriol 185:135–141. https://doi.org/10.1128/JB.185.1.135-141.2003
Muyzer G, De Waal EC, Uitterlinden A (1993) Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700. https://doi.org/10.1128/aem.59.3.695-700.1993
Nurk S, Meleshko D, Korobeynikov A, Pevzner PA (2017) metaSPAdes: a new versatile metagenomic assembler. Genome Res 27:824–834. http://www.genome.org/cgi/doi/https://doi.org/10.1101/gr.213959.116
Qin J, Rosen BP, Zhang Y, Wang G, Franke S, Rensing C (2006) Arsenic detoxification and evolution of trimethylarsine gas by a microbial arsenite S-adenosylmethionine methyltransferase. Proc Natl Acad Sci 103:2075–2080. https://doi.org/10.1073/pnas.0506836103
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. https://doi.org/10.1093/nar/gks1219
Quéméneur M, Cébron A, Billard P, Battaglia-Brunet F, Garrido F, Leyval C, Joulian C (2010) Population structure and abundance of arsenite-oxidizing bacteria along an arsenic pollution gradient in waters of the Upper Isle River Basin, France. Appl Environ Microbiol 76:4566–4570. https://doi.org/10.1128/AEM.03104-09
Rahman Z, Thomas L, Singh VP (2020) Study of As-resistant bacteria from Nadia, India and a survey of two as resistance‐related proteins. J Basic Microbiol 60:47–57. https://doi.org/10.1002/jobm.201900373
Saltikov CW, Newman DK (2003) Genetic identification of a respiratory arsenate reductase. Proc Natl Acad Sci 100:10983–10988. https://doi.org/10.1073/pnas.1834303100
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. https://doi.org/10.1128/AEM.01541-09
Seemann T (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. https://doi.org/10.1093/bioinformatics/btu153
Sun Y, Polishchuk EA, Radoja U, Cullen WR (2004) Identification and quantification of arsC genes in environmental samples by using real-time PCR. J Microbiol Methods 58:335–349. https://doi.org/10.1016/j.mimet.2004.04.015
Tiankao W, Chotpantarat S (2018) Risk assessment of arsenic from contaminated soils to shallow groundwater in Ong Phra Sub-district, Suphan Buri Province, Thailand. J Hydrol-Reg Stud 19:80–96. https://doi.org/10.1016/j.ejrh.2018.08.001
Wright CH (1934) Soil analysis. A handbook of physical and chemical methods. Thomas Murby & Co., London
Xiao KQ, Li LG, Ma LP, Zhang SY, Bao P, Zhang T, Zhu YG (2016) Metagenomic analysis revealed highly diverse microbial arsenic metabolism genes in paddy soils with low-arsenic contents. Environ Pollut 211:1–8. https://doi.org/10.1016/j.envpol.2015.12.023
Yang X, Dai Z, Ge C, Yu H, Bolan N, Tsang DC, Song H, Hou D, Shaheen SM, Wang H, Rinklebe J (2023) Multiple-functionalized biochar affects rice yield and quality via regulating arsenic and lead redistribution and bacterial community structure in soils under different hydrological conditions. J Hazard Mater 443:130308. https://doi.org/10.1016/j.jhazmat.2022.130308
Yeager CM, Gallegos-Graves LV, Dunbar J, Hesse CN, Daligault H, Kuske CR (2017) Polysaccharide degradation capability of Actinomycetales soil isolates from a semiarid grassland of the Colorado Plateau. Appl Environ Microbiol 83:e03020–e03016. https://doi.org/10.1128/AEM.03020-16
Yi K, Li X, Chen D, Yang S, Liu Y, Tang X, Ling G, Zhao Z (2022) Shallower root spatial distribution induced by phosphorus deficiency contributes to topsoil foraging and low phosphorus adaption in sugarcane (Saccharum officinarum L). Front Plant Sci 12:3417. https://doi.org/10.3389/fpls.2021.797635
Zhai W, Wong MT, Luo F, Hashmi MZ, Liu X, Edwards EA, Tang X, Xu J (2017) Arsenic methylation and its relationship to abundance and diversity of arsM genes in composting manure. Sci Rep 7:42198. https://doi.org/10.1038/srep42198
Zhang SY, Zhao FJ, Sun GX, Su JQ, Yang XR, Li H, Zhu YG (2015) Diversity and abundance of arsenic biotransformation genes in paddy soils from southern China. Environ Sci Technol 49:4138–4146. https://doi.org/10.1021/acs.est.5b00028
Zhang SY, Xiao X, Chen SC, Zhu YG, Sun GX, Konstantinidis KT (2021) High arsenic levels increase activity rather than diversity or abundance of arsenic metabolism genes in paddy soils. Appl Environ Microbiol 87:e01383–e01321. https://doi.org/10.1128/AEM.01383-21
Zhao FJ, Harris E, Yan J, Ma J, Wu L, Liu W, McGrath SP, Zhou J, Zhu YG (2013) Arsenic methylation in soils and its relationship with microbial arsM abundance and diversity, and as speciation in rice. Environ Sci Technol 47:7147–7154. https://doi.org/10.1021/es304977m
Zhao C, Zhang Y, Chan Z, Chen S, Yang S (2015) Insights into arsenic multi-operons expression and resistance mechanisms in Rhodopseudomonas palustris CGA009. Front Microbiol 6:986. https://doi.org/10.3389/fmicb.2015.00986
Acknowledgements
This work (Grant No. RGNS 63–173) was supported by Office of the Permanent Secretary, Ministry of Higher Education, Science, Research and Innovation (OPS MHESI), Thailand Science Research and Innovation (TSRI) and Mahidol University. This research project was also supported by the Faculty of Science, Mahidol University. Philip D. Round provided helpful feedback and proofread the English of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sonthiphand, P., Rueangmongkolrat, N., Uthaipaisanwong, P. et al. Soil Microbiomes and their Arsenic Functional Genes in Chronically High-Arsenic Contaminated Soils. Bull Environ Contam Toxicol 112, 49 (2024). https://doi.org/10.1007/s00128-024-03866-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00128-024-03866-1