
Abstract
The chromate ore process residues (COPR) polluted soil was physically separated into coarse sand (2.000–0.425 mm), fine sand (0.425–0.053 mm) and silt to clay (< 0.053 mm) fractions. The Cr speciation was characterized by synchrotron based micro X-ray fluorescence (µ-XRF) and micro X-ray absorption near-edge spectra (µ-XANES). The results indicated that Cr was bearing both in COPR parent minerals and hydrated products and was dominated by Cr(III) in three size-fractions. The synchrotron results indicated that Cr(III) was dominated by chromite, organic matter bound Cr(III) and particle adsorbed Cr3+ in the selected hotspots from the coarse sand, fine sand and silt to clay sized fraction, respectively. While Cr(VI) occurred in the form of CrO42− in the selected hotspots from three size fractions. The difference of Cr(III) species in the size-fractions suggested that higher edaphic effects occurred in the fine size-fractions than in the coarse size-fraction for the weathered COPR.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Chromate ore process residues (COPR) were generated by the process of roasting chromite ore at approximately 1200°C to oxidize the trivalent Cr in the chromite ore to the hexavalent state with the addition of lime. The mineralogical characteristics of COPR have been studied extensively (Hillier et al. 2003, 2007; Chrysochoou et al. 2009a, 2010; Moon and Wazne 2011). The mineral phases are divided into three main categories: unreacted feedstock ore (chromite), high temperature phases produced during Cr extraction (brownmillerite, periclase and larnite), and minerals formed under ambient weathering conditions at the disposal sites (brucite, calcite, aragonite, ettringite, hydrocalumite, hydrogarnet). However, the mineral composition of the weathered COPR may vary greatly among deposition sites and the transformation of mineral phases under ambient condition may therefor affect Cr speciation and its distribution (Hillier et al. 2003; Chrysochoou et al. 2009b). Meanwhile, soil organic matter, Fe(II), and Mn(III,IV) hydroxides and oxides may have an important role in the change of Cr species of weathered COPR (James 1996).
Although chemical extraction is applied widely in the evaluation of heavy metal fractions in soils or sediments, it is limited by only providing information on operationally defined fractions rather than in-situ speciation of heavy metals in solid phase and by only providing an average or “bulk” assessment but not a spatially resolved speciation in a sample (Grafe et al. 2014). In contrast, synchrotron radiation method has a capability in determining in-situ speciation of heavy metals in soils on the micro-scale (Vodyanitskii 2013). For example, synchrotron-based micro X-ray fluorescence (µ-XRF) and micro-X-ray absorption near-edge spectra (µ-XANES) showed that > 50% of the Cr(VI) was hosted by hydrogarnet and hydrotalcite in the weathered COPR materials (Chrysochoou et al. 2009a). Linear combination fitting (LCF) of XANES data has been used to indicate chromium species change from hexavalence to trivalence in the presence of biochar in the Cr polluted water (Rajapaksha et al. 2018). Moreover, µ-XRF mapping coupled with multiscale correlation analysis could be applied to reveal the spatial heterogeneity of heavy metals in the contaminated soil particles (Yu and Lu 2016).
Therefore, the objectives of the present study were to present the in-situ chemical speciation of Cr using synchrotron X-ray analysis in the different size-fractions of a weathered COPR polluted soil. Because in the reality of remediation, COPR polluted soil was usually required to be physically separated based on grain size to concentrate the Cr in a smaller volume prior to subsequent treatment (Hawley et al. 2005), hence this study was expected to be helpful in the selection of appropriate treatment technologies based on the Cr species in the different size-fractions.
Materials and Methods
A bulk COPR polluted soil for the experiment was collected from the surface (0–10 cm) of the former COPR deposition site located in Hangzhou, Zhejiang Province, east China. Approximately 1 kg of the sieved (< 2 mm) dry bulk soil was eventually separated into three size fractions, i.e. F1: coarse sand-sized fraction (2.000–0.425 mm), F2: fine sand-sized fraction (0.425–0.053 mm) and F3: silt-to-clay sized fraction (< 0.053 mm). Each size-fraction was collected and stored dry until analysis. Total Cr content in the COPR polluted soil was analyzed using inductively coupled plasma optical emission spectrometer (ICP-OES) after the soil was digested with mixed acids of HCl, HNO3, HF and HClO4. Total Cr(VI) content was determined by an alkaline digestion procedure based on U.S. EPA method 3036A (USEPA 1996). The hexavalent chromium concentration in the digests was determined using diphenylcarbazide hydrazine spectrophotometry (SEPA 2007). The certified reference materials of GSS-7 and CRM041-30G were added to check the accuracy of the total Cr and Cr(VI) analysis, and the recoveries were within the range of 95%–109% and 91%–106%, respectively. Three replicates were analyzed during the sample analysis for Cr and Cr(VI) and the relative standard deviations (RSD) were all lower than 5%.
The mineralogical characterization for each size-fraction was performed by X-ray diffraction (XRD). The XRD spectra were analyzed using Jade software and phase identification was made by reference to patterns in the International Center for Diffraction Data (ICDD) database released in 1998 and by reference to patterns calculated from crystal structure data in the International Crystal Structure Database (ICSD). The mineral phase was also identified by morphology using scanning election microscope (SEM) and by comparing the atomic ratios of Fe, Ca, Mg and Cr to Al identified by energy dispersive X-ray spectroscopy (EDX) with the theoretical structural formula of a mineral. This method has been successfully used in other studies (Hillier et al. 2003; Chrysochoou et al. 2009a).
Synchrotron-based µ-XRF and µ-XANES were measured with energies ranging from 5 to 20 keV. The sample was adhere to a 3-M plastic film and then fixed onto the sample holder for analysis. The µ-XRF elemental mapping was performed at 10 keV incident energy of X-rays monochromatized with a double crystal Si (111) monochromator which focused on a beam size of 50 × 50 µm2. Fluorescence counts were collected for Cr, Ca and Fe to indicate elemental abundance and distribution. Various hotspots of interest were selected from the maps for Cr K-edge µ-XANES analysis. The hotspots were scanned at a step width of 0.5 eV ranging from 5960 to 6080 eV. A number of reference compounds were analyzed at Cr K-edge to facilitate interpretation of the hotspots data including (1) 0.02M Cr(NO3)3·9H2O solution at pH 3.0 for Cr(III), (2) chromite (GSBD33001.5-94, Beijing), (3) calcium chromate (CaCrO4, 99.9%, J&K Scientific), (4) solid Cr(OH)3, prepared by adding ammonium hydroxide to Cr(NO3)3 solution in a water bath at 90°C (Xu et al. 2004), and (5) aqueous Cr(III)–HA prepared by complexing 0.005M Cr(NO3)3 solution with commercial pure humic acid (technical grade, Sigma-Aldrich) at pH 4.5 as shown in reference (Elzinga and Cirmo 2010). The XANES spectra of Na2CrO4 and Cr2S3 were also used as referential spectra, which were derived from XAFS Spectra Library (http://cars.uchicago.edu/xaslib/search/Cr). All the reference materials were selected based on its possible occurrence in the COPR reported by other studies (Hillier et al. 2003; Chrysochoou et al. 2010; Moon and Wazne 2011). Spectra of the samples and reference compounds were processed using Athena program (version 0.8.056). The ratio of each Cr speciation was calculated using LCF and the goodness of fit was expressed as R-factor (Ohta 2015).
Results and Discussion
Table 1 shows that the total Cr content is up to 23,100 mg kg−1 in the bulk soil and comparable to that in the COPR material from New Jersey (Chrysochoou et al. 2010). The total Cr(VI) content of the bulk soil is 5890 mg kg−1, which accounts for 25.6% of the total Cr content, slightly lower than the Cr(VI) percentage in some typical COPR wastes reported by Dermatas et al. (2006). The Cr and Cr(VI) contents of each soil size fraction are shown in Table 1. Both species show the highest contents occur in the coarse sand sized fraction (2.00–0.45 mm) due to the high amount residue of COPR material in that fraction.
Figure 1 indicates the mineral phases in the different size fractions identified by XRD. In contrast to natural soil containing a high abundance of quartz, the COPR-enriched soil is characterized by a sharp peak of calcite which is corresponding to the high-lime technology used during the roasting of chromite ore in dichromate production in China (Darrie 2001). A difference was obtained from the XRD spectra between the silt-to-clay (< 0.053 mm) sized fraction and sand (2.000–0.425, 0.425–0.053 mm) sized fractions. In the case of soil minerals, a more pronounced peak intensity of quartz is shown in the silt-to-clay sized fraction than in the sand sized fractions, suggesting a higher proportion of natural soil in the smaller grain-size particles. In the case of COPR minerals, brownmillerite, chromatite and periclase (indicative of the COPR parent materials) show more pronounced peak intensities in the coarse sand sized fractions than in the silt-to-clay sized fraction. This indicated that the coarse sand sized fraction was directly impacted by COPR to a greater extent than the fine sized fraction. In addition, ettringite, a type of COPR hydration product, was observed in the silt-to-clay sized fraction but not in the sand sized fractions based on XRD spectra. Ettringite was assumed to be formed in relation to COPR exposure to sulfate within a temperature range of 25–50°C (Moon et al. 2007). The presence of ettringite in the fine-sized fraction was probably in relation to the subsequent short-term electroplating after halt of the dichromate production. Wastewater containing sulfate acid discharged from the electroplating have contaminated the site.

XRD spectra showing the difference of mineral phases among the size-fractions of the COPR-polluted soil
SEM coupled with EDX was assistant to identify Cr bearing mineral phases in the sample. Brownmillerite (Fig. 2b), a typical COPR parent mineral, was found containing a high abundance of Cr. The hydrated minerals including stichtite (Fig. 2a), paraalumhydrocalcite (Fig. 2c) and brucite (Fig. 2f) were also identified to bearing Cr in the sample, which have been reported in other studies as well (Chrysochoou and Dermatas 2006; Hillier et al. 2007; Moon et al. 2007; Moon and Wazne 2011). However Cr bearing in magnesiochromite (Fig. 2d) and hashemite (Fig. 2e) has been rarely reported in the previous studies.

SEM images and EDX spectra showing different Cr bearing minerals from the COPR-polluted soil
The µ-XRF maps of Cr, Fe and Ca for the different size-fractions indicate that Cr hotspots distributions have an analogous feature with Fe and Ca hotspot distributions in the coarse sand sized fraction but not in the other two size-fractions (Fig. 3a). Correlation analysis confirmed that strong positive relationship between Cr intensity count and Fe, Ca intensity count occurred only in the coarse sand fraction. This implied that Cr in the coarse sand sized fraction in the sample was more inclined to be binding with Fe than with Ca on average. That was different to the CORP material from a deposition site in Baltimore, MD, USA, in which Cr was characterized by binding with Ca-rich matrix (Chrysochoou et al. 2009a). Hence the predominant host minerals for Cr might be iron-rich minerals such as brownmillerite and chromite. This was in consistent with the XRD results as shown in Fig. 1. Furthermore, the incorporation of Cr into Fe(III)-hydroxides is another possible explanation for the high correlation between Cr and Fe in the coarse sized fraction (Elzinga and Cirmo 2010).

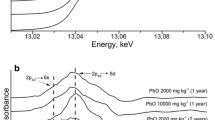
µ-XRF maps a showing heterogeneous distributions of Cr, Fe and Ca in the small particles with different grain size, spot1 (F1), spot2 (F2), spot3 (F2) and spot4 (F3) denoting the selected hotspots for µ-XANES analysis; the Cr near-edge spectra b collected at the various Cr hotspots with LCF line and the spectra of Cr reference materials
The µ-XANES were recorded for determination of the valence state and chemical speciation of Cr at various Cr hotspots on the µ-XRF maps (Fig. 3b). Spot1 (F1) has a pronounced pre-edge peak at 5991.5 eV characteristic of Cr(VI) (Elzinga and Cirmo 2010). A broad peak at around 6028 eV of the post edge position resembles that of CaCrO4 and chromite. The LCF results indicate that the proportion of CaCrO4 and chromite is 40% and 60%, respectively (Table 2). Although this is an indicative result of a single Cr hotspot, the significant correlation of Cr intensity counts with Ca and Fe intensity counts on the µ-XRF maps suggested that the Cr in the coarse sand sized fraction might be dominated by the speciation bearing in the COPR parent mineral phase.
The spots 2 (F2) and spot3 (F2) both from the fine sand sized fraction have quite similar spectrum shape. The LCF results indicate that the Cr(III)–HA speciation accounted for 66% and 73% in the spot2 (F2) and spot3 (F2), respectively, dominating the trivalent chromium in both hotspots, followed by chromite (Table 2). The Cr(VI) could be reduced to Cr(III) in the presence of organic materials and then precipitated as solid state on the surface of organic matter (Rajapaksha et al. 2018). Ohta (2015) and Ding et al. (2016) both found the reduction of Cr(VI) in soil samples and alkaline COPR leachates and the formation of Cr(III)-organic complex or Cr(III) (OH)3 precipitation in the sites with substantial organic matter and ferrous.
The Cr(VI) of spot2 (F2) and spot3 (F2) is dominated by sodium chromate (Na2CrO4), accounting for 16% and 15%, respectively (Table 2). Sodium chromate has a higher solubility than CaCrO4, which was rarely reported in European and North America COPR where only CaCrO4 has been found in some COPR samples (Geelhoed et al. 2002; Chrysochoou et al. 2009b). Matern et al. (2016) has found the occurrence of Na2CrO4 in the pores of COPR samples and speculated that it was caused by poorly drained and high evaporation condition in the deposited sites.
Spot 4 (F3) spectrum presents a pronounced pre-edge peak at 5991.5 eV as spot1 (F1), suggesting the occurrence of Cr(VI). However the LCF results indicate that the Cr mainly presents as trivalent state, which is dominated by Cr3+ and chromite in the hotspot (Table 2). This is reasonable in a fine size-fraction of a long-term weathered COPR polluted soil, in which substantial Cr(VI) has been transformed to Cr(III) and the converted Cr3+ may be immobilized by fine particles of the soil due to its high surface area to volume ratio (Ding et al. 2016; Choppala et al. 2018).
The COPR polluted soil is characterized of high content of Cr an Cr(VI) which decreased with grain size. The SEM-EDX showed that the Cr was bearing both the COPR parent mineral phases (brownmillirite, magnesiochromite and hashemite) and the hydrated products (stichtite, paraalumhydrocalcite and brucite). Synchrotron based µ-XRF mapping and µ-XANES revealed that Cr was primarily in the hexavalent state in the coarse sand sized fraction and occurred in the mixture of chromatite and chromite. In the case of the fine size-fraction, organically bound Cr(III) and adsorbed Cr3+ might occur in some hotspots, suggesting the edaphic effects on the Cr transformation and sequestration after the long-term COPR weathering. The variation in Cr speciation among the size-fractions implied that it was necessary to separate a COPR-enriched soil based on grain size prior to treatment with various methods.
References
Choppala G, Kunhikrishnan A, Seshadri B, Park JH, Bush R, Bolan N (2018) Comparative sorption of chromium species as influenced by pH, surface charge and organic matter content in contaminated soils. J Geochem Explor 184:255–260
Chrysochoou M, Dermatas D (2006) Evaluation of ettringite and hydrocalumite formation for heavy metal immobilization: literature review and experimental study. J Hazard Mater 136:20–33
Chrysochoou M, Fakra SC, Marcus MA, Moon DH, Dermatas D (2009a) Microstructural analyses of Cr(VI) speciation in chromite ore processing residue (COPR). Environ Sci Technol 43:5461–5466
Chrysochoou M, Moon DH, Fakra S, Marcus M, Dermatas D, Christodoulatos C (2009b) Use of micro X-ray absorption spectroscopy and diffraction to delineate Cr(Vi) speciation In COPR. Global Nest J 11:318–324
Chrysochoou M, Dermatas D, Grubb DG, Moon DH, Christodoulatos C (2010) Importance of mineralogy in the geoenvironmental characterization and treatment of chromite ore processing residue. J Geotech Geoenviron 136:510–521
Darrie G (2001) Commercial extraction technology and process waste disposal in the manufacture of chromium chemicals from ore. Environ Geochem Health 23:187–193
Dermatas D, Bonaparte R, Chrysochoou M, Moon DH (2006) Chromite ore processing residue (COPR): hazardous contaminated soil or solid waste? J ASTM Int 3:13313
Ding WX, Stewart DI, Humphreys PN, Rout SP, Burke IT (2016) Role of an organic carbon-rich soil and Fe(III) reduction in reducing the toxicity and environmental mobility of chromium(VI) at a COPR disposal site. Sci Total Environ 541:1191–1199
Elzinga EJ, Cirmo A (2010) Application of sequential extractions and X-ray absorption spectroscopy to determine the speciation of chromium in Northern New Jersey marsh soils developed in chromite ore processing residue (COPR). J Hazard Mater 183:145–154
Geelhoed JS, Meeussen JCL, Hillier S, Lumsdon DG, Thomas RP, Farmer JG, Paterson E (2002) Identification and geochemical modeling of processes controlling leaching of Cr(VI) and other major elements from chromite ore processing residue. Geochimica Cosmochim Acta 66:3927–3942
Grafe M, Donner E, Collins RN, Lombi E (2014) Speciation of metal(loid)s in environmental samples by X-ray absorption spectroscopy: a critical review. Anal Chim Acta 822:1–22
Hawley EL, Deeb RA, Kavanaugh MC, et al. (2005) Treatment technologies for chromium(VI). In: Guertin J, Jacobs JA, Avakian CP (eds) Chromium(VI) handbook. CRC Press, Boca Raton, pp 275–309
Hillier S, Roe MJ, Geelhoed JS, Fraser AR, Farmer JG, Paterson E (2003) Role of quantitative mineralogical analysis in the investigation of sites contaminated by chromite ore processing residue. Sci Total Environ 308:195–210
Hillier S, Lumsdon DG, Brydson R, Paterson E (2007) Hydrogarnet: a host phase for Cr(VI) in chromite ore processing residue (COPR) and other high pH wastes. Environ Sci Technol 41:1921–1927
James BR (1996) The challenge of remediating chromium-contaminated soil. Environ Sci Technol 30:A248–A251
Matern K, Kletti H, Mansfeldt T (2016) Chemical and mineralogical characterization of chromite ore processing residue from two recent Indian disposal sites. Chemosphere 155:188–195
Moon DH, Wazne M (2011) Impact of brownmillerite hydration on Cr(VI) sequestration in chromite ore processing residue. Geosci J 15:287–296
Moon DH, Dermatas D, Wazne M, Sanchez AM, Chrysochoou M, Grubb DG (2007) Swelling related to ettringite crystal formation in chromite ore processing residue. Environ Geochem Health 29:289–294
Ohta A (2015) Speciation study of Cr in a geochemical reference material sediment series using sequential extraction and XANES spectroscopy. Geostand Geoanal Res 39:87–103
Rajapaksha AU, Alam MS, Chen N, Alessi DS, Igalavithana AD, Tsang DCW, Ok YS (2018) Removal of hexavalent chromium in aqueous solutions using biochar: chemical and spectroscopic investigations. Sci Total Environ 625:1567–1573
SEPA (State Environmental Protection Administration of China) (2007) Solid waste-extraction procedure for leaching toxicity sulphuric acid & nitric acid method (HJ/T299-2007). Beijing
US EPA (Unite States Environmental Protection Agency) (1996) Alkaline digestion for hexavalent chromium, Method 3060A. Washington, DC
Vodyanitskii YN (2013) Determination of the oxidation states of metals and metalloids: An analytical review. Eurasian Soil Sci 46:1139–1149
Xu HT, Lou TJ, Li YD (2004) Synthesis and characterize of trivalent chromium Cr(OH)3 and Cr2O3 microspheres. Inorg Chem Commun 7:666–668
Yu XL, Lu SG (2016) Multiscale correlations of iron phases and heavy metals in technogenic magnetic particles from contaminated soils. Environ Pollut 219:19–27
Acknowledgements
This research was funded by Zhejiang Agriculture and Forestry University Research Funding (2017FR021) and the National Natural Science Foundation of China (41771351). We are grateful to the beamline (BL15U1) of Shanghai Synchrotron Radiation Facility (SSRF) for providing µ-XRF and µ-XANES analysis. Thanks are also given to the anonymous reviewers for their suggestive comments on the paper improvement.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhang, H., Zhou, B., Ren, J. et al. Chromium Speciation in the Size-Fractions of a Soil Polluted by Weathered Chromate Ore Process Residue Using Synchrotron X-ray Analysis. Bull Environ Contam Toxicol 103, 3–9 (2019). https://doi.org/10.1007/s00128-018-2399-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00128-018-2399-0