
Abstract
The Matongo carbonatite intrusive body in the Neoproterozoic Upper Ruvubu alkaline plutonic complex (URAPC) in Burundi is overlain by an economic phosphate ore deposit that is present as breccia lenses. The ore exhibits evidence of supergene enrichment but also preserves textures related to the concentration of fluorapatite in the carbonatitic system. Magmatic fluorapatite is abundant in the ore and commonly occurs as millimeter-sized aggregates. It is enriched in light rare earth elements (LREE), which is especially apparent in the final generation of magmatic fluorapatite (up to 1.32 wt% LREE2O3). After an episode of metasomatism (fenitization), which led to the formation of K-feldspar and albite, the fluorapatite-rich rocks were partly brecciated. Oxygen and carbon isotope compositions obtained on the calcite forming the breccia matrix (δ18O = 22.1 ‰ and δ13C = −1.5 ‰) are consistent with the involvement of a fluid resulting from the mixing of magmatic-derived fluids with a metamorphic fluid originating from the country rocks. In a subsequent postmagmatic event, the carbonates hosting fluorapatite were dissolved, leading to intense brecciation of the fluorapatite-rich rocks. Secondary carbonate-fluorapatite (less enriched in LREE with 0.07–0.24 wt% LREE2O3 but locally associated with monazite) and coeval siderite constitute the matrix of these breccias. Siderite has δ18O values between 25.4 and 27.7 ‰ and very low δ13C values (from −12.4 to −9.2 ‰), which are consistent with the contribution of organic-derived low δ13C carbon from groundwater. These signatures emphasize supergene alteration. Finally, the remaining voids were filled with a LREE-poor fibrous fluorapatite (0.01 wt% LREE2O3), forming hardened phosphorite, still under supergene conditions. Pyrochlore and vanadiferous magnetite are other minerals accumulated in the eluvial horizons. As a consequence of the supergene processes and fluorapatite accumulation, the phosphate ore, which contains 0.72 to 38.01 wt% P2O5, is also enriched in LREE (LaN/YbN from 47.1 to 83.5; ΣREE between 165 and 5486 ppm), Nb (up to 656 ppm), and V (up to 1232 ppm). In the case of phosphate exploitation at Matongo, REE could prove to have a subeconomic potential to be exploited as by-products of phosphates.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Carbonatites and related alkaline complexes are of general interest because of their high economic potential (Deans 1966; Mariano 1989; Pell 1996; van Straaten 2002). Phosphate is one of the most important resources associated with carbonatites. It has been mined in South Africa, Zimbabwe, Brazil, Finland, and Sri Lanka (Pell 1996), mainly to produce fertilizer (Verwoed 1986; van Straaten 2002). Phosphates of carbonatite complexes are present either as primary apatite in carbonatites, or as apatite and secondary phosphates in their residual deposits. Hence, they can be the result from different processes, ranging from magmatism to supergene alteration (e.g., De Toledo et al. 2004; Vartiainen and Paarma 1979; Walter et al. 1995).
Carbonatites are widespread throughout Africa, particularly in East Central Africa (Woolley 2001; van Straaten 2002). A phosphate deposit associated with a Neoproterozoic alkaline-carbonatite complex is known in Burundi (Upper Ruvubu alkaline plutonic complex, URAPC). It is located along the western branch of the East African Rift (Tack et al. 1984). The URAPC comprises a subsurface carbonatite, the Matongo carbonatite intrusion, which has been recently characterized from a petrographic and geochemical point of view (Midende et al. 2014). This intrusion, which is known from drilling, is partly overlain by discontinuous breccia lenses with a matrix enriched in P (>15 wt% P2O5, mainly as apatite). The breccias contain an economic concentration of phosphates (Kurtanjek and Tandy 1989; Mariano 1989; Songore 1991). The genesis of the deposit has only been poorly investigated and the characterization of their phosphates has never been properly addressed.
The aim of this paper is to evaluate the genetic processes—magmatic, hydrothermal, and supergene—involved in the formation of the deposit through a comprehensive petrographic and geochemical study of both the phosphate ore and the gangue minerals. For that purpose, the use of cathodoluminescence (CL) constitutes a powerful tool. The luminescence of a mineral is often highly variable depending on the chemical composition and the prevalent physical conditions during its formation (Götze and Kempe 2009). Moreover, CL is particularly well suited to detect the presence and the level of rare earth element (REE) enrichment in apatite (Barbarand and Pagel 2001; Kempe and Götze 2002). Raman spectroscopy was also performed notably to highlight the presence of CO3 2− and its crystallographic position and, therefore, to distinguish carbonate-fluorapatite from fluorapatite in the studied samples.
The carbon and oxygen isotopic compositions of the carbonates (mostly siderite) associated with the phosphates are used to determine the origin of the fluids involved and constrain the mineralizing processes. Finally, the economic potential of the eluvial horizon developed on the Matongo carbonatite is discussed using the whole-rock geochemistry of the phosphates.
Geological setting
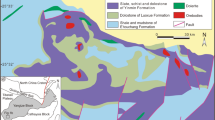
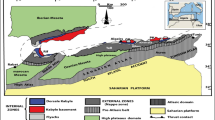
The Neoproterozoic URAPC of Burundi is located along the western branch of the East African Rift (Fig. 1a). It was emplaced in Mesoproterozoic metasedimentary rocks (Akanyaru Supergroup) of the Karagwe-Ankole Belt (KAB; Fernandez-Alonso et al. 2012; Boulvais et al. 2015). The URAPC comprises two major bodies of silicate rocks: (1) feldspathoidal syenite as the “inner unit” and (2) quartz-saturated rocks—from olivine-bearing gabbro and diorite to quartz-bearing syenite and granite—as the “outer unit” (Tack et al. 1984). A subsurface carbonatite body, known as the Matongo carbonatite, has been recognized by drilling.

a Geological sketch map of the studied area (modified and redrawn from Tack et al. 1984 and after Fernandez-Alonso et al. 2012). b Location of the boreholes; the red line corresponds to the section shown in c. c Schematic cross section of the Matongo phosphate deposit along the profile located in b, as schematized from Midende (1984)
For the “outer magmatic unit,” a Rb-Sr whole-rock isochron yielded an age of 707 ± 17 Ma (Tack et al. 1984), which is, within the bounds of uncertainty, in good agreement with both a Rb-Sr isochron age of the “inner magmatic unit” (699 ± 13 Ma; Tack et al. 1984) and with a Pb-Pb isochron of the carbonatite body (690 ± 32 Ma; Demaiffe 2008). Zircon megacrysts are encountered in “pegmatitic-like” veinlets that intrude the supergene weathered rock with preserved original coarse-grained “granitic” texture (Nkurikiye 1989; Tack, unpublished data) in the vicinity of the carbonatite. They are related to the hydrothermal-magmatic evolution of the carbonatite of the URAPC (Decrée et al. 2015). These zircon megacrysts yielded a Pb-Pb evaporation age of 738 ± 4 Ma (Tack et al. 1995) and, more recently, a U-Pb LA-ICP-MS concordant age of 705.5 ± 4.5 Ma (Midende et al. 2014).
The Matongo carbonatite occurs from a depth of about 40–80 m below surface and forms an elongated NNE-oriented intrusion of 2750 by 250 m. Fine- and coarse-grained calciocarbonatite and a ferrocarbonatite have been identified (Demaiffe 2008; Midende 1984; Midende et al. 2014). Fine-grained sövite represents the dominant facies of the carbonatite. It mainly consists of saccharoidal calcite and often contains discrete zones with vanadiferous aegirine and apatite occurring as small grains or small prismatic crystals of up to 5 mm in size. Aegirine and apatite may represent local cumulates, in which pyrochlore [Na,Ca)2Nb2O6(OH,F)] and K-feldspar are also present (Midende 1984). The carbonatite is partially covered by a cap of subsurface brecciated phosphate ore, discovered by airborne geophysical (gamma-ray) surveys in 1971 and 1975 (Songore 1991) and sampled by drilling in the early 1980s (Fig. 1b).
Much of the ore forms horizontal-flattened lenses (Fig. 1c) with a thickness of more than 10 m (Midende 1984). Two main bodies that correspond to eluvial horizons have been encountered in borehole S1 (a 102-m-thick lens with a grade of 13.6 wt% P2O5) and in borehole S2 (a 47.2-m-thick pocket with a grade of 14 wt% P2O5; Midende 1984). The phosphate reserves were estimated at 40 Mt, with an average grade of 4.6 wt% P2O5, and 17.3 Mt of richer ore averaging 11 wt% P2O5 (considering a cutoff grade of 5 wt% and minimal thickness of 1.5 m; Notholt 1999, in DFID 2002). The phosphate mineralization has been ascribed to a series of processes, including decalcification, development of phosphate-rich breccias, hydrothermal phosphate formation, secondary phosphate formation, and development of iron and aluminum phosphates associated with Fe and Mn oxides, clays, and gypsum in near surface and along fractures (Kurtanjek and Tandy 1989; Midende 1984).
Material and methods
Thirteen borehole samples of phosphate ore were selected from the collection of the Royal Museum for Central Africa (RMCA) in Tervuren (Belgium). The location of the drill cores is known (Fig. 1b), but the precise depth of the samples in the core is uncertain.
The petrographic description is based on optical microscopy and scanning electron microscopy (SEM) using a Quanta 20 ESEM (FEI), with energy-dispersive spectroscopy (Apollo 10 silicon drift EDS detector; EDAX) at the Royal Belgian Institute of Natural Sciences. CL studies were performed at the University of Mons using a cold-cathode CL unit model Mk5 operated at 15 kV beam voltage and 500 μA current (Cambridge Image Technology Limited). CL spectra were recorded with a CITL optical spectrometer model COS8200 allowing acquisition from 380 to 1100 nm at 3.7 nm resolution.
Quantitative microanalyses of mineral chemistry were acquired using a Cameca SX51 electron microprobe (EMPA) operated at 15 kV and 20 nA (see Table 1 in the Electronic supplementary material). This microprobe is equipped with four wavelength-dispersive (WDS) spectrometers and standard LIF, PET, TAP, and PC crystals. In Table 1, the reported F contents may be overestimates due to ion diffusion during EPMA analyses (Stormer et al. 1993).
Raman spectroscopy was performed at the Royal Belgian Institute of Natural Sciences to investigate the presence of CO3 2− and its crystallographic position. We used a 532-nm (green) laser Raman spectrometer (Senterra, Olympus BX51, Bruker Optics). The spectra were acquired using a 2-mW excitation power, 5 × 30 s integration time, and with a 50-μm spectrometer slit.
Bulk samples of phosphate ore were selected, ground in an agate mortar, and analyzed for the major and trace element contents (Table 2 in the Electronic supplementary material) at the RMCA (Tervuren). Major element concentrations (Si, Ti, Al, Fe3+, Mg, Mn, Ca) were determined by ICPAES with an average accuracy within 5 %, whereas trace and rare earth element concentrations were obtained by ICP-MS (Thermo X-series). The results conform to international standards regarding the standard deviation (between 5 and 10 %; Fagel et al. 1997).
Carbon and oxygen isotopic compositions of carbonates were obtained at the University of Rennes 1. These isotopic compositions were measured on bulk sample powders, sampled from the carbonate matrix of 12 samples of phosphate ore. The powders were subjected to anhydrous orthophosphoric acid in vessels at 50 °C. Calcite is treated in this fashion for a few hours, whereas siderite requires 60 h of processing. The liberated CO2 was then purified from any possible contributions of H2S contribution due to the presence of sulfides in the whole rock by reaction with Ag3PO4 for 5 min at 70 °C. The isotopic compositions were measured on a VG Optima triple collector mass spectrometer. The following experimental fractionation factors between CO2 and carbonates were used: α CO2-siderite = 1.01075 (Carothers et al. 1988) and α CO2-calcite = 1.009306 (Swart et al. 1991). The analytical uncertainty is ±0.2 ‰ for O and ±0.1 ‰ for C; this was estimated using duplicate analyses on some of the samples and through the reproducibility of standards, both international (NBS 19) and in-house (Prolabo Rennes).
Results
Petrographic description and mineral composition
The phosphate-rich breccias exhibit angular to subrounded clasts that are randomly orientated. The clasts vary in size from a few micrometers to several millimeters (see Figs. 2 and 3 for a general overview of the breccia textures). Aside from the phosphates, the clasts predominantly consist of K-feldspar that is dull red luminescing under CL (Fig. 2a). Such a red emission, which is due to Fe3+ activation, is commonly observed in feldspars occurring in fenitized rocks (Marshall 1988). The K-feldspar clasts show evidence of brecciation and dissolution at the rims (Fig. 2a–c). The resulting fractures and corroded rims are cemented by pink-luminescing albite (up to 200 μm in width; Fig. 2b). Albite crystals are, in turn, variably brecciated and invaded by calcite. Albite is also present as disseminated subhedral to euhedral zoned crystals or clasts in the breccia matrix (Fig. 2a). The pink luminescence of albite is common for unaltered albite and is related to Fe3+ activation (Götte 2009; see the Electronic supplementary material for the CL spectra). Deformed biotite laths (up to ~1 mm in length, Fig. 2c) and calcite clasts are also present in the matrix.

Cathodoluminescence (a, b, d, f) and backscattered electron (c, e) micrographs of the Matongo phosphate ore. Carb-fluorap. carbonate-fluorapatite, Fluorap. fluorapatite, Kfs K-feldspar. a Zoned fluorapatite crystals, two of them being engulfed in a corroded K-feldspar clast. Apatite luminescence is predominantly blue-green to red in the core and blue-violet in the outer rim. Feldspar luminescence is bright pink (albite) and dull red (Kfs). The matrix is nonluminescing siderite and dark green-luminescing carbonate-apatite (see Fig. 3c for a more detailed view of a similar matrix) (sample S2/97.8-98A). b Fluorapatite at the rim of (and enclosed by) K-feldspar clasts in a fine-grained, orange-luminescing calcitic matrix (sample S7/95.16-96.7). c Neoformed monazite, Th-rich monazite, and pyrite in a matrix made up of siderite (light gray) and carbonate-fluorapatite (dark gray) (sample S1/121-121.77). d Closely packed cluster of fluorapatite crystals (see text for further details) (sample S5/115-116). e, f Carbonate-fluorapatite filling—along with monazite—remaining voids within primary fluorapatite aggregates (sample S5/115-116)

Backscattered electron (a, b, g) and cathodoluminescence (c–f) micrographs of the Matongo phosphate ore. a Fluorapatite grains in a matrix comprising siderite and Fe oxide; the insert shows a euhedral Ba-Sr-rich pyrochlore crystal coeval with siderite as an interstitial phase of the fluorapatite (sample S2/117). b Detail of the relation between siderite and Fe oxide in the matrix shown in a (sample S2/117). c Dark green- and brown-luminescing tabular (carbonate-) fluorapatite constituting the matrix cementing the corroded magmatic fluorapatite (sample S2/120.5-121.1). d Fibrous green-luminescing fluorapatite forming the hard lithified phosphorite (sample S1/123-123.7). e Fibrous green-luminescing fluorapatite cementing K-feldpar (comprising small clasts of fluorapatite) and (corroded) zoned fluorapatite (sample S1/123-123.7). f Banded deposit of fibrous green-luminescing fluorapatite and nonluminescing Fe oxide as a replacement of the carbonate matrix (sample S1/123-123.7). g Fluorapatite crystal with a LREE-rich rim, which is partly replaced and overgrown by monazite and LREE-poor carbonate-fluorapatite (sample S2/117)
Fluorapatite and carbonate-fluorapatite are the dominant phosphate minerals. Primary (magmatic) fluorapatite exhibits a complex zoned pattern under CL. In summary, it shows a blue-green to red CL core (up to ~1 mm in diameter) and a blue-violet outer rim (with a typical thickness of ~100 μm; Fig. 2a, d). The crystals are commonly brecciated or traversed by veins. Apatite cores are more heavily affected and may form clasts, which were subsequently cemented by blue-luminescing apatite. This indicates a multistage brecciation and growth history of the apatite crystals. Fluorapatite occurs either as isolated crystals, crystal clasts in the breccia, inclusions within K-feldspar clasts (Fig. 2a, b), or as closely packed clusters of crystals of up to 3 mm in length (Fig. 2d). Such clusters are locally concentrated in centimeter-thick layers/lenses in the breccia (Fig. 2e, f). Regardless of its form, many fluorapatite grains exhibit dissolution features at the rims. Regarding the mineral compositions (Table 1 in the Electronic supplementary material), the blue/violet-luminescing fluorapatite shows a distinct enrichment in REE (0.29 < ΣLREE oxide < 1.88 wt%) coupled with a decrease in Ca (Fig. 4a), and a depletion in SiO2 (up to 0.12 wt%; Fig. 4b) compared to the blue-green CL core (0.13 < ΣLREE oxide < 0.31 wt% and 0.02 < SiO2 < 0.18 wt%). The predominantly blue CL of fluorapatite in carbonatites is well-known (e.g., Mariano 1988; Kempe and Götze 2002; Hayward and Jones 1991; Waychunas 2002) and has been assigned to Ce3+, Eu2+, and Sm3+ emission centers (Kempe and Götze 2002). Eu2+ and Sm3+ activation was observed by spectroscopy together with Dy3+ and Mn2+ activation in the primary fluorapatite (see the Electronic supplementary material). It was not possible to isolate the spectra from the different CL emission colors (blue-green, red, and blue-violet) due to the low spatial resolution of the spectrometer, but they most likely relate to variations in the relative activation of Eu2+ (blue-violet), Sm3+ (red), and Dy3+ or Mn2+ (green).

Correlation between a ΣREE and CaO contents, b SiO2 and P2O5 contents, and c F and P2O5 contents in the different fluorapatite generations in the Matongo phosphate deposits
The breccia matrix contains carbonates and phosphates. The carbonate matrix consists of either orange-luminescing calcite (Fig. 2b), with the orange CL being due to trace Mn2+ substituting for Ca2+ in calcite (Habermann et al. 2000), or, more commonly, siderite. This siderite is finely admixed with Fe oxides (Fig. 3a, b). Siderite is associated with carbonate-fluorapatite in the breccia matrix (identification based on Raman spectra—see below). Two different carbonate-fluorapatite types were observed. The first type forms a fine-grained groundmass of aggregating dark green- and brown-luminescing tabular crystals (Figs. 2c and 3c). This type seems to develop, to some extent, at the expense of the blue-luminescing fluorapatite rim (Fig. 3c). Locally, the rhombic shape of this type of carbonate-fluorapatite suggests the replacement of earlier carbonates. It was not possible to record a CL spectrum due to their weak luminescence. The green luminescence could be related to Mn2+ activation with a small contribution by REE (Waychunas 2002). The second type of carbonate-fluorapatite is nonluminescing and is found overgrowing and cementing primary fluorapatite (Fig. 2e, f). The lack of luminescence in this carbonate-fluorapatite most likely results from the absence of activator elements (REE and Mn2+). Similar nonluminescent fluorapatite has been observed in the supergene environment in the Catalão alkaline-carbonatitic complex (De Toledo et al. 2004) and in laterites formed on carbonatites (Mariano 1988). The chemical composition of nonluminescing carbonate-fluorapatite shows a depletion in light rare earth elements (LREE) and Ca (0.05 < ΣLREE oxide < 0.24 wt%, 52.96 < CaO < 53.63 wt%, Table 1 in the Electronic supplementary material; Fig. 4a) and a decrease in the P content (36.06 < P2O5 < 37.04; Fig. 4c) compared to the primary fluorapatite. Although Mn was not measured in the nonluminescing carbonate-apatite, it is likely that the concentration is extremely low due to its oxidation in supergene conditions. This prevented its incorporation in the apatite lattice.
Lastly, a bright green-luminescing fibrous fluorapatite occurs as filling the remaining voids, locally as ribbons and colloform concretions in alternation with Fe oxides (variously admixed with silica) (Fig. 3d–f). This fluorapatite facies macroscopically constitutes a hard carvernous boxwork. Boxwork structures resembling a cleavage pattern are also observed on a microscopic scale. This points to the replacement of a former carbonate by phosphate. This secondary fluorapatite is the last generation of apatite in the studied samples. Its green luminescence is due to Mn2+ activation as indicated by the emission spectrum (in the Electronic supplementary material). Assuming a reconcentration of the primary fluorapatite, there was a significant loss of REE during the dissolution and precipitation process (as proposed by Bouzari et al. 2014; Lypaczewski et al. 2013). As predicted, the fibrous fluorapatite is depleted in LREE (ΣLREE oxide = 0.01 wt%) and is characterized by an increase of the Ca, P, and F contents (CaO = 55.25 wt%, P2O5 = 40.60 wt%, F = 5.86 wt%; Fig. 4).
Monazite is another phosphate that is commonly encountered in these breccias. It is usually present as an interstitial phase of the primary fluorapatite grains (forming patches of up to 300 μm across; Fig. 2c, e), in association with carbonate-fluorapatite. Monazite also occurs as small crystals (from a few micrometers up to 15 μm) at the corroded LREE-rich rims of fluorapatite crystals, still in close association with carbonate-fluorapatite (although deposited slightly earlier, Fig. 3g). In this case, both minerals seem to form, at least partly, at the expense of fluorapatite. In situ microprobe analyses show that monazite is characterized by variable degrees of enrichment in REE (48.1 < ΣLREE oxide < 57.7 wt%), Ca (1.6–12.5 wt% CaO), Th (0.2–9.6 wt% ThO2), Sr (up to 5.2 wt% SrO), and U (up to 1.9 wt% U2O3) and a slight increase in Fe, Y, and Si (0.3–1.4 wt% FeO, up to 0.6 wt% Y2O3, and 0.1–0.6 wt% SiO2, Table 1 in the Electronic supplementary material). A particular Th-monazite has also been found in the breccias (37.8–42.4 wt% ThO2, 8.7–16.8 wt% ΣLREE oxide, 5.8–7.7 wt% CaO, 3.2–4 wt% SiO2, ~0.9 wt% BaO, 0.2–0.8 wt% SrO).
The breccia matrix also includes numerous clasts (up to several millimeters in size) of vanadiferous magnetite, pyrite, ilmenite, and pyrochlore. Two different types of pyrochlore have been recognized. The first type is enriched in U and Pb (locally, it shows a Sr-Ba enrichment at the rims) and occurs as clasts in the breccia. The clast habit and U-Pb enrichment strongly favor its magmatic origin (Nasraoui and Bilal 2000; Chakhmouradian 2006; Bambi et al. 2008; Mitchell 2014). The second type of pyrochlore is Sr-Ba rich and forms zoned euhedral crystals (up to 100 μm in width; insert in Fig. 3a), which are coeval with the siderite in the breccia matrix. In these breccias, the fissures and remaining voids are filled by a mixture of Fe oxides and silica (Fig. 3f). Mn oxides have also been observed.
Raman spectra
Most of the Raman spectra exhibit well-defined peaks, with the exception of the tabular prismatic and fibrous fluorapatite, which is characterized by a distinctive fluorescence (see Electronic supplementary material for the complete Raman spectra). The Matongo fluorapatite grains show the best-defined peak around 963 cm−1 (from 961 to 965 cm−1), which corresponds to the ν1 vibrational mode of the PO4 3−. In all of the studied fluorapatite grains, the ν2 mode of this anion is observed in the 424–432-cm−1 range.
As a general rule, the ν4 vibration mode of PO4 3− in fluorapatite is characterized by four wave numbers at 581, 592, 608, and 617 cm−1, with a decrease of the first and last bands as the fluoride content increases (Penel et al. 1997). This is the case for the Matongo fluorapatites where a peak centered on ~590 cm−1 (with a range from 587 to 592 cm−1) exhibits a shoulder at ~606 cm−1 (range from 603 to 610 cm−1). In most of the fluorapatites, the ν3 vibration mode of PO4 3− is expressed by two bands at 1050–1053 cm−1 and in the wide range 1070–1081 cm−1 (Fig. 5). In the tabular carbonate-fluorapatite, only the second peak between 1078 and 1081 cm−1 is present. The shift on the 1076 to 1070 cm−1 (observed in the nonluminescent carbonate-fluorapatite) is typically attributed to the presence of the CO3 2− anion (CO3 2−–PO4 3− substitution; Penel et al. 1998), whereas the shift from the 1076-cm−1 band to higher wavenumbers (up to 5 cm−1) is related to fluoride substitution (Penel et al. 1997). Therefore, the nonluminescing fluorapatite can be regarded as carbonate-fluorapatite, which is also supported by the low sum in electron microprobe analyses (average of 92.6 wt%; Table 1).

Raman spectra of magmatic fluorapatites and hydrothermal-supergene fluorapatites from the Matongo phosphate deposit. Acquired using λ excitation = 532 nm, power 2 mW, acquisition time 5 × 30 s. Only the 1000–1100-cm−1 range is shown to detail ν3 vibration mode of PO4 3− and CO3 2−–PO4 3− substitution: 1000–1100 cm−1
The tabular fluorapatite with a dark green to brown CL does not exhibit the Raman peak shift related to the carbonate substitution. It is characterized by a lack of anions in the phosphate site, as observed for the nonluminescing carbonate-fluorapatite (microprobe sum of 94.5 wt%; Table 1). This could be explained by CO3 2− substitution for PO4 3−. Such a hypothesis is likely because (i) the tabular fluorapatite is coeval with the nonluminescing carbonate-fluorapatite and (ii) both minerals have likely replaced a carbonate matrix, which is additionally supported by the rhombic morphology of some crystals. Thus, the tabular fluorapatite is interpreted as a carbonate-fluorapatite. The absence of a peak shift in the Raman spectra could be related to a crystallographic orientation effect, which is known to influence the intensity and even the position of Raman peaks in apatite (Tsuda and Arends 1994).
Other unresolved peaks of the Raman spectra in the ~1500–3500-cm−1 range are far more intense for the magmatic fluorapatite. They are typical of REE-induced fluorescence and can be related to the presence of these elements in the studied fluorapatite, as confirmed by in situ analyses of magmatic fluorapatite (Table 1 in the Electronic supplementary material).
Whole-rock geochemistry of the phosphate deposits
In the breccias, the Fe2O3 (2.39–46.51 wt%), P2O5 (0.72–38.01 wt%), CaO (12.69–51.00 wt%), and SiO2 (0.79–15.45 wt%) contents are strongly variable (Table 2 in the Electronic supplementary material). Moreover, CaO and P2O5 contents show a negative correlation with the Fe2O3 content. These geochemical characteristics reflect the heterogeneity of the clasts and the variable proportion of clasts and matrix in whole-rock powders. The samples are rich in Al2O3 (up to 4.59 wt%), MnO (up to 4.31 wt%), and K2O (up to 3.16 wt%) but poor in MgO (0.01–0.76 wt%) and Na2O (up to 0.94 wt%). High concentrations of S are related to the presence of pyrite in some of the samples (up to 15.73 wt% S) (Table 2).
The chondrite-normalized REE patterns of the Matongo phosphate ore (Fig. 6a) show an enrichment in LREE (LaN/YbN from 47.1 to 83.5), with high ΣREE contents (707–5486 ppm), low negative Ce anomalies (0.7 < Ce*/Ce < 0.9) and Eu anomalies (0.8 < Eu*/Eu < 0.9, Table 2). These patterns are quite similar to those of the primary fine-grained sövite of the Matongo carbonatite (Midende et al. 2014). The total REE content of the phosphate ore, on the other hand, is higher in comparison to the sövite (up to 7.5×). The calcite-rich sample (S7/95.16-96.7) has lower ΣREE (165 ppm) and LaN/YbN (6.1) (Table 2, Fig. 6a). The primitive mantle (P.M.)-normalized spidergrams of the Matongo phosphate ore (Fig. 6b) display enrichment in several HFSE (high-field-strength elements), like Nb, Ta (up to 920 times and 655 times the P.M. contents, respectively; Table 2), REE, and LILE (U, which is up to 15,261 times enriched compared to the P.M.) and depletion in Ti, Hf, Zr, and K. In addition, these rocks can exhibit a significant enrichment in V, up to 1232 ppm (~15 times the P.M. content for sample S5/115-116, Table 2). As a whole, these spidergrams display a similar shape as those of the fine-grained Matongo sövite (Fig. 6b). In general, the Th, U, Pb, Nb, and Ta contents are higher than the mean content of the sövite (up to 7.9, 36.2, 8.6, 2.5, and 4.1×, respectively; Midende et al. 2014).

REE patterns (a) and multielement diagram (b) of the Matongo phosphate ore, compared to the fine-grained sövite II facies from the Matongo carbonatite (Midende et al. 2014). REE patterns are normalized to the chondrites (values from Sun 1982) and multielement diagrams are normalized to the primitive mantle (values from McDonough et al. 1992)
Stable isotopes
The C and O isotopic composition of the Matongo phosphate ore is given in Table 2 and Fig. 7. The δ18O ratios of siderite are within a narrow range between 25.4 and 27.7 ‰ (mean = 26.5 ± 0.7 ‰, n = 11), the corresponding δ13C values vary between −9.2 and −12.4 ‰ (mean = −11.3 ± 1.1, n = 11; Table 2 in the Electronic supplementary material). As a group, siderite-bearing samples form a distinctive group separate from both primary carbonatitic values and secondary values acquired during superposed hydrothermal alterations (Fig. 7). The calcite-bearing sample S7/95.16-96.7 on the other hand, displays isotopic compositions (δ18O = 22.1 ‰ and δ13C = −1.5 ‰) indistinguishable from the calcite veins that crosscut the Matongo carbonatite (Decrée et al. 2015).

δ13C versus δ18O for the calcite and siderite forming the breccia matrix of the phosphate ore. Samples from the Matongo carbonatite are plotted for comparison purposes. Trend 1 corresponds to an alteration of the Matongo carbonatite due to interactions with moderate-temperature hydrothermal fluids (estimated at 150–300 °C; Decrée et al. 2015). Trend 2 relates to low-temperature fluids (~100 °C) originating from (and interacting with) carbonated lithologies of the country rocks around the URAPC (Decrée et al. 2015)
Discussion
Inherited magmatic fluorapatite and the involvement of hydrothermal fluids
The primary, predominantly blue CL, fluorapatite occurs as inherited ovoid/prismatic crystals, reminiscent of those found in the carbonatitic environment of Qasiarsuk (Hayward and Jones 1991, see their Fig. 6), Lemitar Mountain (McLemore and Barker 1987, see their Fig. 2a), and Jacupiranga (Alves 2008, see her Fig 4.5 and Plate B1.p). Similarly, the blue-violet-luminescing LREE-rich fluorapatite encountered is commonly interpreted as a magmatic (or late magmato-hydrothermal) mineral phase. The blue-luminescing fluorapatite also occurs locally as a rim of the red-green-luminescing fluorapatite, in optical continuity with the core. This can be interpreted as the result of the sequential crystallization of fluorapatite during carbonatite emplacement (as already proposed by Walter et al. 1995). The increase of REE in the rim is correlated with a decrease in CaO (Fig. 4a), confirming the coupled substitution of these elements.
In the immediate vicinity of the Matongo carbonatite, fenites mainly consist of K-feldspar (Midende et al. 2014). Partially dissolved fluorapatite together with the presence of K-feldspar clasts (themselves corroded, brecciated, and crosscut by albite veins) suggests early crystallization during carbonatite emplacement, probably before metasomatism and fenitization. At a later stage, this mineral assemblage became brecciated. Red-luminescing calcite constitutes, at least locally, the matrix of the so-formed breccia (as observed in sample S7/95.16-96.7). The δ18O and δ13C values of this calcite (22.1 ‰ vs. SMOW and −1.5 vs. PDB, respectively) are significantly displaced from primary carbonatitic values. They point to the crystallization of calcite at medium (150–300 °C; Decrée et al. 2015) temperatures during hydrothermal alteration, together with the formation of calcitic veins that crosscut the carbonatite (trend 2 in Fig. 7). Its high δ13C value points to a contribution of marine-derived carbon, which was likely incorporated in the hydrothermal fluid as a result of interaction with country rock.
Early supergene carbonate-fluorapatite
In the breccia matrix, carbonate-fluorapatite is coeval with siderite. The siderite’s δ18O and δ13C values (25.4 to −27.7 and −9.2 to −12.4 ‰, respectively) differ from primary or hydrothermally altered carbonatites (Fig. 7) by their light C isotopic composition. These very low δ13C values may result from several different processes, the most probable being the incorporation of low δ13C organic CO2 derived from the surface through the circulation of meteoric/groundwater fluids. The incorporation of low δ13C carbon in the fluid by interaction with organic matter-rich metasedimentary rocks cannot be excluded. Nevertheless, the genetic relationship with fluids from the surface is confirmed by the coexistence of iron hydroxides with siderite at the microscale, because iron hydroxides form under oxidizing conditions as encountered near the surface. The high δ18O value of siderite (near 26–27 ‰) is consistent with the precipitation at near-surface temperature. For example, siderite formed under meteoric water conditions (δ18O between −1 and −5 ‰) and temperatures between 30 and 55 °C (estimate using the siderite-H2O fractionation factor of Zheng 1999) would acquire a δ18O value of 26–27 ‰. The involvement of such a low-temperature and low δ13C surface fluid is obviously not connected to the carbonatitic/magmatic event.
Considering that surface fluids may induce the partial dissolution of the magmato-hydrothermal carbonates hosting the primary fluorapatite, we can ascribe the brecciation of the primary magmato-hydrothermal facies to groundwater circulation, triggering the carbonate-fluorapatite precipitation in close association with the sideritic breccia matrix. At Juquiá, a weathering profile developed on a phosphate-rich carbonatite. The carbonate-fluorapatite crystals there are comparable and have been referred to as the first secondary supergene apatite forming after the primary magmatic fluorapatite (Walter et al. 1995). Moreover, at Matongo, the Ba-Sr-rich pyrochlore (coeval with siderite) crystallizing as euhedral crystals in the matrix is considered as a good indicator of supergene processes. By comparison, pyrochlore rich in U-Pb indicates the involvement of magmatic processes in alkaline complexes (Chakhmouradian 2006; Bambi et al. 2008; Mitchell 2014; Nasraoui and Bilal 2000).
In the studied samples, carbonate-fluorapatite is commonly found in association with monazite, developed at the LREE-rich rim of magmatic fluorapatite (as shown in Fig. 3g). Corrosion/replacement textures suggest that monazite and carbonate-fluorapatite form partly at the expense of the primary phosphate. Extensive dissolution of primary (i.e., magmatic) apatite in the uppermost part of the weathering profile is well-known from similar carbonatitic phosphate ores (e.g., Walter et al. 1995). Moreover, in several natural occurrences and experiments, fluorapatite, in association with monazite, has been suggested to form from a precursor fluorapatite (Harlov et al. 2005 and references therein). The nucleation of monazite in apatite involves the reaction (Harlov et al. 2002):
In our study, apatite 1 corresponds to the blue-violet-luminescing LREE-enriched fluorapatite and apatite 2 to the recrystallized carbonate-fluorapatite showing a slightly decreased/lower Si content compared to fluorapatite 1 (see Fig. 4b). The partial dissolution of phosphate and carbonate led to the presence of P, Ca, and CO3 2− ions in the fluid phase and to the formation of secondary corrosion cavities. The related increase of the pH also decreased the solubility of REE (e.g., Gieré 1990; Wood 1990), favoring monazite precipitation. Similarly, a decrease in pressure in open pore space decreases the solubility of monazite, whereas it increases the solubility of apatite (Ayers and Watson 1991; Gieré 1996). This could explain why monazite precipitated before carbonate-fluorapatite (as observed on Fig. 3g) at the dissolved rim of primary fluorapatite. In addition, the common microcrystalline shapes of carbonate-fluorapatite suggest a highly saturated environment where many crystallization nuclei are present (Bonev 1972). Finally, after the precipitation of hydrogen-free salts (as Ca5(PO4)3F), the fluids, previously enriched in HCO3− and H2PO4 −, became more acidic, resulting in an enhanced corrosion and permeability of the rock (Andrade et al. 1999).
Late supergene fibrous fluorapatite and hardened phosphorite
Secondary green-luminescing fibrous fluorapatite forms coatings around primary fluorapatite grains and, in association with iron hydroxide crusts, forms hard lithified phosphorite. It formed during the final stages of ore genesis in remaining voids. Similar features (i.e., colloform and fibrous fluorapatite forming hardened phosphorite) are common in phosphate deposits related to alkaline complexes (Davies 1956; Walter et al. 1995; Vartiainen and Paarma 1979) and typically result from supergene alteration (De Toledo et al. 2004; Mariano 1989; Vartiainen and Paarma 1979). By analogy, the fibrous fluorapatite in the Matongo phosphate ore is interpreted in the same way.
Aluminum phosphates, commonly reported in a near-surface supergene environment above carbonatites (Barros de Oliveira and Liguori Imbernon 1998; Freyssinet et al. 2005), have not been observed in the Matongo samples. This is probably due to the fact that our samples were taken at deep levels (from ~95 to ~124 m below the present surface). Indeed, closer to the surface, an association of crandallite, goyazite, and wavellite has been reported in Matongo (Kurtanjek and Tandy 1989; Midende 1984).
Economic potential of the phosphate deposit at Matongo
The economic potential of the phosphates in the breccia lenses overlying the Matongo carbonatite has been assessed by Kurtanjek and Tandy (1989). Enrichments in Nb, V, and REE are observed in the phosphate deposit.
Two generations of pyrochlore (magmatic U-Pb rich and supergene Ba-Sr rich) can be identified in the Matongo phosphate ore. The average Nb content is ~350 ppm (~500 ppm Nb2O5), corresponding to about 1.3 times the original content in the primary carbonatite. The concentration is comparable to the Nb enrichment of the phosphate horizons at Catalão, in Brazil (350 to ca. 5000 ppm; Barros de Oliveira and Liguori Imbernon 1998). It is, however, far below the well-known Nb deposits of Lueshe, in DRC (a weathered cap containing an average of 1.34 wt% Nb2O5; Notholt et al. 1990), or Mabounie, in Gabon (Nb enrichment up to 1.5 wt% in the phosphate-rich horizons; Edou-Minko 2003). Vanadium (average concentration of ~890 ppm V2O5) occurs mainly in clasts of magnetite and is concentrated in the phosphate deposit compared to the primitive mantle (values taken from McDonough et al. 1992).
Rare earth elements are concentrated by a factor of 3 to 4 compared to the primary carbonatite. This level of concentration is comparable to Catalão (0.15–0.75 wt% REE2O3; Barros de Oliveira and Liguori Imbernon 1998) or Mabounie (~600 ppm Ce; Edou-Minko 2003). Most of the Matongo samples are characterized by P2O5 contents varying from 8.08 to 14.89 wt% (average content of 11.7 wt% P2O5), with corresponding REE2O3 contents ranging between 2025 and 5486 ppm (average content of ~3720 ppm REE2O3; samples RG 140281, S2/97.8-98 A, S2/97.8-98 B, S2/99-100, S2/100-101 A, S2/100-101 B, S2/101-106, and S2/117). These samples are most likely comparable to the “rich ore” at Matongo, which averages 11 wt% P2O5 and constitutes 17.3 Mt of the reserves (Notholt 1999, in DFID 2002). In addition, 40 Mt of less enriched phosphate ore is present, grading around 4.6 wt% P2O5 (Notholt 1999, in DFID 2002). The reference values obtained on our samples (11.7 wt% P2O5 with a corresponding content of ~3720 ppm REE2O3) are used to roughly estimate the REE reserves at Matongo. Based on this data (17.3 Mt at 11 wt% P2O5, with an estimated REE content of ~3500 ppm REE2O3, and 40 Mt at 4.6 wt% P2O5, with an estimated REE content of ~1500 ppm REE2O3), the REE reserves could approximate ~120,000 t of REE2O3 in the phosphate deposit. A large part of the REE must be present in fluorapatite that contains up to 1.3 wt% LREE2O3, even though monazite, pyrochlore, and possibly Fe oxides also host REE.
The exploitation of the REE as by-product of the Matongo fluorapatite could be economically viable, as is the case in other alkaline massifs. For instance, in the Khibina and the Lovozero alkaline massifs (Kola Peninsula, Russia), one of the possible ways to increase the production of REE is to extract the REE from the apatite (containing ~1 wt% REE2O3 at Khibina and 5–6 wt% REE2O3 in Lovozero). This treatment could provide 200–450 additional tons of REE2O3 in the Lovozero massif alone (Zaitsev and Kogarko 2012). Recent studies actually show that the hydrochloric acid treatment of apatite related to carbonatites leads to high recovery rates of REE (~80 %; Pereira and Bilal 2012). In other types of phosphate deposits, the exploitation of REE as by-product of the apatite would be interesting as well. A particular case is that of the sedimentary phosphate deposits in the USA. Emsbo et al. (2015) recently proposed that the REE reserves in these deposits—and their exploitation as by-product of francolite (carbonate-fluorapatite)—could partially resolve part of the world supply shortage in REE. Apart from the endowment of these deposits, the key factor of such prospects is the extractability of the REE from carbonate-fluorapatite (900–1200 ppm as median ΣREE values of pure francolite in US phosphate deposits). This extraction is relatively easy compared to most traditional REE prospects (Emsbo et al. 2015). In Iran (Esfordi and Lakeh Siyah areas), phosphate deposits related to metasomatism and containing apatite enriched in REE (up to 2.5 wt% REE) are also considered as targets for the exploitation of REE as by-product of the phosphate mine (Ghorbani 2013). Therefore, considering the REE potential reserves at Matongo (~120,000 t of REE2O3), assuming that a large part of it is comprised within fluorapatite, and considering the relatively easy extraction of REE from this mineral, the exploitation of REE as by-product of the Matongo phosphates could prove to be of economic interest.
Conclusions
The Matongo phosphate ore shows a diversity of fluorapatite mineralization stages, which are related to various processes ranging from carbonatitic/carbonaceous magmatism to supergene alteration.
In the Matongo carbonatite intrusion, the primary igneous formations are enriched in fluorapatite, which is associated with calcite (+ pyroxene) in the fine-grained sövite or with pyroxenes in cumulate facies (Midende et al. 2014). This magmatic fluorapatite is characterized by several growth zones (highlighted by different CL colors, with a last generation enriched in LREE) and occurs as aggregates of up to 3 mm in size. After an episode of metasomatism-fenitization, which led to the formation of K-feldspar and albite, the fluorapatite-rich formations were partly brecciated. Stable isotope analyses of the calcite that constitutes the breccia matrix suggest that the fluids responsible for this event are similar to the hydrothermal fluids that altered the carbonatite soon after its emplacement.
The circulation of surface waters or groundwater, unrelated to the magmatic history of the URAPC, induced carbonate dissolution, brecciation, and corrosion of the fluorapatite. Siderite (finely admixed with Fe oxides) and carbonate-fluorapatite forms the matrix of the resulting breccia. This supergene event led to the concentration of the heavy minerals, among them fluorapatite, pyrochlore, and V-rich magnetite, in eluvial horizons (here lenses overlying the Matongo carbonatite body). This event is also responsible for the partial weathering of several minerals, such as magmatic fluorapatite and U-Pb-rich pyrochlore. These became corroded and recrystallized to a carbonate-fluorapatite + monazite assemblage and Sr-Ba-rich pyrochlore, respectively. Finally, at a late stage of supergene weathering, fibrous fluorapatite crystallized in the remaining voids to form lithified phosphorite. The latter represents the most evolved facies linked to these processes. In the East African Rift, denudation events and associated weathering are known during the Triassic, at the Jurassic-Cretaceous transition, and during the Paleogene (Delvaux et al. 2012; Van der Beek et al. 1998). At the margin of the Congo River Basin, however, the Neogene seems to be a likely period for a weathering/lateritization episode and the subsequent formation of secondary ore (De Putter et al. 2013; Decrée et al. 2010). From an economic point of view, the subeconomic REE concentrations of the Matongo deposit are potential by-products if and when the phosphate ore becomes exploited.
References
Alves PR (2008) The carbonatite-hosted apatite deposit of Jacupiranga, SE Brazil: styles of mineralization, ore characterization, and association with mineral processing. Unpublished PhD thesis, Missouri University of Science and Technology
Andrade FRD, Möller P, Lüders V, Dulski P, Gilg HA (1999) Hydrothermal rare earth elements mineralization in the Barra do Itapirapuã carbonatite, southern Brazil: behaviour of selected trace elements and stable isotopes (C, O). Chem Geol 155:91–113
Ayers JC, Watson EB (1991) Solubility of apatite, monazite, zircon, and rutile in supercritical aqueous fluids with implications for subduction zone geochemistry. Philos Trans R Soc Lond Ser A Phys Eng Sci 335:365–375
Bambi AJCM, Costanzo A, Malgarejo JC, Conçalves AO, Alfonso P, Buta Neto A, Manuel J (2008) Evolution of pyrochlore in carbonatites: the Angola case. Revista de la sociedad española de mineralogía, pp 43–44
Barbarand J, Pagel M (2001) Cathodoluminescence study of apatite crystals. Am Mineral 86:473–484
Barros de Oliveira SM, Liguori Imbernon RA (1998) Weathering alteration and related REE concentration in the Catalão I carbonatite complex, central Brazil. J S Am Earth Sci 11:379–388
Bonev I (1972) On the terminology of the phenomena of mutual crystal orientation. Acta Crystallogr Sect A Cryst Phys, Diffr, Theor Gen Crystallogr 28:508–512
Boulvais P, Cobert C, Baele JM, Tack L, Nimpagaritse G, Gardien V, Demaiffe D, Decrée S (2015) A syntectonic alkaline massif in Burundi: geometry, fluid-rock interaction and element (REE, HFSE) mobility. 13th SGA biennal meeting, Nancy, France. Abstract book
Bouzari F, Hart C, Barker S, Bissig T (2014) Porphyry indicator minerals (PIMS): a new exploration tool for concealed deposits in south-central British Columbia. University of British Columbia. Geoscience BC Report 2011–17, 31 p
Carothers WW, Lanford HA, Rosenbauer RJ (1988) Experimental oxygen isotope fractionation between siderite-water and phosphoric acid liberated CO2-siderite. Geochim Cosmochim Acta 52:2445–2450
Chakhmouradian AR (2006) High-field-strength elements in carbonatitic rocks: geochemistry, crystal chemistry and significance for constraining the sources of carbonatites. Chem Geol 235:138–160
Davies KA (1956) The geology of part of south-east Uganda, with special reference to the alkaline complexes. Geol Surv Uganda Mem 8:62–76
De Putter T, Mees F, Ruffet G, Delvaux D, Decrée S, Dewaele S, Nimpagaritse G, Nkurunziza P, Tack L (2013) Exploring the Neogene as a period for supergene ore formation and exposure in the Great Lakes Region (Wester Great Rift Valley). 24th Colloquium of African Geology, Addis Ababa, Ethiopia. Abstract book: p 140
De Toledo MCM, Lenharo SL, Ferrari VC, Fontan F, De Parseval P, Leroy G (2004) The compositional evolution of apatite in the weathering profile of the Catalão I alkaline-carbonatitic complex, Goiás, Brazil. Can Mineral 42:1139–1158
Deans T (1966) Economic mineralogy of African carbonatites. In: Tuttle OF, Gittins J (eds) Carbonatites. Interscience Wiley, New York, pp 385–413
Decrée S, Deloule É, Ruffet G, Dewaele S, Mees F, Marignac C, Yans J, De Putter T (2010) Geodynamic and climate controls in the formation of Mio–Pliocene world-class oxidized cobalt and manganese ores in the Katanga province, DR Congo. Mineral Deposits 45(7):621–629
Decrée S, Boulvais P, Cobert C, Baele JM, Midende G, Gardien V, Tack L, Nimpagaritse G, Demaiffe D (2015) Structurally-controlled hydrothermal alteration in the Upper Ruvubu Alkaline Plutonic Complex (Burundi): implications for REE and HFSE mobilities. Precambrian Res 269:281–295
Delvaux D, Kervyn F, Macheyeki AS, Temu EB (2012) Geodynamic significance of the TRM segment in the East African Rift (W-Tanzania): active tectonics and paleostress in the Ufipa plateau and Rukwa basin. J Struct Geol 37:161–180
Demaiffe D (2008) Le magmatisme alcalin et carbonatitique: synthèse sur la province paléozoique de Kola (Russie) et caractéristiques générales du massif protérozoique de Matongo (Burundi). Bull Séances Acad R Sci O-M 54:171–196
Edou-Minko A (2003) Geochemical profile of the Mabounie niobiferous residual deposit (Gabon). J Sci 3:56–62
Emsbo P, McLaughlin PI, Breit GN, du Bray EA, Koenig AE (2015) Rare earth elements in sedimentary phosphate deposits: solution to the global REE crisis? Gondwana Res 27:776–785
Fagel N, André L, Debrabant P (1997) Multiple “seawater” derived geochemical signature in the Indian Ocean pelagic clays? Geochim Cosmochim Acta 61:989–1008
Fernandez-Alonso M, Cutten H, De Waele B, Tack L, Tahon A, Baudet D, Barritt SD (2012) The Mesoproterozoic Karagwe-Ankole Belt (formerly the NE Kibara Belt): the result of prolonged extensional intracratonic basin development punctuated by two short-lived far-field compressional events. Precambrian Res 216:63–86
Freyssinet P, Butt CRM, Morris RC, Piantone P (2005) Ore-forming processes related to lateritic weathering. Econ Geol 100th anniversary volume, pp 681–722
Ghorbani M (2013) The economic geology of Iran mineral deposits and natural resources. Springer Geology, Springer, 569 p
Gieré R (1990) Hydrothermal mobility of Ti, Zr and REE: examples from the Bergell and Adamello contact aureoles (Italy). Terra Nov. 2:60–67
Gieré R (1996) Formation of rare earth minerals in hydrothermal systems. In: Jones AP, Wall F, Williams CT (eds) Rare earth minerals, vol 7, The mineralogical series. Chapman & Hall, London, pp 105–150
Götte J (2009) Petrological modifications in continental target rocks from terrestrial impact structures: evidence from cathodoluminescence. In: Gucsik A (ed) Cathodoluminescence and its application in the planetary sciences, pp 45–60
Götze J, Kempe U (2009) Physical principles of cathodoluminescence and its applications to geosciences. In: Gucsik A (ed) Cathodoluminescence and its application in the planetary sciences. Springer, Heidelberg, pp 1–22
Habermann D, Neuser RD, Richter DK (2000) Quantitative high resolution spectral analysis of Mn2+ in sedimentary calcite. In: Pagel M, Barbin V, Blanc P, Ohnenstetter D (eds) Cathodoluminescence in geosciences. Springer, Heidelberg, pp 331–358
Harlov DE, Andersson UB, Förster HJ, Nyström JO, Dulski P, Broman C (2002) Apatite-monazite relations in the Kiirunavaara magnetite-apatite ore, northern Sweden. Chem Geol 191:47–72
Harlov DE, Wirth R, Förster HJ (2005) An experimental study of dissolution-reprecipitation in fluorapatite: fluid infiltration and the formation of monazite. Contrib Miner Petrol 150:268–286
Hayward CL, Jones AP (1991) Cathodoluminescence petrography of Middle Proterozoic extrusive carbonatite from Qasiarsuk, South Greenland. Min Mag 55:591–603
Kempe U, Götze J (2002) Cathodoluminecence (CL) behaviour and crystal chemistry of apatite from rare-metal deposits. Min Mag 66:151–172
Kurtanjek MP, Tandy BC (1989) The igneous phosphate deposits of Matongo-Bandaga, Burundi. In: Notholt AJG, Sheldon RP, Davidson DF (eds) Phosphate deposits of the world, vol 2, Phosphate rock resources. Cambridge University Press, Cambridge, pp 262–266
Lypaczewski P, Normandeau PX, Paquette J, McMartin I (2013) Petrographic and cathodoluminescence characterization of apatite from the Sue-Dianne and Brooke IOCG mineralization systems, Great Bear magmatic zone, Northwest Territories. Geol Surv Can, Open File 7319. doi:10.4095/292369
Mariano AM (1988) Some further geological application of cathodoluminescence. In: Marshall DJ (ed) Cathodoluminescence of geological materials. Unwin Hyman, Boston, pp 94–123
Mariano AN (1989) Nature of economic mineralization in carbonatites and related rocks. In: Bell K (ed) Carbonatites: genesis and evolution. Unwin Hyman, London, pp 149–176
Marshall DJ (1988) Cathodoluminescence of geological materials. Hyman, Boston, 146 p
McDonough WF, Sun SS, Ringwood AE, Jagoutz E, Hofmann AW (1992) Potassium, rubidium and cesium in the Earth and moon and the evolution of the mantle of the Earth. Geochim Cosmochim Acta 56:1001–1012
McLemore VT, Barker JM (1987) Some geological application of cathodoluminescence, examples from the Lemitar Mountains and Riley travertine, Socorro County, New Mexico. N M Geol, 37–40
Midende G (1984) La carbonatite de Matongo (Burundi). Unpublished PhD thesis, Université Libre de Bruxelles, Bruxelles
Midende G, Boulvais P, Tack L, Melcher F, Gerdes A, Dewaele S, Demaiffe D, Decrée D (2014) Petrography, geochemistry and U-Pb zircon age of the Matongo carbonatite Massif (Burundi): implication for the Neoproterozoic geodynamic evolution of Central Africa. J Afr Earth Sci 100:656–674
Mitchell RH (2014) Primary and secondary niobium mineral deposits associated with carbonatites. Ore Geol Rev 64:626–641
Nasraoui M, Bilal E (2000) Pyrochlores from the Lueshe carbonatite complex (Democratic Republic of Congo); a geochemical record of different alteration stages. J Afr Earth Sci 18:237–251
Nkurikiye L (1989) Le kaolin au Burundi: Contribution à sa caractérisation, sa genèse et son utilisation. Unpublished M. Sc. thesis, Burundi University
Notholt AJG (1999) Phosphate: a world monograph (a compilation of unfinished paper). British Geological Survey
Notholt AJG, Highley DE, Deans T (1990) Economic minerals in carbonatites and associated alkaline igneous rocks. Trans Inst Min Metall 99:B59–B80
Pell J (1996) Mineral deposits associated with carbonatites and related alkamine igneous rocks. In: Mitchell RH (ed) Undersaturated alkaline rocks: mineralogy, petrogenesis, and economic potential, vol 24, Short Course Series. Mineralogical Association of Canada, Winnipeg, pp 271–310
Penel G, Leroy G, Rey C, Sombret B, Huvenne JP, Bres E (1997) Infrared and Raman microspectrometry study of fluor-fluor-hydroxy and hydroxy-apatite powders. J Mater Sci 8:271–276
Penel G, Leroy G, Rey C, Bress E (1998) MicroRaman spectral study of the PO4 and CO3 vibrational modes in synthetic and biological apatites. Calcif Tissue Int 63:475–481
Pereira F, Bilal E (2012) Phosphoric acid extraction and rare earth recovery from apatites of the Brazilian phosphatic ores. Rom J Mineral Deposits 85:49–52
Songore T (1991) The Matongo phosphate deposits in Burundi. Fertil Res 30:151–153
Stormer JC, Pierson ML, Tacker RC (1993) Variation of F and Cl X-ray intensity due to anisotropic diffusion in apatite. Am Mineral 78:641–648
Sun SS (1982) Chemical composition and origin of the Earth’s primitive mantle. Geochim Cosmochim Acta 46:179–192
Swart PK, Burns SJ, Leder JJ (1991) Fractionation of the stable isotopes of oxygen and carbon in carbon dioxide during the reaction of calcite with phosphoric acid as a function of temperature and technique. Chem Geol 86:89–96
Tack L, De Paepe P, Deutsch S, Liégeois JP (1984) The alkaline plutonic complex of the Upper Ruvubu (Burundi): geology, age, isotopic geochemistry and implications for the regional geology of the Western rift. In: Klerkx J, Michot J (eds) African geology, Tervuren, pp 91–114
Tack L, Liégeois JP, André L, Navez J (1995) The Upper Ruvubu alkaline plutonic complex (Burundi). Annual Report of the Royal Museum for Central Africa, Tervuren, p 9
Tsuda H, Arends J (1994) Orientational micro-Raman spectroscopy on hydroxyapatite single crystals and human enamel crystallites. J Dent Res 73:1703–1710
Van der Beek P, Mbede E, Andriessen P, Delvaux D (1998) Denudation history of the Malawi and Rukwa Rift flanks (East African Rift System) from apatite fission track thermochronology. J Afr Earth Sci 26(3):363–385
Van Straaten (2002) Rocks for crops. Agrominerals of sub-Saharan Africa. International Centre for Research in Agroforestry, Nairobi, p 338
Vartiainen H, Paarma H (1979) Geological characteristics of the Sokli Carbonatite Complex, Finland. Econ Geol 74:1296–1306
Verwoed WJ (1986) Mineral deposits associated with carbonatites and alkaline rocks. In: Anhaeusser C, Maske S (eds) Mineral deposits of Southern Africa. Geol Soc S Afr, pp 2173–2191
Walter AV, Nahon D, Flicoteaux R, Girard JP, Melfi A (1995) Behaviour of major and trace elements and fractionation of REE under tropical weathering of a typical apatite-rich carbonatite from Brazil 136:591–602
Waychunas GA (2002) Apatite luminescence. In: Kohn MJ, Rakovan J, Hughes JM (eds) Reviews in mineralogy and geochemistry, vol 48, Phosphates. Mineralogical Society of America, Washington, pp 701–742
Wood SA (1990) The aqueous geochemistry of the rare-earth elements and yttrium: 1. Review of available low-temperature data for inorganic complexes and the inorganic REE speciation of natural waters. Chem Geol 82:159–186
Woolley AR (2001) Alkaline rocks and carbonatites of the world. Part 3: Africa. Geological Society of London, 372 p
Zaitsev V, Kogarko L (2012) Sources and perspectives of REE in the Lovozero massif (Kola Peninsula, Russia). European Mineralogical Conference. Abstract book, vol. 1, EMC2012-290, http://meetingorganizer.copernicus.org/EMC2012/EMC2012-290.pdf
Zheng YF (1999) Oxygen isotope fractionation in carbonate and sulfate minerals. Geochem J 33:109–126
Acknowledgments
The authors would like to warmly thank the MRAC for providing the study samples. Laurence Monin (MRAC) is thanked for her role in providing whole-rock analyses. Rénovat Nyandwi is thanked for his help and the preparation of the polished section. Chris Harris and an anonymous reviewer are sincerely thanked for their help to improve the quality of the manuscript. The authors are also grateful to Hartwig Frimmel and Bernd Lehmann for the editorial handling of this paper. Thomas Goovaerts is thanked for the English review of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editorial handling: Lawrence P. Rozas
Electronic supplementary material
Below is the link to the electronic supplementary material.
Electronic supplementary material 1
Representative cathodoluminescence spectra of minerals in the Matongo phosphatic ore (operating conditions: 15 kV–500 μA, defocussed beam of approx. 4 mm diameter, spectra not corrected for system response). Upper spectra: Mn2+-activated calcite and Fe3+-activated albite and K-feldspar. The observed luminescence color is orange, red and pink, respectively. Lower spectra: Mn2+- and REE-activated primary fluorapatite (dominant blue-CL color) and Mn2+-activated secondary fluorapatite (green CL). The spectrum for the primary fluorapatite is representative for an average of its emission (blue-green and red-CL core and blue-violet rim). (JPEG 639 kb)
Electronic supplementary material 2
Raman spectra obtained on magmatic fluorapatites (a) and hydrothermal-supergene fluorapatites (b) from the Matongo phosphate deposit. Acquired using λ = 532 nn, power: 2 mW, acquisition time: 5 × 30 s, spectral range: 0–4500 cm−1. Peaks that are not directly related to the fluorapatite structure are present on the Raman spectra at the following frequencies (presented by decreasing intensities): ~2080 cm−1, ~3290 cm−1, in the 2500–2700 cm−1 range, in the 1750–1820 cm−1, and in the 1480–1530 cm−1 range. When the Raman shift is converted to wavelength (in nm), these unresolved peaks correspond fairly well to the REE emission lines. The most important are those of Sm3+ (at ~599, 644 and 652 nm), others would correspond to Eu3+ (multiple peaks in the 614–618 nm range), Tb3+ (one or two peaks at 587–588 nm), and Dy3+ (multiple peaks in the 577–579 m range). These wavelengths, which are typical of the fluorescence induced by the REE, can be related to the presence of these elements in the studied fluorapatite, what has been confirmed by in situ analyses on magmatic fluorapatites (Table 1) (PDF 2611 kb)
Table 1
Representative microprobe analyses (oxides in wt%) of apatite and monazite from the Matongo phosphate ore (XLSX 16 kb)
Table 2
Major element contents (in wt%), REE and trace elements (in ppm) of samples from the Matongo phosphate ore (XLSX 18 kb)
Rights and permissions
About this article

Cite this article
Decrée, S., Boulvais, P., Tack, L. et al. Fluorapatite in carbonatite-related phosphate deposits: the case of the Matongo carbonatite (Burundi). Miner Deposita 51, 453–466 (2016). https://doi.org/10.1007/s00126-015-0620-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00126-015-0620-1