
Abstract
Aims/hypothesis
Variations in FTO are the strongest common genetic determinants of adiposity, and may partly act by influencing dopaminergic signalling in the brain leading to altered reward processing that promotes increased food intake. Therefore, we investigated the impact of such an interaction on body composition, and peripheral and brain insulin sensitivity.
Methods
Participants from the Tübingen Family study (n = 2245) and the Malmö Diet and Cancer study (n = 2921) were genotyped for FTO SNP rs8050136 and ANKK1 SNP rs1800497. Insulin sensitivity in the caudate nucleus, an important reward area in the brain, was assessed by fMRI in 45 participants combined with intranasal insulin administration.
Results
We found evidence of an interaction between variations in FTO and an ANKK1 polymorphism that associates with dopamine (D2) receptor density. In cases of reduced D2 receptor availability, as indicated by the ANKK1 polymorphism, FTO variation was associated with increased body fat and waist circumference and reduced peripheral insulin sensitivity. Similarly, altered central insulin sensitivity was observed in the caudate nucleus in individuals with the FTO obesity-risk allele and diminished D2 receptors.
Conclusions/interpretation
The effects of variations in FTO are dependent on dopamine D2 receptor density (determined by the ANKK1 polymorphism). Carriers of both risk alleles might, therefore, be at increased risk of obesity and diabetes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The prevalence of obesity and diabetes is increasing dramatically worldwide. In addition to behavioural factors, such as changes in eating habits and lack of physical activity, hereditary factors strongly determine body weight [1]. Variations in the fat mass and obesity-associated gene (FTO) are currently the strongest known genetic factors that predispose humans to non-monogenic obesity [2–4]. Carriers of the risk allele are more liable to have a higher BMI due to increased energy intake [5, 6] and decreased satiety [7]. Underlying mechanisms may include altered postprandial responses in reward- and appetite-regulating brain areas [8, 9], and impaired postprandial suppression of the hunger hormone ghrelin [9]. In addition, FTO polymorphisms are a common genetic determinant for type 2 diabetes, particularly via their effects on adiposity [2, 10].
Interestingly, recent data indicate that FTO variations determine mitochondrial activity via a network that involves expressional changes in IRX3 and IRX5 [11, 12]. In addition to associations with adipocyte function [12], FTO polymorphisms influence the expression of IRX3 in the human brain [11]. FTO variations have also been shown to coincide with altered regulation of a molecular network including RPGRIP1L in neuronal cells that ultimately leads to altered cellular function and potentially promotes adiposity [13].
Recent work in rodent models suggests that variations in Fto may be linked to increased food intake and obesity through an influence on dopamine signalling [14]. Dopamine neurons express Fto. Conditional loss of Fto in dopamine neurons specifically impairs dopamine receptor type 2/3 (D2/D3)-mediated neuronal activation. Accordingly, Fto-deficient mice show altered D2/D3 receptor-dependent regulation of locomotor activity and reward sensitivity. These findings also highlight the possibility that Fto influences food intake by altering D2 receptor-dependent feeding behaviour.
In terms of metabolism, dopamine signalling is crucial for the regulation of whole-body insulin sensitivity [15]. Deep brain stimulation of dopaminergic areas in the brain modulates glucose metabolism and body weight in rodents [16, 17]. Accordingly, the dopamine receptor agonist bromocriptine improves insulin sensitivity and induces weight loss in humans [18], and is therefore approved for the treatment of type 2 diabetes in some countries [18]. On a cellular level, there is clear evidence for an interaction between insulin and dopamine signalling [19–21] in the regulation of reward and food intake [22, 23].
Therefore, one possible hypothesis is that variations in FTO affect dopaminergic regulation of insulin sensitivity, thereby influencing body weight. Indeed, the FTO risk allele is linked to central insulin resistance [24]. Furthermore, there is evidence that central insulin regulates peripheral insulin sensitivity [25–29]. However, the well-known associations of FTO variants with whole-body insulin sensitivity [30–32] and diabetes risk [2, 10] are mainly driven by associations with BMI [2, 10, 31].
Alterations in dopamine signalling via D2 receptors are linked to common genetic variations. The TaqIA restriction fragment length polymorphism (rs1800497) is located in the ankyrin repeat and protein kinase domain-containing protein (ANKK1) gene approximately 9 kb downstream of the D2 receptor gene [33]. In candidate gene studies, carriers of the risk allele (T allele) show diminished D2 receptor density [34, 35], reduced D2 receptor binding potential [36] and an increased risk of substance abuse [37]. With regard to food intake, carriers of the ANKK1 risk allele have reduced responses to palatable and energy-dense food in brain regions involved in reward processing [38]. While the polymorphism is not a major determinant of body weight per se [39], carriers of the risk allele have difficulty losing or maintaining body weight [39, 40]. Specifically, an attenuated response to food in the striatum of risk-allele carriers was a predictor of further weight gain [41]. A recent study in humans identified an interaction between FTO and ANNK1 polymorphisms that determines midbrain activity during reward learning [42].
Since there is evidence that both FTO and ANKK1 confer risk for obesity by their influence on D2 receptors, we investigated whether common variations in these two genes interact to increase the risk for high body weight, unfavourable body fat distribution, altered glucose metabolism and decreased insulin sensitivity of the whole body, as well as of the brain, specifically the striatum (including the putamen, pallidum and caudate nucleus). Such an interaction would be relevant given that approximately 30% of all populations carry at least one risk allele of both variants (mean minor allele frequency of FTO and ANKK1 in various populations is approximately 33% according to 1000 genomes data [www.1000genomes.org; accessed 20 May 2015]). Moreover, the presence of an interaction would highlight the importance of the role of FTO in dopamine signalling in humans.
Methods
Participants of the Tübingen Family cohort
The Tübingen Family (TUEF) study is an ongoing project that includes Europid volunteers from southern Germany who have an elevated risk of type 2 diabetes but do not have manifest diabetes [28]. Subjects are recruited by personal contact as well as via local advertisements, for example in newspapers. All participants are metabolically characterised by a 75 g OGTT (see details below). Less than 5% of the cohort are related. Exclusion criteria include major medical and psychiatric diseases, as well as treatment with medication for weight loss or blood glucose control. Informed written consent is obtained from all participants, and the local ethics committee have approved the protocol.
We included 2245 TUEF study participants with a BMI <40 kg/m2 in the current analyses. Based on ADA criteria for glucose tolerance, 1588 had normal glucose tolerance, 224 had impaired fasting glucose, 189 had impaired glucose tolerance, 124 displayed both impaired fasting glucose and impaired glucose tolerance, and 120 had type 2 diabetes. The clinical characteristics of the TUEF study population are presented in Table 1. For all participants, total body fat and lean body mass were measured by bioelectrical impedance (BIA-101; RJL Systems, Detroit, MI, USA).
Participants of the MDC CA reinvestigation cohort
The Malmö Diet and Cancer (MDC) study is a population-based cohort collected in Malmö, southern Sweden. Between 1991 and 1996, 30,000 individuals born between 1923 and 1950 were recruited to the study, from which, 6103 non-related individuals were randomly selected to take part in the MDC CA (cardiovascular arm) study. All selected individuals who were alive and had not emigrated from Sweden (n = 4924) were invited to a re-examination starting in 2007. Of these, 3734 accepted and underwent a standard 75 g OGTT. All participants provided written informed consent, and the study was approved by the Lund University ethics review committee.
Genotyping
For the TUEF cohort, DNA from whole blood was isolated with a commercial DNA isolation kit (NucleoSpin, Macherey – Nagel, Düren, Germany). Genotyping for FTO SNP rs8050136 and ANKK1 SNP rs1800497 was performed by mass spectrometry (Sequenom, Hamburg, Germany). The genotypes validated by bidirectional sequencing in 50 participants were 100% identical. Minor allele frequencies were 43% for the FTO A risk allele and 19% for the ANKK1 T risk allele. Both SNPs were in Hardy–Weinberg equilibrium (HWE; FTO SNP p = 0.7, ANKK1 SNP p = 0.2).
The MDC cohort was genotyped at the Broad genotyping facility (Cambridge, MA, USA) using the Illumina OmniExpressExome Beadchip v1.0 B array (Illumina, San Diego, CA, USA). Genotypes were called using the Gencall algorithm in Genomestudio (Illumina) with refinement of rare variants with missing calls undertaken using zCall [43]. Standard quality control filters were applied. In brief, SNPs were excluded if they were monomorphic, had a low call rate (<99%) or deviated from HWE (p < 10−4). Samples were excluded if they were duplicates, unexpected first/second relatives, sex mismatched, heterozygosity outliers (>3 SD from mean), ancestral outliers or had a low call rate (<95%). No principal component adjustment was used since the population was homogeneous and participants were recruited at a single centre. Both SNPs were in HWE (FTO SNP p = 0.6, ANKK1 SNP p = 0.8).
OGTT
A 75 g OGTT was performed as previously described [44]. In the TUEF participants, glucose, insulin and C-peptide were measured five times during the experiment (0, 30, 60, 90 and 120 min). For the MDC cohort, blood samples were obtained at 0, 30 and 120 min.
Hyperinsulinaemic–euglycaemic clamp
In a subgroup of 499 participants, insulin sensitivity was assessed using a hyperinsulinaemic-euglycaemic glucose clamp as previously described [44]. The clamp-derived insulin sensitivity index was calculated by dividing the mean glucose infusion rate during steady state by the plasma insulin concentrations during this time [44].
Analytical procedures
In the TUEF study, plasma glucose was measured by the glucose-oxidase method (Yellow Springs Instruments, Yellow Springs, OH, USA). Insulin and C-peptide concentrations were measured using commercial chemiluminescence assays for ADVIA Centaur (Siemens Medical Solutions, Fernwald, Germany). In the MDC study, plasma glucose was measured using a glucose dehydrogenase method (Hemocue, Ängelholm, Sweden) and serum insulin was measured using ELISA (Dako, Glostrup, Denmark).
fMRI
Forty-five healthy volunteers (19 women, 26 men; mean age 26.4 ± 3.4 years) underwent fMRI to assess regional insulin action in the brain. They were retrospectively genotyped for the two SNPs of interest. Eleven individuals were homozygous for both non-risk major alleles (FTO rs8050136 and ANKK1 rs1800497), 20 were homozygous major allele carriers of the ANKK1 SNP and homozygous minor allele carriers of the FTO SNP, five were homozygous minor allele carriers of the ANKK1 SNP and homozygous for the FTO SNP major allele, and nine were homozygous for the minor alleles of both polymorphisms.
Experiments were conducted after an overnight fast of at least 10 h and commenced at 07:00 hours with a ‘resting state’ fMRI measurement under basal conditions. Following the basal measurement, 160 U of human insulin was administered via intranasal spray as previously described [28, 45]. After 30 min, a second ‘resting state’ fMRI measurement was performed.
fMRI data acquisition
Whole-brain fMRI data was obtained with a 3.0 T scanner (Siemens Tim Trio, Erlangen, Germany). Functional data were collected by echo-planar imaging sequence: repetition time 2 s, echo time 30 ms, field of view 210 mm2, matrix 64 × 64, flip angle 90°, voxel size 3.3 × 3.3 × 3.6 mm3, slice thickness 3.6 mm, 1 mm gap and the images were acquired in ascending order. Each brain volume comprised 26 axial slices and each functional run contained 176 image volumes, resulting in a total scan time of 6.04 min. In addition, high-resolution T1 weighted anatomical images (MPRage: 192 slices, matrix: 256 × 224, 1 × 1 × 1 mm3) of the brain were obtained.
Fractional amplitude of low-frequency fluctuation
Low-frequency (0.01–0.08 Hz) fluctuations (LFF) of the blood oxygenation level-dependent (BOLD) signal in resting state fMRI data are believed to reflect intrinsic neural activity. We carried out the fractional amplitude of LFF (fALFF) analysis on the preprocessed functional data using REST (http://resting-fmri.sourceforge.net), as previously described [45]. The fALFF maps of the basal fMRI measurement were subtracted from the fALFF maps of the 30 min fMRI measurement for each participant. We took a region-of-interest approach by using the WFU PickAtlas tool (http://fmri.wfubmc.edu/software/PickAtlas, accessed 13 February 2015) to extract mean fALFF values of the striatum, a critical component of the dopaminergic reward system. Areas of the striatum include the caudate nucleus, putamen and pallidum. The extracted fALFF values were then further analysed in a linear regression model.
Calculations
Insulin sensitivity during the OGTT was estimated as 10,000/(G0 × I0 × Gmean × Imean)1/2, where G = glucose (in mmol/l) and I = insulin (in pmol/l). AUC was calculated according to the trapezoid method.
Statistical analysis
Data that were not normally distributed were log e -transformed to approximate a normal distribution prior to analysis. ANCOVA was used to identify interactions between SNPs. Associations were analysed by multiple linear regression analysis. A p value <0.05 was considered to be statistically significant; a p value <0.1 was considered to indicate a trend. The statistical software package JMP (SAS Institute, Cary, NC, USA) was used for statistical analysis.
Results
Associations with body composition and metabolic traits in the TUEF cohort
Interactions between the FTO SNP rs8050136 and ANKK1 SNP rs1800497 on body composition and metabolic traits were investigated by ANCOVA for both polymorphisms. There were interactions between the two SNPs for percentage body fat, waist circumference and waist-to-hip ratio, as well as for the insulin sensitivity index and fasting insulin in models adjusted for sex and age (all p < 0.05, Table 2). No significant interactions were detected for lean body mass, fasting and post-load glycaemia, or insulin secretion (assessed as insulin30min and AUCC-peptide/AUCglucose). Repeating the analyses in unadjusted models revealed comparable results (all p < 0.05), except for waist-to-hip ratio and the insulin sensitivity index which only showed trends towards significance (p = 0.1 and p = 0.08, respectively).
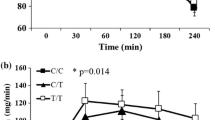
The group was then stratified according to ANKK1 rs1800497 genotype in homozygous carriers of the C allele vs those carrying at least one copy of the T risk allele (see Table 2). Associations of FTO rs8050136 were then tested for both ANKK1 groups separately. The FTO SNP was associated with body weight and BMI independent of the ANKK1 SNP; however, the effect sizes were markedly larger in carriers of the ANKK1 T allele than in CC allele carriers (Table 2). While the FTO SNP was not associated with body fat content or waist-to-hip ratio in homozygous carriers of the ANKK1 C allele (p for additive inheritance model [p add] 0.1, β 0.017 ± 0.012, Fig. 1a; p add 0.5, β −0.003 ± 0.004, respectively), associations were found in participants harbouring the T allele (p add 0.0006, β 0.056 ± 0.016, Fig. 1a; p add 0.03, β 0.017 ± 0.008, respectively). Furthermore, it was only against the ANKK1 T allele background that the FTO obesity-risk allele carriers had a lower insulin sensitivity index and higher fasting insulin than the FTO non-risk allele carriers (p add 0.0012, β −0.121 ± 0.037, Fig. 1b; p add 0.0002, β 0.126 ± 0.034, respectively). This association was not present in the ANKK1 SNP CC carriers (Table 2, Fig. 1b).

The obesity-risk polymorphism in FTO and the D2 receptor polymorphism in ANKK1 interact to affect (a) body fat content and (b) peripheral insulin sensitivity in the TUEF cohort. Participants were stratified according to their ANKK1 rs1800497 genotype in CC and XT carriers. FTO rs8050136 genotype is indicated by bar colour: white bars, FTO CC allele carriers; black bars, FTO XA obesity-risk allele carriers. With regard to body fat content, no association with the FTO SNP was found in ANKK1 CC carriers, while carriers of the FTO non-risk allele had lower body fat than obesity-risk allele carriers under the ANKK1 XT background. Accordingly, the FTO SNP was not associated with OGTT-derived peripheral insulin sensitivity index in ANKK1 CC carriers, while peripheral insulin sensitivity was better in FTO non-risk than in FTO obesity-risk allele carriers under the ANKK1 XT background. Data are sex- and age-adjusted means ± SEM. AU, arbitrary units; NS, non-significant (p > 0.05), ***p < 0.001
Associations with body composition and metabolic traits in the MDC cohort
In the MDC cohort, interactions between the ANKK1 and FTO polymorphisms were detected for waist circumference and waist-to-hip ratio (Table 3).
Corresponding with the results from the TUEF cohort, waist circumference was associated with the FTO polymorphism in participants with the ANKK1 T allele (p add 0.025, β 0.019 ± 0.008), while no such association was detected in ANKK1 CC allele carriers (p add 0.2, β −0.005 ± 0.006). Also the insulin sensitivity index and fasting insulin levels were only associated with the FTO SNP in ANKK1 T allele carriers (p add 0.02, β −0.084 ± 0.005; p add 0.04, β 0.078 ± 0.04, respectively), but not in participants with the ANKK1 CC allele (p add 0.7, β 0.027 ± 0.04; p add 0.9, β −0.001 ± 0.03, respectively; see Table 3).
Associations with insulin action in the caudate nucleus
Since FTO rs8050136 is known to be linked to brain insulin sensitivity [24], we investigated how the two polymorphisms relate to brain insulin action in the striatum, a major dopaminergic area in the brain in which the ANKK1 SNP is known to be important [41], following intranasal insulin administration.
To increase statistical power in this small group, we only tested dominant inheritance models. As for peripheral insulin sensitivity, we detected an interaction between the FTO rs8050136 and ANKK1 rs1800497 SNPs on insulin response in the caudate nucleus (ANCOVA p for dominant inheritance model [p dom] 0.0060). While the reaction to insulin did not differ between FTO genotypes in carriers of the ANKK1 CC alleles, it differed in the ANKK1 T allele carriers. Therefore, insulin reduced activity in the caudate nucleus in FTO non-risk allele carriers, but increased activity in obesity-risk allele carriers (Fig. 2).

Insulin sensitivity in the caudate nucleus of the striatum (a) is determined by the obesity-risk polymorphism in FTO and the D2 receptor polymorphism in ANKK1. Insulin effects on the caudate nucleus were assessed by fMRI combined with intranasal insulin administration in 45 healthy volunteers. (b) Insulin-induced change in caudate activity was determined and participants were stratified according to their ANKK1 rs1800497 genotype in CC and XT carriers. FTO rs8050136 genotype is indicated by bar colour: white bars, FTO CC allele carriers; black bars, FTO XA obesity-risk allele carriers. There was no association with the FTO SNP in ANKK1 CC carriers, whereas FTO differed in the striatal response to insulin under the ANKK1 XT background. Here, FTO non-risk allele carriers had a reduction in activity while FTO obesity-risk allele carriers showed an increase. This figure shows unadjusted means ± SEM. p values are taken from an unadjusted additive inheritance model. AU, arbitrary units; NS, non-significant (p > 0.05), *p < 0.05
Discussion
In this study, we have demonstrated an interaction between variations in FTO and the ANKK1/TaqIA polymorphism that affects body composition, as well as peripheral and central insulin sensitivity. While the FTO SNP was not associated with body fat distribution or insulin sensitivity in persons carrying the ANKK1 non-risk allele, it was associated with both variables in participants carrying the ANKK1/TaqIA polymorphism.
These findings, that were replicated in the independent MDC cohort, provide further evidence that variations in FTO influence dopamine signalling in humans in the same way as predicted from rodents [14]. Specifically, on the basis of the associations between ANKK1 variations and D2 receptor availability [34, 35], as well as D2 receptor binding potential in humans [36]. Our results indicate that the FTO polymorphism investigated here may be less critical when sufficient D2 receptors are available but it may have adverse effects when dopaminergic transmission is limited. Given the high frequency of both polymorphisms in the general population, a substantial number of people will carry the risk alleles for both polymorphisms, i.e. in our study population >20% of the participants carried both risk alleles. Our results demonstrate that these individuals will be at increased risk for obesity, insulin resistance and type 2 diabetes. Our data also highlight the possibility that in addition to other known environmental perturbations, the identified interaction contributes to the known association of the FTO polymorphism with both obesity and diabetes [2, 4].
Complementing our findings of peripheral markers of metabolism and obesity, our neuroimaging results show that the effect of the detected interaction between FTO and ANKK1 on insulin sensitivity is not limited to peripheral tissues, but that it also exists in the D2-receptor rich caudate nucleus. The ANKK1 genotype influenced D2 receptor binding and BOLD responses to the receipt of palatable and energy-dense food in this region [41, 46]. This is of particular interest since both BOLD response [47] and dopamine release [48] in the caudate nucleus following palatable food intake was related to ratings of meal pleasantness. This demonstrates the importance of D2 receptor signalling in the caudate nucleus in eating behaviour and food reward. Interactions between insulin and dopaminergic networks contribute to both these brain functions accordingly [20]. Even though brain insulin sensitivity was not specifically tested in animals lacking Fto in dopaminergic neurons [14], it is tempting to speculate that altered insulin responses contributed to the observed phenotype.
There are at least two plausible explanations as to how the detected interactions in the brain could lead to the observed peripheral associations with regard to body fat distribution and systemic metabolism. Dopamine signalling in the brain is a determinant of peripheral insulin sensitivity [18]. Accordingly, circulating prolactin, a proxy for dopaminergic tone, has an age-dependent association with peripheral insulin sensitivity and glycaemia [49]. Based on our current results, dopaminergic control over peripheral glucose metabolism could be affected by FTO genotype.
However, we observed that the known association of the FTO SNP rs8050136 with brain insulin resistance depends on functional dopamine signalling, as determined by the ANKK1 polymorphism. The FTO SNP was not associated with insulin responsiveness in the caudate nucleus in persons carrying the ANKK1 non-risk allele, while it was associated with regional brain insulin sensitivity in carriers of the risk allele. In addition to various other functions, such as memory and reward, insulin delivery to the brain induced weight loss [29, 50]. Brain insulin sensitivity even predicts loss of metabolically unhealthy visceral fat during a lifestyle intervention [51]. Furthermore, insulin in the brain was recently shown to modulate peripheral insulin sensitivity in humans [28, 29]. Impaired brain insulin signalling could therefore contribute to increased body fat content and increased waist circumference, as well as to reduced peripheral insulin sensitivity in carriers of both risk alleles. In addition, our results in combination with the finding of an altered midbrain response in participants carrying both polymorphisms during a reward learning task [42] indicate that the interaction of FTO and ANNK1 affects metabolic and behavioural brain networks.
The alterations that we observed in the brain in carriers of both risk alleles might trigger the alterations in peripheral metabolism, but we cannot rule out the possibility that the detected associations in regional brain activity are secondary to peripheral changes, e.g. altered body fat content. How FTO variations influence IRX3/5 or RPGRIP1L expression in dopaminergic neurons and how this relates to their functional properties has not yet been addressed. Further studies are required to gain a better understanding of the mechanisms underlying our results. Both cohorts that were analysed in this work only included individuals of European descent, and SNP rs8050136 may not be a good proxy for the association of the FTO locus with obesity in cohorts of different ethnicity. Therefore, our results cannot necessarily be transferred to another population. Thus, replication studies are needed, especially with participants of different genetic origin. Since we used a candidate gene approach, additional contributions of other genetic variants will be missed.
In summary, we report an interaction between the obesity-risk polymorphism in FTO and the ANKK1/TaqIA SNP indicating that the ∼20% of the population carrying both risk alleles have a markedly higher risk for obesity and diabetes. Our data also suggest that the ANKK1 risk allele may be a major contributor to the previously reported influence of FTO on obesity and metabolism.
Abbreviations
- BOLD:
-
Blood oxygenation level-dependent
- D2/D3:
-
Dopamine receptor type 2/3
- fALFF:
-
Fractional amplitude of low-frequency fluctuation
- HWE:
-
Hardy–Weinberg equilibrium
- LFF:
-
Low-frequency fluctuations
- MDC CA:
-
Malmö Diet and Cancer study cardiovascular arm
- p add :
-
p for additive inheritance model
- p dom :
-
p for dominant inheritance model
- TUEF:
-
Tübingen Family study
References
Walley AJ, Asher JE, Froguel P (2009) The genetic contribution to non-syndromic human obesity. Nat Rev Genet 10:431–442. doi:10.1038/nrg2594
Frayling TM, Timpson NJ, Weedon MN et al (2007) A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316:889–894. doi:10.1126/science.1141634
Dina C, Meyre D, Gallina S et al (2007) Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet 39:724–726. doi:10.1038/ng2048
Loos RJF, Yeo GSH (2014) The bigger picture of FTO: the first GWAS-identified obesity gene. Nat Rev Endocrinol 10:51–61. doi:10.1038/nrendo.2013.227
Haupt A, Thamer C, Staiger H et al (2009) Variation in the FTO gene influences food intake but not energy expenditure. Exp Clin Endocrinol Diabetes Off J Ger Soc Endocrinol Ger Diabetes Assoc 117:194–197. doi:10.1055/s-0028-1087176
Cecil JE, Tavendale R, Watt P et al (2008) An obesity-associated FTO gene variant and increased energy intake in children. N Engl J Med 359:2558–2566. doi:10.1056/NEJMoa0803839
Wardle J, Carnell S, Haworth CMA et al (2008) Obesity associated genetic variation in FTO is associated with diminished satiety. J Clin Endocrinol Metab 93:3640–3643. doi:10.1210/jc.2008-0472
Heni M, Kullmann S, Veit R et al (2013) Variation in the obesity risk gene FTO determines the postprandial cerebral processing of food stimuli in the prefrontal cortex. Mol Metab 3:109–113. doi:10.1016/j.molmet.2013.11.009
Karra E, O’Daly OG, Choudhury AI et al (2013) A link between FTO, ghrelin, and impaired brain food-cue responsivity. J Clin Invest 123:3539–3551. doi:10.1172/JCI44403
Staiger H, Machicao F, Fritsche A, Häring H-U (2009) Pathomechanisms of type 2 diabetes genes. Endocr Rev 30:557–585. doi:10.1210/er.2009-0017
Smemo S, Tena JJ, Kim K-H et al (2014) Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 507:371–375. doi:10.1038/nature13138
Claussnitzer M, Dankel SN, Kim K-H et al (2015) FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N Engl J Med 373:895–907. doi:10.1056/NEJMoa1502214
Stratigopoulos G, Burnett LC, Rausch R et al (2016) Hypomorphism of Fto and Rpgrip1l causes obesity in mice. J Clin Invest 126:1897–1910. doi:10.1172/JCI85526
Hess ME, Hess S, Meyer KD et al (2013) The fat mass and obesity associated gene (Fto) regulates activity of the dopaminergic midbrain circuitry. Nat Neurosci 16:1042–1048. doi:10.1038/nn.3449
Luo S, Luo J, Meier AH, Cincotta AH (1997) Dopaminergic neurotoxin administration to the area of the suprachiasmatic nuclei induces insulin resistance. Neuroreport 8:3495–3499
Diepenbroek C, van der Plasse G, Eggels L et al (2013) Alterations in blood glucose and plasma glucagon concentrations during deep brain stimulation in the shell region of the nucleus accumbens in rats. Front Neurosci 7:226. doi:10.3389/fnins.2013.00226
Halpern CH, Tekriwal A, Santollo J et al (2013) Amelioration of binge eating by nucleus accumbens shell deep brain stimulation in mice involves D2 receptor modulation. J Neurosci Off J Soc Neurosci 33:7122–7129. doi:10.1523/JNEUROSCI.3237-12.2013
Defronzo RA (2011) Bromocriptine: a sympatholytic, d2-dopamine agonist for the treatment of type 2 diabetes. Diabetes Care 34:789–794. doi:10.2337/dc11-0064
Figlewicz DP, Szot P, Chavez M et al (1994) Intraventricular insulin increases dopamine transporter mRNA in rat VTA/substantia nigra. Brain Res 644:331–334
Figlewicz DP, Sipols AJ (2010) ENERGY REGULATORY SIGNALS AND FOOD REWARD. Pharmacol Biochem Behav 97:15–24. doi:10.1016/j.pbb.2010.03.002
Speed NK, Matthies HJG, Kennedy JP et al (2010) Akt-dependent and isoform-specific regulation of dopamine transporter cell surface expression. ACS Chem Neurosci 1:476–481. doi:10.1021/cn100031t
Palmiter RD (2007) Is dopamine a physiologically relevant mediator of feeding behavior? Trends Neurosci 30:375–381. doi:10.1016/j.tins.2007.06.004
Könner AC, Hess S, Tovar S et al (2011) Role for insulin signaling in catecholaminergic neurons in control of energy homeostasis. Cell Metab 13:720–728. doi:10.1016/j.cmet.2011.03.021
Tschritter O, Preissl H, Yokoyama Y et al (2007) Variation in the FTO gene locus is associated with cerebrocortical insulin resistance in humans. Diabetologia 50:2602–2603. doi:10.1007/s00125-007-0839-1
Obici S, Feng Z, Karkanias G et al (2002) Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci 5:566–572. doi:10.1038/nn861
Pocai A, Lam TKT, Gutierrez-Juarez R et al (2005) Hypothalamic K(ATP) channels control hepatic glucose production. Nature 434:1026–1031. doi:10.1038/nature03439
Pocai A, Obici S, Schwartz GJ, Rossetti L (2005) A brain-liver circuit regulates glucose homeostasis. Cell Metab 1:53–61. doi:10.1016/j.cmet.2004.11.001
Heni M, Wagner R, Kullmann S et al (2014) Central insulin administration improves whole-body insulin sensitivity via hypothalamus and parasympathetic outputs in men. Diabetes 63:4083–4088. doi:10.2337/db14-0477
Heni M, Kullmann S, Preissl H et al (2015) Impaired insulin action in the human brain: causes and metabolic consequences. Nat Rev Endocrinol 11:701–711. doi:10.1038/nrendo.2015.173
Do R, Bailey SD, Desbiens K et al (2008) Genetic variants of FTO influence adiposity, insulin sensitivity, leptin levels, and resting metabolic rate in the Quebec Family Study. Diabetes 57:1147–1150. doi:10.2337/db07-1267
Haupt A, Thamer C, Machann J et al (2008) Impact of variation in the FTO gene on whole body fat distribution, ectopic fat, and weight loss. Obes Silver Spring Md 16:1969–1972. doi:10.1038/oby.2008.283
Scott LJ, Mohlke KL, Bonnycastle LL et al (2007) A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 316:1341–1345. doi:10.1126/science.1142382
Neville MJ, Johnstone EC, Walton RT (2004) Identification and characterization of ANKK1: a novel kinase gene closely linked to DRD2 on chromosome band 11q23.1. Hum Mutat 23:540–545. doi:10.1002/humu.20039
Jönsson EG, Nöthen MM, Grünhage F et al (1999) Polymorphisms in the dopamine D2 receptor gene and their relationships to striatal dopamine receptor density of healthy volunteers. Mol Psychiatry 4:290–296
Zhang Y, Bertolino A, Fazio L et al (2007) Polymorphisms in human dopamine D2 receptor gene affect gene expression, splicing, and neuronal activity during working memory. Proc Natl Acad Sci U S A 104:20552–20557. doi:10.1073/pnas.0707106104
Gluskin BS, Mickey BJ (2016) Genetic variation and dopamine D2 receptor availability: a systematic review and meta-analysis of human in vivo molecular imaging studies. Transl Psychiatry 6:e747. doi:10.1038/tp.2016.22
Comings DE, Muhleman D, Ahn C et al (1994) The dopamine D2 receptor gene: a genetic risk factor in substance abuse. Drug Alcohol Depend 34:175–180
Felsted JA, Ren X, Chouinard-Decorte F, Small DM (2010) Genetically determined differences in brain response to a primary food reward. J Neurosci Off J Soc Neurosci 30:2428–2432. doi:10.1523/JNEUROSCI.5483-09.2010
Benton D, Young HA (2016) A meta-analysis of the relationship between brain dopamine receptors and obesity: a matter of changes in behavior rather than food addiction? Int J Obes 40:S12–S21. doi:10.1038/ijo.2016.9
Winkler JK, Woehning A, Schultz J-H et al (2012) TaqIA polymorphism in dopamine D2 receptor gene complicates weight maintenance in younger obese patients. Nutr Burbank Los Angel Cty Calif 28:996–1001. doi:10.1016/j.nut.2011.12.018
Stice E, Spoor S, Bohon C, Small DM (2008) Relation between obesity and blunted striatal response to food is moderated by TaqIA A1 allele. Science 322:449–452. doi:10.1126/science.1161550
Sevgi M, Rigoux L, Kühn AB et al (2015) An Obesity-Predisposing Variant of the FTO Gene Regulates D2R-Dependent Reward Learning. J Neurosci Off J Soc Neurosci 35:12584–12592. doi:10.1523/JNEUROSCI.1589-15.2015
Goldstein JI, Crenshaw A, Carey J et al (2012) zCall: a rare variant caller for array-based genotyping: genetics and population analysis. Bioinforma Oxf Engl 28:2543–2545
Stefan N, Machicao F, Staiger H et al (2005) Polymorphisms in the gene encoding adiponectin receptor 1 are associated with insulin resistance and high liver fat. Diabetologia 48:2282–2291. doi:10.1007/s00125-005-1948-3
Kullmann S, Frank S, Heni M et al (2013) Intranasal insulin modulates intrinsic reward and prefrontal circuitry of the human brain in lean women. Neuroendocrinology 97:176–182. doi:10.1159/000341406
Roth CL, Hinney A, Schur EA et al (2013) Association analyses for dopamine receptor gene polymorphisms and weight status in a longitudinal analysis in obese children before and after lifestyle intervention. BMC Pediatr 13:197. doi:10.1186/1471-2431-13-197
Small DM, Zatorre RJ, Dagher A et al (2001) Changes in brain activity related to eating chocolate: from pleasure to aversion. Brain J Neurol 124:1720–1733
Small DM, Jones-Gotman M, Dagher A (2003) Feeding-induced dopamine release in dorsal striatum correlates with meal pleasantness ratings in healthy human volunteers. Neuroimage 19:1709–1715
Wagner R, Heni M, Linder K et al (2014) Age-dependent association of serum prolactin with glycaemia and insulin sensitivity in humans. Acta Diabetol 51:71–78. doi:10.1007/s00592-013-0493-7
Hallschmid M, Benedict C, Schultes B et al (2004) Intranasal insulin reduces body fat in men but not in women. Diabetes 53:3024–3029
Tschritter O, Preissl H, Hennige AM et al (2012) High cerebral insulin sensitivity is associated with loss of body fat during lifestyle intervention. Diabetologia 55:175–182. doi:10.1007/s00125-011-2309-z
Acknowledgements
We thank all study participants for their cooperation in this project. We gratefully acknowledge the excellent technical assistance of Maike Borutta, Anna Bury, Anja Dessecker, Dr Louise Fritsche, Alke Guirguis, Ellen Kollmar, Corinna Sailer, Andreas Vosseler and Roman-Georg Werner (all University of Tübingen, Germany).
Parts of this study were presented in abstract form at the 75th Scientific Sessions of the American Diabetes Association in June 2015.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was supported in part by a grant (01GI0925) from the Federal Ministry of Education and Research (BMBF) to the German Center for Diabetes Research (DZD e.V.) and the Helmholtz Alliance ICEMED-Imaging and Curing Environmental Metabolic Diseases and through an EFSD Rising Star award supported by Novo Nordisk to MH. The MDCS was supported by the Swedish Research Council’s grant, a Linné grant and an ERC Advanced Research Grant (269045) to LG. EA was supported by the Albert Påhlsson Foundation and the Bo & Kerstin Hjelt Diabetes Foundation.
Author contributions
MH performed the study, analysed the data and wrote the manuscript. SK performed the study, analysed the fMRI data and contributed to discussions. EA and PA analysed the MDC data and contributed to discussions. RW, FM, HS and AF acquired data and contributed to discussions. DMS contributed to the interpretation of the data and contributed to discussions. HUH, LCG, AF and HP designed the studies and contributed to discussions. All authors revised the manuscript and approved the final version before submission. HP is the guarantor for the data from Tübingen and LCG is the guarantor for the data from Malmö.
Duality of interest
All authors declare that there are no conflicts of interest that are directly relevant to the content of this study.
Rights and permissions
About this article

Cite this article
Heni, M., Kullmann, S., Ahlqvist, E. et al. Interaction between the obesity-risk gene FTO and the dopamine D2 receptor gene ANKK1/TaqIA on insulin sensitivity. Diabetologia 59, 2622–2631 (2016). https://doi.org/10.1007/s00125-016-4095-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-4095-0