
Abstract
Aims/hypothesis
The accumulation of extracellular matrix (ECM) is a characteristic of diabetic nephropathy, and is partially caused by profibrotic proteins TGF-β and connective tissue growth factor (CTGF). We aimed to identify microRNAs (miRNAs) targeting CTGF on podocytes in diabetic nephropathy.
Methods
We investigated miRNAs targeting CTGF on podocytes with miRNA array analysis and identified a candidate miRNA, miR-26a. Using overexpression and silencing of miR-26a in cultured podocytes, we examined changes of ECM and its host genes. We further investigated glomerular miR-26a expression in humans and in mouse models of diabetic nephropathy.
Results
miR-26a, which was downregulated by TGF-β1, was expressed in glomerular cells including podocytes and in tubules by in situ hybridisation. Glomerular miR-26a expression was downregulated by 70% in streptozotocin-induced diabetic mice. Transfection of miR-26a mimics in cultured human podocytes decreased the CTGF protein level by 50%, and directly inhibited CTGF expression in podocytes, as demonstrated by a reporter assay with the 3′-untranslated region of the CTGF gene. This effect was abolished by a mutant plasmid. miR-26a mimics also inhibited TGF-β1-induced collagen expression, SMAD-binding activity and expression of its host genes CTDSP2 and CTDSPL. Knockdown of CTDSP2 and CTDSPL increased collagen expression in TGF-β-stimulated podocytes, suggesting that host genes also regulate TGF-β/SMAD signalling. Finally, we observed a positive correlation between microdissected glomerular miR-26a expression levels and estimated GFR in patients with diabetic nephropathy.
Conclusions/interpretation
The downregulation of miR-26a is involved in the progression of diabetic nephropathy both in humans and in mice through enhanced TGF-β/CTGF signalling.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetic nephropathy is a major microvascular complication of diabetes mellitus and the leading cause of end-stage renal disease worldwide [1]. The condition is characterised by microalbuminuria, glomerular hypertrophy, mesangial expansion and an accumulation of extracellular matrix (ECM) proteins [2]. The accumulation of ECM proteins, such as collagen, is one of the main hallmarks of diabetic nephropathy [3]. Evidence has shown that TGF-β plays a key role in these pathological changes [4]. Podocyte injury and loss are found in the very early stages of diabetic nephropathy and may be a major starting point of glomerular injury in the disease [5]. Metabolic and haemodynamic dysregulation in diabetes can cause structural and functional changes in podocytes, leading to podocyte injury or loss [5]. Several cytokines or growth factors, such as TGF-β and connective tissue growth factor (CTGF, also known as CCN2) [6, 7], have been proposed to be involved in podocyte injury. The precise mechanisms of their role, however, remain unclear.
CTGF belongs to the CCN family of cysteine-rich growth factors, which consists of six related genes with four conserved domains [8]. Under normal conditions, CTGF mRNA is mainly expressed in podocytes and detected in some parietal epithelial cells of the glomeruli in the kidneys of humans and rats [9, 10]. Under the diabetic milieu, CTGF expression is markedly upregulated in podocytes, mesangial cells and tubulointerstitial cells, which are associated with fibrotic lesions [6, 9, 10]. CTGF gene expression is strongly induced by TGF-β in a SMAD3/SMAD4-dependent manner [11], and recombinant CTGF potentially enhances the synthesis of ECM proteins, such as collagen or fibronectin, in cultured podocytes [12], mesangial cells [13], tubular epithelial cells [14] and fibroblasts [15]. In addition to a direct fibrogenic effect, CTGF can exacerbate TGF-β-induced fibrosis by activating TGF-β and SMAD signalling through promoting the association of TGF-β with its receptor [16]. TGF-β-induced collagen production is attenuated by a CTGF neutralising antibody or antisense oligonucleotide targeting CTGF [17–19]. We demonstrated that in streptozotocin (STZ)-induced diabetes, the podocyte-specific CTGF transgenic mice had marked worsening of proteinuria and mesangial expansion accompanied by podocyte loss compared with wild-type mice [7]. Although these observations are consistent with CTGF as a key mediator of diabetic nephropathy progression, the precise regulation of CTGF in glomeruli in the presence of diabetes, especially in podocytes, remains elusive.
MicroRNAs (miRNAs) are small (approximately 22-nucleotide long), noncoding RNAs that downregulate gene expression by modulating the stability and/or translation of target mRNAs. These molecules play critical roles in various cellular functions. Dysregulation of miRNAs is involved in numerous pathological conditions, including renal disease [20, 21]. miR-192 [22], miR-200 [23], and miR-215 [24] are reported to be involved in the TGF-β-dependent pathogenesis of diabetic nephropathy. The mechanism of how they modulate the disease seem very complex and further investigations are required to clarify the role of these miRNAs in diabetic nephropathy [25]. A number of studies have investigated the regulation of CTGF expression at the promoter level [11, 26], but few have examined the post-transcriptional regulation of CTGF by miRNAs. miRNA targeting CTGF in diabetic nephropathy has not been reported. The present study was performed to identify miRNAs targeting CTGF and to investigate the role of miRNAs in the progression of diabetic nephropathy.
Methods
Human biopsy samples and laser microdissection
This study was conducted according to the principles expressed in the Declaration of Helsinki and was approved by the Ethics Committee on Human Research at Kyoto University Graduate School of Medicine (Kyoto, Japan). Patients with type 2 diabetes who underwent renal biopsy were enrolled in the study under informed consent. Renal biopsy samples were obtained from 11 patients with diabetic nephropathy, as diagnosed by renal biopsy. The estimated GFR (eGFR) was calculated using the equation adopted by the Japanese Society of Nephrology [27].
Two to ten glomeruli on a slide were microdissected and collected using the Leica 6500 Laser Microdissection System (Leica Microsystems, Wetzlar, Germany) and then RNA was extracted (for further details, please refer to the electronic supplementary material [ESM] Methods).
Diabetic animal models
All animal experiments were approved by the Animal Experimentation Committee of Kyoto University Graduate School of Medicine. Twelve 8-week-old male C57BL/6J mice were used in this study (Clea Japan, Tokyo, Japan). Mouse models of diabetes were generated by intraperitoneal injection of STZ (100 mg/kg body weight for three consecutive days [7]; for further details, please refer to the ESM Methods).
In situ hybridisation
In situ hybridisation was performed as described previously [18] (for further details, please refer to the ESM Methods).
Cell culture
Conditionally immortalised human podocytes were developed by transfection with the temperature-sensitive SV40 T-gene, as described previously [28]. Cells were stimulated with recombinant human TGF-β1 (R&D Systems, Minneapolis, MN, USA; for further details, please refer to the ESM Methods).
MicroRNA microarray
Differentiated podocytes were treated with 5 ng/ml TGF-β1 (R&D Systems) for 24 h in RPMI 1640 medium under serum starvation. Labelled samples were hybridised to the Human miRNA Microarray Kit 8x15K V2 (Agilent Technologies, Palo Alto, CA, USA) containing probes for 723 mature human miRNAs. The microarray images were analysed using Feature Extraction Software (Agilent Technologies). Data are uploaded on Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo, Accession no. GSE66133; for further details, please refer to the ESM Methods).
Real-time RT-PCR analysis
Real-time RT-PCR was performed as described previously [7] (for further details, please refer to the ESM Methods).
Transfection
Differentiated podocytes were transfected with miR-26a mimics or mimic negative control (NC; 30 nmol/l), miR-26a inhibitors or inhibitor NC (30 nmol/l), and siRNA for CTGF, CTDSP2 and CTDSPL (5 nmol/l) by a pulse of electroporation with Nucleofector (Amaxa Biosystems, Cologne, Germany) using the Basic Nucleofector Kit for Primary Mammalian Epithelial cells (Amaxa Biosystems) [29] (for further details, please refer to the ESM Methods).
Immunocytochemical study
Immunocytochemical analyses of SMAD4 was performed with the stimulation of TGF-β1 with miR-26a mimic or NC transfection (for further details, please refer to the ESM Methods).
Western blotting
Western blotting was performed as previously described [30] (for further details, please refer to the ESM Methods).
Plasmid construction and luciferase assay
The pMirTarget-CTGF 3′-untranslated region (UTR) vector was purchased from OriGene Technologies (Rockville, MD, USA). Mutant constructs were generated by replacing the seed regions of the miR-26a binding sites (two to seven) with 5′-TTGGTT-3′ (Fig. 5f, ESM Table 1) by PCR-based, site-directed mutagenesis [31]. The SMAD binding element (SBE4)-luciferase plasmid contains four copies of SBE (taaGTCTAGACggcaGTCTAGACgtac) in the pBV-luciferase plasmid (Addgene, Cambridge, MA, USA) [32] (for further details, please refer to the ESM Methods).
Statistical analyses
Data are expressed as the mean ± SE. Statistical analysis was performed using one-way ANOVA or Student’s unpaired t test as appropriate. Spearman’s correlation coefficients were used to examine the correlation between two variables. A p value <0.05 was considered statistically significant. All data were analysed using StatView 5.0 software (SAS Institute, Cary, NC, USA) or JMP7.1 (SAS Institute).
Results
Downregulation of miR-26a by TGF-β stimulation in cultured human podocytes
CTGF is a major downstream mediator of TGF-β that promotes ECM accumulation and fibrosis by directly interacting with TGF-β in diabetic nephropathy. To identify miRNAs regulating CTGF downstream of TGF-β, we analysed expression profiles of 723 miRNAs in immortalised human podocytes after TGF-β1 (5 ng/ml) stimulation using microRNA array analysis. We identified 46 miRNAs in which expression levels were altered by >1.5-fold between PBS- and TGF-β1-treated podocytes (Fig. 1a, Table 1). Of these 46 miRNAs, a microRNA database (http://microrna.org, last accessed 20 January 2015) identified miR-26a as being the only microRNA targeting CTGF. miR-26a has a putative target site in the 3′- UTR of the CTGF gene (Fig. 1b). The seed regions of miR-26a are highly conserved across species (Fig. 1c). To validate microarray data, we further examined the expression of miR-26a in TGF-β1-treated podocytes by real-time RT-PCR. Stimulation with TGF-β1 decreased miR-26a expression on podocytes in a dose-dependent manner (Fig. 1d). Expression of another internal control, small nuclear RNA U6B, did not change following stimulation with TGF-β1 (ESM Fig. 1).

miR-26a is downregulated by TGF-β1 stimulation in human podocytes. (a) Differentiated podocytes were treated with TGF-β1 (5 ng/ml) or PBS for 24 h. The expression profiles of 723 miRNAs were analysed by microRNA array, and 46 miRNAs showed >1.5-fold differences in the expression levels between PBS- and TGF-β1-treated podocytes. miR-26a is underlined. (b) miR-26a binding sites in the 3′-UTR of the CTGF transcripts of various species. Seven nucleotides in the 5′ region of miR-26a contain a perfect match with 3′-UTR of CTGF sequences. The miRNA seed regions are in bold. (c) The mature sequences of miR-26a are conserved across various species. The seed sequences are highlighted by grey shading. (d) The expression of miR-26a in 5 or 20 ng/ml TGF-β1-treated podocytes was examined by real-time RT-PCR at 24 h after stimulation. RNU48 was used as an internal control. n = 5 for each group. *p < 0.05. Data are mean ± SE
Expression and localisation of miR-26a in glomeruli of diabetic mice
We analysed miR-26a expression in glomeruli of diabetic mice because podocytes are thought to be injured at a very early stage in diabetic nephropathy. First, we studied the localisation of miR-26a in the kidney by ISH. miR-26a expression was detected in glomerular cells, including podocytes and in tubules and collecting ducts of control mice (Fig. 2a, b). Next, we examined miR-26a expression in the glomeruli of STZ-induced diabetic mice. Compared with control mice, glomerular miR-26a expression was significantly lower (by 70%) at 3 weeks after STZ administration (Fig. 3a). By contrast, glomerular Tgfb1, Ctgf and Col1a1 mRNA expressions were increased in diabetic mice compared with controls (Fig. 3b–d). In addition, the expression of host genes encoding miR-26a, Ctdsp2 and Ctdspl, was increased (Fig. 3e, f). Thus, miR-26a is expressed in the kidney, including in podocytes, and is downregulated in diabetic nephropathy.

Localisation of miR-26a in the kidney. ISH for miR-26a in the kidneys of control mice at (a) lower magnification (×40) and (b) higher magnification (×400). miR-26a is detected in glomerular cells, including podocytes, and in tubules and collecting ducts of control mice

miR-26a is downregulated in glomeruli of diabetic mice. Real-time RT-PCR analyses of (a) miR-26a, (b) Tgfb1, (c) Ctgf, (d) Col1a1, (e) Ctdsp2 and (f) Ctdspl mRNA expression in glomeruli of control or STZ-treated mice (n = 6 in each group) at 3 weeks after vehicle or STZ injection. *p < 0.05, **p < 0.01. Data are mean ± SE. sno202, small nucleolar RNA MB11-202
Glomerular miR-26a expression level is associated with eGFR in renal biopsy specimens from patients with diabetic nephropathy
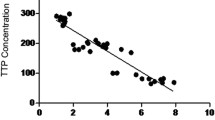
We investigated glomerular miR-26a expression in optimal cutting temperature compound-embedded, laser-microdissected renal biopsy specimens with diabetic nephropathy by real-time RT-PCR (Fig. 4a). Table 2 summarises the clinical characteristics of the patients. We examined the association between glomerular miR-26a expression and clinical variables at the time of renal biopsy. Figures 4b and c show representative views of renal biopsy specimens with diabetic nephropathy at the early and the advanced stages, respectively. Of note, glomerular miR-26a expression in human diabetic nephropathy positively correlated with eGFR (Fig. 4d; p = 0.025; r = 0.659). Glomerular miR-26a expression did not correlate with proteinuria (Fig. 4e).

Correlation between glomerular miR-26a expression and clinical variables in human biopsy samples. (a) Representative views of glomerular extraction in OCT-embedded renal biopsy specimens before and after laser-micro dissection (magnification ×200). Representative views of renal biopsy specimens with diabetic nephropathy at the (b) early stages in PAS stain (serum creatinine [Cr] level, 70.7 mmol/l; urinary protein, 0.6 g/g Cr, magnification ×100) and (c) advanced stages in PAS stain (serum Cr level, 256 mmol/l; urinary protein, 7.9 g/g Cr, magnification ×200). Dot plot analysis of glomerular miR-26a with (d) eGFR (r = 0.66, p = 0.025) and (e) proteinuria (r = −0.47, p = 0.15). Line in each graph shows the regression. n = 11. p values <0.05 were considered statistically significant. LMD, laser microdissection; OCT, optimal cutting temperature; PAS, periodic acid–Schiff
CTGF is a direct target of miR-26a
To investigate whether or not miR-26a targets the CTGF gene, we transfected miR-26a mimics (30 nmol/l) into podocytes by nucleofection and then examined CTGF expression by real-time RT-PCR and western blotting. Transfection of miR-26a mimics did not change CTGF mRNA level (Fig. 5a) but significantly reduced CTGF protein level by approximately 50% (Fig. 5b, c), suggesting that miR-26a negatively regulates CTGF in a post-transcriptional manner. To determine whether or not CTGF is a direct target of miR-26a, we performed luciferase reporter assays using the pMirTarget-CTGF 3′-UTR plasmid, which contains the CTGF 3′-UTR sequence downstream of the firefly luciferase gene (Fig. 5d). Co-transfection of the reporter plasmids with miR-26a mimics significantly reduced luciferase activity by approximately 40% compared with NC (Fig. 5e), and miR-26a inhibitors significantly increased luciferase activity by 30% (Fig. 5e). These results suggest that binding of miR-26a to the 3′-UTR of CTGF mRNA inhibits translation. To confirm these results, we generated a corresponding miR-26a mutant plasmid by replacing the seed regions of the miR-26a-binding sites with 5′-TTGGTT-3′ (Fig. 5f, ESM Table 1). Such mutation abolished the ability of miR-26a to suppress luciferase expression (Fig. 5g). These results demonstrate that CTGF is a direct target of miR-26a.

CTGF is a direct target of miR-26a. (a) Real-time RT-PCR analysis of CTGF and (b, c) western blots of CTGF in cultured human podocytes transfected with miR-26a mimics or mimic NC at 48 h after transfection. Arrowheads indicate 36 and 38 kDa CTGF of which a larger band indicates a glycosylated form. GAPDH was used as an internal control (n = 6 in each group). (d) Schematic representation of luciferase reporter constructs which contain CTGF 3′-UTR sequence downstream of the firefly luciferase gene (pMirTarget-CTGF 3′-UTR plasmid). (e) Podocytes were transfected with pMirTarget-CTGF 3′-UTR plasmid in the absence or the presence of miR-26a mimics or miR-26a inhibitors (30 nmol/l) and analysed at 8 h after transfection (n = 6 in each group). (f) Site-directed mutagenesis was performed to mutate the seed regions within the CTGF 3′-UTR. Lower case letters indicate mutated nucleotides. (g) Podocytes were transfected with a luciferase reporter plasmids containing wild or mutant CTGF 3ʹ-UTR and analysed at 8 h after transfection. miR-26a mimics inhibited luciferase activity, but a mutant plasmid abolished the inhibitory effect of miR-26a. White bars, mimic NC; black bars, miR-26a mimic (n = 6 in each group). CMV, cytomegalovirus; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; Luc, luciferase. *p < 0.05, **p < 0.01. Data are mean ± SE
Inhibition of TGF-β-induced ECM accumulation and TGF-β/SMAD signalling by miR-26a
We further examined whether miR-26a can inhibit TGF-β-induced ECM accumulation by inhibiting CTGF. Transfection of miR-26a mimics significantly decreased TGF-β1-induced upregulation of COL1A1 and COL4A3 expression (Fig. 6a, b). This effect was consistent with the findings that knockdown of CTGF by siRNA decreased TGF-β1-induced COL1A1 and COL4A3 expression (Fig. 6c–f). These observations demonstrate that miR-26a can inhibit TGF-β/CTGF-induced ECM accumulation. We also analysed SMAD activity in response to stimulation by TGF-β1. TGF-β augmented phosphorylation of SMAD3, and this phosphorylation was significantly abolished by miR-26a (Fig. 6g). Nuclear translocation of SMAD4 induced by TGF-β was attenuated by miR-26a (Fig. 6h). The activation of SBEs by TGF-β was also significantly inhibited (by 90%) by treatment with miR-26a mimics (Fig. 6i). Interestingly, the miR-26a mimics led to an approximately 75% decrease in TGFB1 autoinduction (Fig. 6j). Next, we examined whether miR-26a inhibited putative miR-26a-target genes, SMAD2 and SMAD4, by western blotting. SMAD2 and SMAD4 protein levels were not altered by miR-26a mimics (ESM Fig. 2). These results suggest miR-26a suppresses TGF-β-induced ECM accumulation by directly inhibiting CTGF in podocytes.

miR-26a inhibits TGF-β/SMAD signalling in podocytes. Cultured human podocytes were transfected with mimics or plasmid and then stimulated with TGF-β1 (20 ng/ml) at 2 days after the transfection. The expression levels of (a) COL1A1 and (b) COL4A3 were examined at 24 h after stimulation with transfection of miR-26a mimics or mimic NC by real-time RT-PCR (n = 3 in each group). (c–f) Human podocytes were transfected with siRNA for CTGF or NC (5 nmol/l). The expression levels of (c) CTGF, (d) TGFB1, (e) COL1A1 and (f) COL4A3 were examined (n = 4 in each group). (g) Western blots for phospho-SMAD3 (arrow head) and total SMAD3 in cultured podocytes. Podocytes transfected with miR-26 mimics or NC were stimulated with TGF-β and then analysed at 30 min after stimulation. 26a, miR-26a mimics. (h) Immunocytochemical analyses of SMAD4 (red) in TGF-β-stimulated podocytes transfected with miR-26a mimics or NC (magnification ×400). DAPI, blue. (i) SMAD activity was measured using luciferase reporter plasmids containing the SBE. Podocytes were transfected with SBE plasmids and then stimulated with TGF-β1 (20 ng/ml) for 48 h. Luciferase activity was measured (n = 6 in each group). (j) The expression levels of TGFB1 were examined at 24 h after stimulation with transfection of miR-26a mimics or mimic NC (n = 3 in each group). *p < 0.05, **p < 0.01. Data are mean ± SE
Reduction of CTDSP2 and CTDSPL enhances TGF-β signalling
Mature miR-26a is generated from two distinct loci, miR-26a-1 and miR-26a-2, which are located in the intron of CTDSPL and CTDSP2, respectively (Fig. 7a). The targeting sequences of miR-26a for CTDSPL and CTDSP2 are highly conserved among species (Fig. 7b). We examined expression levels of CTDSP2 and CTDSPL at 24 h after stimulation with TGF-β1. The expression of CTDSP2 and CTDSPL was significantly downregulated by TGF-β1 stimulation (Fig. 7c, d), indicating that miR-26a is expressed in parallel with its host gene. siRNA knockdown of CTDSP2 or CTDSPL enhanced TGF-β1-induced ECM accumulation in cultured podocytes (Fig. 7e–h), suggesting that CTDSP2 and CTDSPL have inhibitory effects on TGF-β/SMAD signalling in podocytes. Furthermore, miR-26a mimics suppressed CTDSP2 and CTDSPL mRNA (Fig. 7i, j). These data suggest that miR-26a is involved in the complex regulation of its host gene (Fig. 8).

Regulation and function of CTDSP2 and CTDSPL in podocytes. (a) Schematic genomic maps of host genes of miR-26a. Mature miR-26a is generated from two distinct loci, miR-26a-1 and miR-26a-2, which are located in the intron of CTDSPL and CTDSP2, respectively. (b) miR-26a binding sites in the 3′-UTR of the CTDSP2 and CTDSPL transcripts across various species. Six or seven nucleotides in the 5′ region of miR-26a contain a perfect match with 3′-UTR of CTDSP2 or CTDSPL sequences, respectively. The miRNA seed regions are in bold. Expression of (c) CTDSP2 and (d) CTDSPL in podocytes stimulated with TGF-β1 for 24 h were examined by real-time RT-PCR. GAPDH was used as internal control (n = 9 in each group). (e–h) Human podocytes were transfected with siRNA for 5 nmol/l (e, f) CTDSP2 or (g, h) CTDSPL, and then stimulated with TGF-β1 (20 ng/ml) at 2 days after the transfection. The expression levels of (e, g) COL1A1 and (f, h) COL4A3 were examined at 3 days after stimulation (n = 4 in each group). Real-time RT-PCR analyses of (i) CTDSP2 and (j) CTDSPL in cultured human podocytes transfected with miR-26a mimics or mimic NC. (n = 3 in each group). *p < 0.05, **p < 0.01. Data are mean ± SE

Proposed mechanism of the role of miR-26a targeting CTGF in diabetic conditions. TGF-β downregulates miR-26a expression and the reduction of miR-26a upregulates CTGF at the post-transcriptional level, which promotes the downstream activation of TGF-β leading to ECM accumulation. miR-26a is coded in the intron of host genes, CTDSP2 and CTDSPL. miR-26a targets CTDSP2 and CTDSPL genes forming an autoregulatory feedback loop
Discussion
We identified that miR-26a, which targets the CTGF gene, is a potent suppressor of TGF-β/SMAD signalling in cultured human podocytes. Overactivity of TGF-β/SMAD signalling stimulates the production of ECM and plays an important role in the pathogenesis of diabetic nephropathy [33, 34]. CTGF is a major downstream profibrotic factor of TGF-β [13, 16] and the inhibition of CTGF could be a promising therapeutic strategy against diabetic nephropathy [35, 36]. Duisters et al reported that miR-133 and miR-30 suppress CTGF expression post-transcriptionally in myocytes [37]. In the current study, we showed that miR-26a negatively regulated CTGF at the post-transcriptional level in cultured human podocytes. A previous report showed that miR-26a is highly expressed in the kidneys of 3-week-old mice and displays a pan-glomerular pattern based on ISH [21]. We also revealed that miR-26a was highly expressed in glomeruli within podocytes, tubules and collecting ducts of kidneys from control mice. Previous studies showed that miR-26a inhibits SMAD1 and SMAD4 in smooth muscle cells [38] and that miR-26a represses TGF-β1-induced fibrogenesis in MRC-5 cells by inhibiting CTGF and SMAD4 [39]. Inhibition of miR-26a causes pulmonary fibrosis in vivo and deposition of collagen I and III in vitro [39]. Wei et al reported that miR-26a, which is regulated by nuclear factor κB, inhibits both CTGF and collagen I in cardiac fibroblasts [40]. However, the mechanism underlying miR-26a regulation including its host genes has not yet been elucidated. We revealed that miR-26a decreases CTGF and ECM proteins and that phosphorylation of SMAD3 and miR-26a is expressed in parallel with its host genes, CTDSP2 and CTDSPL in podocytes. However, we could not confirm that miR-26a targets SMAD2 and SMAD4 in podocytes (ESM Fig. 2). Direct evidence that CTGF is the candidate gene targeted by miR-26a in TGF-β1-induced ECM accumulation has not been provided in this study. However, the following points are consistent with the notion that miR-26a decreases ECM thorough CTGF inhibition: first, miR-26a mimics decrease CTGF protein levels in podocytes by 50%, which could significantly decrease ECM. Second, our study indicates that SMAD2 and SMAD4 are not the direct targets of miR-26a, but that CTGF is the only gene targeted by miR-26a in the pathway of TGF-β-induced ECM expression (ESM Fig. 2, www.mirbase.org, last accessed 20 January 2015). Taken together, these observations indicate that miR-26a ameliorates TGF-β-induced ECM accumulation by inhibiting CTGF in podocytes (Fig. 8).
Podocyte dysfunction plays a pivotal role in the progression of diabetic nephropathy and is partly caused by increased levels of intraglomerular TGF-β in diabetic nephropathy [41]. TGF-β upregulates α1(1) and α3(4) collagens, contributing to the thickening of glomerular basement membrane in diabetes [34]. CTGF is also reported to increase collagen IV and fibronectin in cultured podocytes [12]. We identified miR-26a targeting genes relevant to TGF-β-induced ECM accumulation. Treatment of miR-26a inhibited COL1A1 and COL4A3 gene expression in podocytes and decreased the expression of CTGF, suggesting that miR-26a works as an anti-fibrotic factor in podocytes. A recent study has shown that miR-26a regulates genes associated with podocyte differentiation and formation of the cytoskeleton, and that the level of glomerular miR-26a is lower in patients with lupus nephritis and IgA nephropathy compared with healthy controls [42]. These results are consistent with our observation that glomerular miR-26a is lower in patients with diabetic nephropathy than controls. Because the expression of miR-26a in glomeruli is not confined to podocytes by ISH, miR-26a may function in other glomerular cells such as mesangial cells.
Overactivity of TGF-β in the kidneys is observed within days of diabetes onset, and Kato et al reported that the glomerular expression level of miR-192, which suppresses TGF-β signalling, begins increasing at a very early stage in STZ mice [22]. Our in vitro study findings indicate that the downregulation of miR-26a results in the upregulation of the CTGF gene, leading to overactive TGF-β signalling. The downregulation of miR-26a is thus likely to be involved in the upregulation of TGF-β signalling at the onset of diabetic nephropathy. Kato et al recently reported that miR-192 expression is induced by complex mechanisms [43]; initially, by SMAD transcription factors followed by sustained expression by epigenetic histone acetylation in mesangial cells of diabetic model mice. We speculate that miR-26a expression is also regulated by several different mechanisms in different phases of the disease. Investigation of miRNA in human renal biopsy specimens, especially in glomeruli, is not fully performed in diabetic nephropathy. Although we had a limited number of biopsy specimens, we investigated miR-26a expression in glomeruli of human diabetic nephropathy and observed a positive correlation between miR-26a and eGFR; miR-26a was lower in advanced diabetic nephropathy patients. This reduction in miR-26a may lead to dysregulation in TGF-β signalling in the progression of diabetic nephropathy.
miR-26a-1, miR-26a-2 and miR-26b are coded in the introns of their host genes, CTDSPL, CTDSP2 and CTDSP1, respectively [44]. The arrangement between miR-26a family genes and host genes is well conserved across vertebral species. In the present study, miR-26a reduced the mRNA levels of its host genes. CTDSP2 and CTDSPL are widely expressed in the human tissues, including the kidneys [45, 46]. CTDSPs act as phosphatases and dephosphorylate the linker region of regulatory SMADs leading to activation of the SMAD pathway [47]. Blockade of CTDSPs is reported to decrease the activity of TGF-β/SMAD signalling [47]. However, we observed that inhibition of CTDSPs augmented TGF-β/SMAD signalling in podocytes. miR-26a reduced the mRNA levels of its host genes. These results suggest the role of CTDSPs is cell type/context-dependent and that miR-26a autoregulates the expression of its host genes. miRNAs act as buffers against fluctuations in gene expression [48]. Our results indicate that miR-26a acts as a buffer against TGF-β/SMAD signalling by attenuating the transcriptional noise of CTGF. This action may confer robustness to TGF-β/SMAD signalling. Furthermore, we observed that TGF-β downregulated miR-26a. This observation suggests that TGF-β transduces its downstream signals by abolishing suppressive effects of miR-26a.
In conclusion, we determined that miR-26a targets the CTGF gene and attenuates TGF-β signalling and ECM accumulation in podocytes under diabetic condition. The downregulation of miR-26a was observed at the onset of diabetic nephropathy in mice and in advanced diabetic nephropathy in humans. The augmentation of miR-26a could be a novel therapeutic strategy for the treatment of diabetic nephropathy.
Abbreviations
- CCN:
-
Cysteine-rich, angiogenic inducer, 61, CTGF, nephroblastoma overexpressed
- CTDSP:
-
Small carboxy-terminal domain phosphatase
- CTGF:
-
Connective tissue growth factor
- ECM:
-
Extracellular matrix
- eGFR:
-
Estimated GFR
- ISH:
-
In situ hybridisation
- miRNA:
-
MicroRNA
- NC:
-
Negative control
- SBE:
-
SMAD binding element
- STZ:
-
Streptozotocin
- UTR:
-
Untranslated region
References
Kato M, Natarajan R (2014) Diabetic nephropathy-emerging epigenetic mechanisms. Nat Rev Nephrol 10:517–530
Mauer SM, Steffes MW, Ellis EN, Sutherland DE, Brown DM, Goetz FC (1984) Structural-functional relationships in diabetic nephropathy. J Clin Invest 74:1143–1155
Kanwar YS, Sun L, Xie P, Liu FY, Chen S (2011) A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu Rev Pathol 6:395–423
Yamamoto T, Nakamura T, Noble NA, Ruoslahti E, Border WA (1993) Expression of transforming growth factor beta is elevated in human and experimental diabetic nephropathy. Proc Natl Acad Sci U S A 90:1814–1818
Pagtalunan ME, Miller PL, Jumping-Eagle S et al (1997) Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest 99:342–348
Roestenberg P, van Nieuwenhoven FA, Joles JA et al (2006) Temporal expression profile and distribution pattern indicate a role of connective tissue growth factor (CTGF/CCN-2) in diabetic nephropathy in mice. Am J Physiol Ren Physiol 290:F1344–F1354
Yokoi H, Mukoyama M, Mori K et al (2008) Overexpression of connective tissue growth factor in podocytes worsens diabetic nephropathy in mice. Kidney Int 73:446–455
Gupta S, Clarkson MR, Duggan J, Brady HR (2000) Connective tissue growth factor: potential role in glomerulosclerosis and tubulointerstitial fibrosis. Kidney Int 58:1389–1399
Ito Y, Aten J, Bende RJ et al (1998) Expression of connective tissue growth factor in human renal fibrosis. Kidney Int 53:853–861
Makino H, Mukoyama M, Sugawara A et al (2003) Roles of connective tissue growth factor and prostanoids in early streptozotocin-induced diabetic rat kidney: the effect of aspirin treatment. Clin Exp Nephrol 7:33–40
Holmes A, Abraham DJ, Sa S, Shiwen X, Black CM, Leask A (2001) CTGF and SMADs, maintenance of scleroderma phenotype is independent of SMAD signaling. J Biol Chem 276:10594–10601
Fuchshofer R, Ullmann S, Zeilbeck LF, Baumann M, Junglas B, Tamm ER (2011) Connective tissue growth factor modulates podocyte actin cytoskeleton and extracellular matrix synthesis and is induced in podocytes upon injury. Histochem Cell Biol 136:301–319
Riser BL, Denichilo M, Cortes P et al (2000) Regulation of connective tissue growth factor activity in cultured rat mesangial cells and its expression in experimental diabetic glomerulosclerosis. J Am Soc Nephrol 11:25–38
Burns WC, Twigg SM, Forbes JM et al (2006) Connective tissue growth factor plays an important role in advanced glycation end product-induced tubular epithelial-to-mesenchymal transition: implications for diabetic renal disease. J Am Soc Nephrol 17:2484–2494
Frazier K, Williams S, Kothapalli D, Klapper H, Grotendorst GR (1996) Stimulation of fibroblast cell growth, matrix production, and granulation tissue formation by connective tissue growth factor. J Investig Dermatol 107:404–411
Abreu JG, Ketpura NI, Reversade B, de Robertis EM (2002) Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-β. Nat Cell Biol 4:599–604
Duncan MR, Frazier KS, Abramson S et al (1999) Connective tissue growth factor mediates transforming growth factor β-induced collagen synthesis: down-regulation by cAMP. FASEB J 13:1774–1786
Yokoi H, Mukoyama M, Sugawara A et al (2002) Role of connective tissue growth factor in fibronectin expression and tubulointerstitial fibrosis. Am J Physiol Ren Physiol 282:F933–F942
Yokoi H, Mukoyama M, Nagae T et al (2004) Reduction in connective tissue growth factor by antisense treatment ameliorates renal tubulointerstitial fibrosis. J Am Soc Nephrol 15:1430–1440
Harvey SJ, Jarad G, Cunningham J et al (2008) Podocyte-specific deletion of dicer alters cytoskeletal dynamics and causes glomerular disease. J Am Soc Nephrol 19:2150–2158
Ho J, Ng KH, Rosen S, Dostal A, Gregory RI, Kreidberg JA (2008) Podocyte-specific loss of functional microRNAs leads to rapid glomerular and tubular injury. J Am Soc Nephrol 19:2069–2075
Kato M, Zhang J, Wang M et al (2007) MicroRNA-192 in diabetic kidney glomeruli and its function in TGF-β-induced collagen expression via inhibition of E-box repressors. Proc Natl Acad Sci U S A 104:3432–3437
Kato M, Arce L, Wang M, Putta S, Lanting L, Natarajan R (2011) A microRNA circuit mediates transforming growth factor-β1 autoregulation in renal glomerular mesangial cells. Kidney Int 80:358–368
Mu J, Pang Q, Guo YH et al (2013) Functional implications of microRNA-215 in TGF-β1-induced phenotypic transition of mesangial cells by targeting CTNNBIP1. PLoS One 8, e58622
Kasinath BS, Feliers D (2011) The complex world of kidney microRNAs. Kidney Int 80:334–337
Okada H, Inoue T, Kikuta T et al (2008) Poly(ADP-ribose) polymerase-1 enhances transcription of the profibrotic CCN2 gene. J Am Soc Nephrol 19:933–942
Matsuo S, Imai E, Horio M et al (2009) Revised equations for estimated GFR from serum creatinine in Japan. Am J Kidney Dis 53:982–992
Saleem MA, O'Hare MJ, Reiser J et al (2002) A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol 13:630–638
Noguchi M, Hosoda K, Fujikura J et al (2007) Genetic and pharmacological inhibition of Rho-associated kinase II enhances adipogenesis. J Biol Chem 282:29574–29583
Ogawa Y, Mukoyama M, Yokoi H et al (2012) Natriuretic peptide receptor guanylyl cyclase-A protects podocytes from aldosterone-induced glomerular injury. J Am Soc Nephrol 23:1198–1209
Kanamoto N, Tagami T, Ueda-Sakane Y et al (2012) Forkhead box A1 (FOXA1) and A2 (FOXA2) oppositely regulate human type 1 iodothyronine deiodinase gene in liver. Endocrinology 153:492–500
Zawel L, Dai JL, Buckhaults P et al (1998) Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell 1:611–617
Ziyadeh FN, Hoffman BB, Han DC et al (2000) Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci U S A 97:8015–8020
Iglesias-de la Cruz MC, Ziyadeh FN, Isono M et al (2002) Effects of high glucose and TGF-β1 on the expression of collagen IV and vascular endothelial growth factor in mouse podocytes. Kidney Int 62:901–913
Adler SG, Schwartz S, Williams ME et al (2010) Phase 1 study of anti-CTGF monoclonal antibody in patients with diabetes and microalbuminuria. Clin J Am Soc Nephrol 5:1420–1428
Guha M, Xu ZG, Tung D, Lanting L, Natarajan R (2007) Specific down-regulation of connective tissue growth factor attenuates progression of nephropathy in mouse models of type 1 and type 2 diabetes. FASEB J 21:3355–3368
Duisters RF, Tijsen AJ, Schroen B et al (2009) miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res 104:170–178
Leeper NJ, Raiesdana A, Kojima Y et al (2011) MicroRNA-26a is a novel regulator of vascular smooth muscle cell function. J Cell Physiol 226:1035–1043
Liang H, Xu C, Pan Z et al (2014) The antifibrotic effects and mechanisms of microRNA-26a action in idiopathic pulmonary fibrosis. Mol Ther 22:1122–1133
Wei C, Kim IK, Kumar S et al (2013) NF-κB mediated miR-26a regulation in cardiac fibrosis. J Cell Physiol 228:1433–1442
Wolf G, Chen S, Ziyadeh FN (2005) From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes 54:1626–1634
Ichii O, Otsuka-Kanazawa S, Horino T et al (2014) Decreased miR-26a expression correlates with the progression of podocyte injury in autoimmune glomerulonephritis. PLoS One 9, e110383
Kato M, Dang V, Wang M et al (2013) TGF-β induces acetylation of chromatin and of Ets-1 to alleviate repression of miR-192 in diabetic nephropathy. Sci Signal 6:ra43
Zhu Y, Lu Y, Zhang Q et al (2012) MicroRNA-26a/b and their host genes cooperate to inhibit the G1/S transition by activating the pRb protein. Nucleic Acids Res 40:4615–4625
Su YA, Lee MM, Hutter CM, Meltzer PS (1997) Characterization of a highly conserved gene (OS4) amplified with CDK4 in human sarcomas. Oncogene 15:1289–1294
Kashuba VI, Li J, Wang F et al (2004) RBSP3 (HYA22) is a tumor suppressor gene implicated in major epithelial malignancies. Proc Natl Acad Sci U S A 101:4906–4911
Sapkota G, Knockaert M, Alarcon C, Montalvo E, Brivanlou AH, Massague J (2006) Dephosphorylation of the linker regions of Smad1 and Smad2/3 by small C-terminal domain phosphatases has distinct outcomes for bone morphogenetic protein and transforming growth factor-β pathways. J Biol Chem 281:40412–40419
Ebert MS, Sharp PA (2012) Roles for microRNAs in conferring robustness to biological processes. Cell 149:515–524
Acknowledgements
We gratefully acknowledge M. Fujimoto, Y. Sakashita, C. Kimura, N. Igarashi and E. Nishimura (Department of Nephrology, Kyoto University Graduate School of Medicine, Kyoto, Japan) and other lab members for their technical assistance, and A. Yamamoto and S. Ogino (Department of Nephrology, Kyoto University Graduate School of Medicine) for their secretarial assistance.
Funding
This work was supported in part by research grants from the Japanese Ministry of Education, Culture, Sports, Science and Technology, the Japanese Ministry of Agriculture, Forestry and Fisheries, the Japanese Ministry of Health, Labour and Welfare, Japan Agency for Medical Research and Development (AMED), Fujiwara Memorial Foundation and Takeda Science Foundation.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
KK, HY and MM designed the study, wrote the paper and approved its final version. KM, MK, TK, HI, AI, KPM, YK, SO, NT, KN and MY contributed to the study design, acquisition of data, interpretation of the article, revision of the manuscript and approved its final version. MAS and AS contributed materials and analysis and interpretation of data, revised the article’s intellectual content and approved the final version. HY is responsible for the integrity of the work as a whole.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 77 kb)
ESM Table1
(PDF 75 kb)
ESM Table2
(PDF 25 kb)
ESM Fig. 1
(PDF 180 kb)
ESM Fig. 2
(PDF 383 kb)
Rights and permissions
About this article

Cite this article
Koga, K., Yokoi, H., Mori, K. et al. MicroRNA-26a inhibits TGF-β-induced extracellular matrix protein expression in podocytes by targeting CTGF and is downregulated in diabetic nephropathy. Diabetologia 58, 2169–2180 (2015). https://doi.org/10.1007/s00125-015-3642-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-015-3642-4