
Abstract
The interactions between c-Kit and its ligand, stem cell factor (SCF), play an important role in haematopoiesis, pigmentation and gametogenesis. c-Kit is also found in the pancreas, and recent studies have revealed that c-Kit marks a subpopulation of highly proliferative pancreatic endocrine cells that may harbour islet precursors. c-Kit governs and maintains pancreatic endocrine cell maturation and function via multiple signalling pathways. In this review we address the importance of c-Kit signalling within the pancreas, including its profound role in islet morphogenesis, islet vascularisation, and beta cell survival and function. We also discuss the impact of c-Kit signalling in pancreatic disease and the use of c-Kit as a potential target for the development of cell-based and novel drug therapies in the treatment of diabetes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus, characterised primarily by a disturbance in glucose homeostasis, is widely recognised as a global epidemic. Type 1 and type 2 diabetes mellitus constitute the two main types of diabetes. Type 1 diabetes mellitus is associated with absolute insulin deficiency as a consequence of autoimmune-mediated destruction of beta cells in the pancreas [1]. In contrast, type 2 diabetes mellitus is characterised by a failure of beta cells to compensate for systemic insulin resistance [1]. In both diabetic states, beta cells are exposed to a hyperglycaemic environment that results in the progressive deterioration of beta cell function and the induction of beta cell apoptosis. Of note, insulin resistance is neither necessary nor sufficient to cause diabetes, whereas beta cell dysfunction is the primary cause of both types of diabetes mellitus. It follows that a major aim for diabetes research is to determine how to restore and preserve beta cell function. Therefore, understanding the factors that govern beta cell expansion and survival in the pancreas is essential. One such factor currently under examination is c-Kit, a receptor tyrosine kinase, and its ligand, stem cell factor (SCF). The binding of SCF to c-Kit results in its activation, which mediates survival, migration and proliferation in multiple cell types, including pancreatic beta cells. This review provides a brief overview of SCF–c-Kit biochemistry and their role in organ development. The current understanding of c-Kit in the developing pancreas, especially with regard to islet formation in both humans and rodents, is summarised. Furthermore, we discuss c-Kit as a marker and maintenance factor for pancreatic stem/progenitor cells, providing new insights into the signal transduction machinery by which c-Kit regulates beta cell survival and function under normal and diabetic physiological conditions.
c-Kit and SCF
Structure of c-Kit and SCF
c-Kit is a cellular homologue of the v-Kit oncogene. It is a member of the type III group of receptor tyrosine kinases, encoded by the W locus on chromosome 4 (4q11-21) in humans, and on chromosome 5 in mice [2, 3]. Structurally, c-Kit is closely related to platelet-derived growth factor receptor (PDGFR), sharing 63% homology with the tyrosine kinase sequences and 53% homology with the amino terminus of the kinase domain [4]. Similar to PDGFR, c-Kit consists of an extracellular region comprising five Ig-like domains, a single membrane-spanning region and a cytoplasmic region containing a hydrophilic kinase insert domain (Fig. 1a). The first three domains form the ligand-binding pocket, while the fourth and fifth domains play a critical role in c-Kit monomer positioning and dimerisation [5]. A similar ligand-binding mechanism has been proposed for PDGFR. Like PDGFR, the intracellular portion of c-Kit consists of a juxtamembrane domain with an ATP-binding region, a phosphotransferase region split into two domains by a kinase insert and a COOH-terminal tail. Most of the phosphorylation sites are located in the cytoplasmic region and are important for transducing intracellular activation signals.

Structure of c-Kit and SCF. (a) c-Kit has extracellular, transmembrane and intracellular regions. The extracellular region consists of five Ig-like domains. The transmembrane region keeps c-Kit attached to the cell membrane. The intracellular region contains two kinase domains split into two parts by the kinase insert domain [4, 5, 10–13]. (b) sSCF is generated by cleavage of mSCF (220 or 248) [6–9, 14, 15]. Primary proteolytic cleavage sites are indicated by the arrows
SCF is a product of the Sl locus, mapped to chromosome 12 in humans and chromosome 10 in mice [6]. There are six known SCF transcripts in humans and four in mice, but two alternative transcripts are predominantly synthesised in the pancreas, as membrane-bound forms of 220 or 248 (mSCF) amino acids (Fig. 1b). The protein structure of both mSCF forms includes an extracellular domain, a transmembrane domain and an intracellular region. In humans, SCF 248 contains a proteolytic cleavage site, possibly accelerating the production of soluble SCF (sSCF) 165 by post-transcriptional modification. However, both SCF 220 and 248 can be cleaved in mice to generate monomeric sSCF (Fig. 1b) [7]. Dimerisation of mSCF makes it more biologically active than its monomeric counterpart [8]. mSCF also results in more persistent c-Kit activation and prolonged receptor lifespan, whereas sSCF induces relatively transient receptor activation and enhances receptor degradation [9]. SCF is produced and released by various cells, including fibroblasts, keratinocytes and endothelial cells [10, 11]. It has also been reported that both Scf (also known as Kitl) mRNA and protein expression occur in adult mouse islets [12].
SCF and c-Kit signalling pathways
Receptor activation
c-Kit exists as a monomer and is stimulated to form homodimers following binding of SCF to the first three Ig-like domains of c-Kit [13]. The subsequent interaction between Ig-like domains 4 and 5 in adjacent c-Kit monomers works to further stabilise c-Kit dimerisation, allowing correct positioning and efficient trans-phosphorylation of its cytoplasmic kinase domains [14, 15]. The juxtamembrane domain plays a critical role in regulating c-Kit activity [16] (Fig. 1a). SCF stimulation promotes the release of the juxtamembrane domain from the activation loop, enabling catalytic function of the receptor tyrosine kinase to transduce its downstream signal [5].
Molecular signal transduction
Nine tyrosine phosphorylation sites have been identified on c-Kit (Fig. 1a). Trans-phosphorylation occurs on tyrosine kinase residues, which act as docking sites for signalling kinase molecules containing a Src homology 2 domain and a phosphotyrosine-binding domain (Fig. 2). c-Kit phosphorylation at Y721 activates the phosphoinositide 3-kinase (PI3K) pathway, resulting in enhanced cell survival and proliferation [17]. Phosphorylation of c-Kit at Y703 and Y936 activates the mitogen-activated protein kinase (MAPK) pathway [18], which affects gene transcription, cell differentiation and proliferation [13]. c-Kit can be phosphorylated at Y568, Y570 and Y936 to enhance interaction with the SH2 domain of the Src family of tyrosine kinases (SFK). Activation of this kinase family is associated with cell proliferation and survival via Akt phosphorylation. Also, one of the mechanisms of activation of the Janus kinase–signal transducers and activators of transcription (JAK–STAT) pathway requires the action of both SFK and PI3K [13, 19, 20]. There are also reports suggesting that SFK directly phosphorylates focal adhesion kinase, which is important for cell migration [21]. The phospholipase C-γ pathway can interact with the tyrosine kinase residue Y730 of c-Kit [22], and has been found to play an important role in suppressing cell apoptosis [23]. The phosphorylation of Y823 is a ligand-activated event required for sustaining phosphorylation of downstream signalling molecules (e.g. PI3K, MAPK) of c-Kit. Mutation of Y823 leads to increased c-Kit internalisation and degradation, suggesting a role for this site in stabilising the active kinase conformation [24]. CrkII is an adaptor protein of the Crk family, which can be phosphorylated by SCF and can interact with Y900 in c-Kit. However, the functional consequence of Crk binding with Y900 in c-Kit via the p85 subunit of PI3K is not clear [25], but may provide possible link to the c-Jun N-terminal kinase (JNK) pathway. c-Kit signalling pathways are not simple linear reactions, but, rather, integrated inputs from different pathways that determine the biological consequences in different cellular contexts. For instance, c-Kit crosstalks with the erythropoietin receptor or interleukin receptors to recruit common downstream signalling molecules, creating a method for modulating diverse physiological responses [26–29].

c-Kit signalling pathways. Binding of SCF triggers c-Kit downstream signalling pathways, including the MAPK (blue), PI3K (green), phospholipase C-γ (PLCγ) and Janus kinase–signal transducers and activators of transcription (JAK–STAT) pathways (orange). Activation of these pathways is implicated in numerous cellular processes, such as gene transcription, cellular proliferation, differentiation, survival and adhesion [16–27]. Src homology region 2 domain-containing phosphatase-1 (SHP-1), Casitas b-lineage lymphoma (c-Cbl), SOCS and the protein kinase C signals (brown) are involved c-Kit downregulation. BAD, Bcl-2-associated death promoter; DAG, diacylglycerol; GRB2, growth factor receptor-bound protein 2; MEK1/2, mitogen-activated protein kinase/extracellular-regulated kinase kinase 1/2; mTOR, mechanistic target of rapamycin
Receptor downregulation
Tight regulation of c-Kit signalling is crucial for maintaining proper cellular function. c-Kit signalling can be attenuated through several routes, including receptor internalisation, tyrosine dephosphorylation and kinase domain inactivation by serine phosphorylation. It has been shown that c-Kit internalisation can be initiated by the association of the E3 ubiquitin ligase, Casitas b-lineage lymphoma (c-Cbl) at Y568 and Y936 on c-Kit [30, 31]. The signalling proteins, suppressor of cytokine signalling isoforms 1 (SOCS1) and 6 (SOCS6), can also induce c-Kit endocytosis and its subsequent lysosomal degradation [32]. Furthermore, protein kinase C signalling may exert negative feedback on c-Kit activity through serine phosphorylation at S741 and S746 in the receptor, which results in shedding of the c-Kit extracellular domain from the cell surface [13, 33]. Lymphocyte-specific adaptor protein Lnk also plays an important role in c-Kit downregulation. Lnk preferentially binds to the juxtamembrane region of c-Kit, suppressing the intrinsic c-Kit catalytic activity [34]. In addition, c-Kit signalling can be negatively regulated by Src homology region 2 domain-containing phosphatase-1 (SHP-1) through receptor tyrosine dephosphorylation [35].
c-Kit function in organ development
A strong correlation between c-Kit expression and the pluripotency of embryonic stem cells (ESCs) indicates that c-Kit is a critical factor for the differentiation and survival of these cells. c-Kit-null murine ESCs die when induced to differentiate, and apoptosis also occurs upon differentiation of normal ESCs treated with a c-Kit-neutralising antibody [36]. Similarly, combinations of SCF and other growth factors have been reported to promote efficient haematopoietic differentiation from human ESCs [37], and other reports indicate that SCF-c-Kit interactions are essential for haematopoietic stem cell proliferation, survival and adhesion via the PI3K pathway [38]. Indeed, investigation of the physiological roles of SCF and c-Kit in mice with various naturally occurring mutations in the W and Sl loci, revealed that SCF-c-Kit interactions play a crucial functional role in a wide variety of tissues. c-Kit signalling plays an indispensable role in melanocyte survival and pigmentation, demonstrated by a lack of hair pigmentation, associated with loss of melanocytes, in W or Sl mutant mice [39]. SCF-c-Kit interactions are important for gametogenesis, as evidenced in men by an SCF mutation that causes idiopathic male infertility [40]. Furthermore, W and Sl mutant mice display increased germ cell apoptosis, with a corresponding degree of sterility [41]. More importantly, accumulating evidence suggests that c-Kit expression is also linked to the development of the endocrine pancreas, as well as to beta cell survival and function [11, 12, 42–53].
c-Kit expression and function in the pancreas
c-Kit in the developing rodent pancreas
c-Kit expression was first detected in RINm5F rat insulinoma cell lines and fetal rat islets (Table 1) [43]. Subsequent immunohistochemistry studies revealed c-Kit localisation to pancreatic ducts [42], and c-Kit (also known as Kit) mRNA was detectable by embryonic day 13 in the fetal rat pancreas [48]. One cell lineage tracing study utilising lacZ transgenic murine embryos showed that c-Kit expression was restricted in a subpopulation of endocrine and epithelial cells [46]. The abundance of c-Kit expression in early rodent pancreatic development indicates that it may be involved in maintaining the endocrine cell precursor pool in fetal rodents. We investigated c-Kit expression in the rodent pancreas during the transition from prenatal to postnatal life at our laboratory. c-Kit was localised to the ductal region and in 40% of beta cells at embryonic day 18, but c-Kit+ cells progressively declined in the pancreas postnatally [47]. These observations suggest that decreased c-Kit expression may correlate with islet endocrine cell maturation. Two distinct putative stem cell-like populations expressing either c-Kit or Sca-1, another stem cell antigen, in the developing rodent pancreas were recently characterised. Flow cytometry analyses revealed that the isolated c-Kit+ cell population co-expressed markers related to islet differentiation, including pancreatic and duodenal homeobox 1 (PDX-1) and neurogenin 3 (NGN3), but isolated Sca-1+ cells lacked expression of these markers [54]. These findings imply that c-Kit can be used as a marker to identify a putative islet precursor cell population in the developing rodent endocrine pancreas.
Few studies have focused on the effects of c-Kit mutations with regard to pancreatic endocrine morphogenesis. One study reported that islets were still present in the absence of c-Kit in W lacZ /W lacZ embryos, although few morphometric details were examined, and there was no indication of functional status [55]. These data raise two possibilities: (1) c-Kit is not critical for the determination and specification of islet formation, but is more involved in islet survival and function; or, more likely, (2) there are redundant pathways, such as PDGFR signalling, compensating for the loss of c-Kit during pancreatic development in rodents.
c-Kit in the developing human pancreas
The developing human pancreas and multiple human pancreatic cancer cell lines express both c-Kit and SCF (Table 1) [11, 49, 53, 56]. c-Kit+ cells were detected at the earliest stage of human pancreatic development, as single endocrine cells differentiated from ducts [49]. The expression of c-Kit was restricted to ductal regions and adjoining neogenic islet clusters, whereas SCF expression was scattered throughout the developing human pancreas [11]. The majority of c-Kit+ cells displayed the ductal cell marker, cytokeratin 19, and transcription factors associated with islet differentiation, including PDX-1, sex determining region Y-box 9 (SOX9), NGN3, and homeobox protein Nkx6.1, between 8 and 12 weeks of human fetal age. However, as age progressed to 19–21 weeks, co-expression between c-Kit and these markers declined [11]. These results coincide with the findings of studies in rodents, indicating that cell populations expressing c-Kit may: (1) represent endocrine precursors participating in islet neogenesis; and/or (2) serve as an instructive signal, directing islet differentiation and proliferation in the developing human pancreas.
SCF–c-Kit in islet differentiation
SCF–c-Kit interactions mediate beta cell differentiation and proliferation and have been demonstrated across multiple species in vitro. Stimulation of c-Kit activity promoted gene transcription and proliferation in INS-1 rat insulinoma cells [12, 46, 51]. Furthermore, treatment by exogenous SCF induced differentiation of human pancreatic carcinoma, epithelial-like cells (PANC-1) into islet-like clusters [53]. Using isolated primary rodent islet cultures, fetal islet clusters treated with SCF exhibited augmented insulin and total DNA content [42]. Our previous study demonstrated that rat islet epithelial monolayers that were expanded on collagen I were highly proliferative, with 45% of cells expressing c-Kit [57]. These c-Kit+ cells co-expressed PDX-1, NGN3, paired box 4 (PAX4), and multiple undifferentiated cell markers, and could form islet-like clusters under specific culture conditions [57]. SCF–c-Kit interactions also have a direct physiological effect on neonatal porcine islets. SCF released from microencapsulated Sertoli cells significantly accelerated the differentiation and maturation of neonatal porcine islets in vitro [58]. Our group has demonstrated, by SCF and Wortmannin inhibition experiments, that SCF enhances the differentiation of c-Kit-expressing immature endocrine cells in the human fetal pancreas, likely through stimulation of the PI3K pathway [11]. These findings suggest the potential importance of c-Kit in promoting early beta cell differentiation and survival.
c-Kit in the regenerating pancreas
Although there is no consensus on the identity and origin of pancreatic stem/progenitor cells, c-Kit-expressing cells exhibit many stem/progenitor cell features in the regenerating pancreas. In a study of islet regeneration performed with the pancreatic duct-ligated rat model, we found that c-Kit expression was activated only in the ductal cells of the ligated tail portion during islet neogenesis, along with upregulation of PDX-1 and Nkx2.2 expression, suggesting that islet neogenesis may arise from these ductal progenitor cells [59]. Increased c-Kit and PDX-1 expression was also observed in islets of the streptozotocin (STZ)-induced diabetic rat pancreases, suggesting that c-Kit may play a role during beta cell regeneration [60]. In a model of rat pancreatitis in which extensive pancreatic cellular damage was induced by cerulean, beta cell replenishment occurred due to replication of the highly proliferative pre-existing c-Kit-expressing beta cells [61]. c-Kit-expressing cells from other cell sources may also be involved in promoting beta cell regeneration. Transplanted adult bone marrow-derived c-Kit-expressing cells reduce hyperglycaemia in diabetic mice [62]. The frequency of insulin co-expression with bone marrow-derived c-Kit+ cells was low, and there was no evidence of an increase in PDX-1 expression in these cells. This suggests that the bone marrow-derived c-Kit+ cells did not, themselves, immediately replace the damaged beta cell population. However, they may play a paracrine role in promoting proliferation of the existing beta cells and differentiation of precursor cells. This observation was verified by transplantation of purified (ALDHhighc-Kit+CD133+CD34+) multipotent stromal cells, which showed significantly enhanced islet cell proliferation, insulin production and revascularisation in vivo [63].
c-Kit in pancreatic disease and other metabolic disorders
Despite the necessity of c-Kit as a developmental regulator, it is also characterised as a proto-oncogene [3]. Indeed, inappropriate expression of SCF or c-Kit leads to a wide range of pancreatic diseases, ranging from chronic pancreatitis to pancreatic cancer [64]. It was reported that SCF-c-Kit interactions had an influential effect on mast cells, in which increased mast cell presence correlated with inflammation and fibrosis in chronic pancreatitis [65]. c-Kit is found in ducts during chronic pancreatitis, consistent with other studies reporting increased ductal c-Kit expression after pancreatic ductal ligation in rodents [59]. Pancreatic cancer is amongst the most lethal and least common human cancers and has been linked to diabetes and chronic pancreatitis [66]. High levels of c-Kit expression are also found on pancreatic neoplastic cells and throughout the duct of the cancerous pancreas [67], indicating that SCF–c-Kit interactions have profound implications for pancreatic cancer cell proliferation and invasiveness. While cancer aetiology is varied, both somatic and germline mutations in the SCF allele have been linked to testicular cancer. The findings of these genome-wide association studies (GWAS) are notable because it is unusual to find these risk alleles with per-allele odds ratios larger than 2 [68]. The single nucleotide polymorphism (SNP) variants in signalling events mediated by SCF were also associated with advanced cirrhosis, indicating that SCF–c-Kit interactions could play a role in other complex metabolic diseases [69]. SH2B1 was identified as a SH2 domain-containing adaptor protein and directly associates with the Y568 binding site in c-Kit. Interestingly, the SH2B1 risk alleles are genetically associated with human obesity in GWAS on large populations [70, 71]. Aside from SH2B1 SNPs, several SH2B1 non-synonymous variants were also genetically linked to insulin resistance and type 2 diabetes [72], implying that SCF–c-Kit pathways may be one of the causal factors for the development of diabetes in humans.
Altered c-Kit expression affects islet function and survival
c-Kit mutation and overexpression in islets
The first reported in vivo evidence linking c-Kit with beta cell survival and function utilised mice carrying the viable dominant spotting (W v) mutation [50]. The W v point mutation causes a threonine to methionine substitution at the first catalytic region of the c-Kit kinase domain, greatly diminishing its kinase activity to ~10–20% of that observed in normal c-Kit monomers. More importantly, the c-Kit W v mutant monomer can act in a trans-dominant manner to prevent tyrosine phosphorylation of normal c-Kit by dimerisation, reflected by a ~60% reduction in c-Kit transduction activity [73]. Phenotypic analysis of mice heterozygous for the W v mutation (c-Kit Wv/+) showed impaired glucose tolerance and a marked insulin secretory defect by 8 weeks of age [50]. The development of a diabetic phenotype, particularly in male mice, was due to a severe loss of beta cell mass and reduced proliferative capacity. Interestingly, these changes were not observable in female mice until 40 weeks of age, indicating that the delay of diabetes was possibly due to differences between sexes in sex hormone function [50]. Furthermore, downregulation of the PI3K–Akt–glycogen synthase kinase 3β (GSK3β) pathway, which connects c-Kit activity to physiological changes in beta cells of c-Kit Wv/+ mice, was demonstrated [52]. Inhibition of activated GSK3β with 1-azakenpaullone, a GSK3β inhibitor, was shown to rescue c-Kit Wv/+ mice from defective glucose metabolism and increase islet cyclin D1 and PDX-1 expression [52]. This observation suggests that dysregulation of the Akt–GSK3β pathway, downstream of c-Kit, is responsible for the onset of diabetes in c-Kit Wv/+ mice.
The most convincing evidence supporting the importance of the impact of c-Kit on beta cell function and survival was determined using a transgenic mouse model with overexpression of the human c-KIT gene specifically in beta cells directed by the rat insulin promoter (c-KitβTg mice) [12]. Our group showed that c-KitβTg mice displayed improved glucose tolerance, which was associated with expansion of beta cell mass as a result of increased beta cell proliferation. Overexpression of c-KIT also led to a profound effect on insulin secretion in response to glucose challenge, and insulin content in beta cells of these mice was increased compared with that in their control littermates [12]. Increased c-Kit activity upregulated the Akt–GSK3β–cyclin D1 pathway, which was associated with enhanced expression of islet-specific transcription factors, including PDX-1 and MafA [12]. Notably, c-KitβTg mice also counteracted the detrimental effects of a 4 week high-fat diet (HFD) challenge, in which beta cell function and mass were maintained. Furthermore, overexpression of c-KIT in beta cells of c-Kit Wv/+ mice could partially reverse the diabetic phenotype, demonstrating that c-Kit has a beneficial effect on beta cells and directly influences beta cell health [12].
c-Kit and Fas receptor interplay in islets
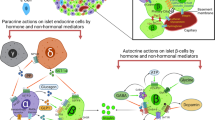
Accumulating evidence has demonstrated that Fas receptor (FasR) signalling contributes to cell apoptosis, which is accompanied by the absence of c-Kit signalling in melanocytes [74], gametes [75], oocytes [76] and haematopoietic stem cells [77]. FasR is known as apoptosis antigen 1, CD95, or tumour necrosis factor receptor superfamily member 6. Fas ligand binding and subsequent activation of FasR leads to programmed cell death in many systems via activation of the caspase 8-mediated downstream cell death machinery. We recently reported that deficient c-Kit signalling resulted in increased beta cell death in c-Kit Wv/+ mice, which was associated with upregulation of p53 levels and induction of FasR activity in c-Kit Wv/+ mouse islets [51] (Fig. 3). It is clear that p53 is an important checkpoint protein that promotes cell cycle arrest [78]. Several lines of evidence have indicated that FasR expression can be upregulated by p53 activation through promotion of FasR gene transcription and trafficking of FasR to the cell surface via the Golgi apparatus [79, 80]. The interplay between c-Kit and FasR signalling in beta cell survival was further verified by a double mutant mouse model (c-Kit Wv/+ mice with the lymphoproliferation [lpr] mutation, which disrupts expression of the cell surface FasR). Lack of functional FasR in c-Kit Wv/+ mice reduced the extent of beta cell apoptosis via downregulation of the caspase 8-mediated extrinsic apoptotic pathway, and enhanced FLICE-like inhibitor protein/nuclear factor κB pro-survival signalling [51]. These in vivo results were further supported by in vitro observations using INS-1 cells, whereby c-Kit activation negatively modulated both p53 and FasR in a PI3K-dependent manner. In particular, knockdown of c-Kit in INS-1 cells led to increased p53 and FasR levels, reversed by treatment with a p53 inhibitor or Fas small interfering (si)RNA, ultimately resulting in a reduction in apoptosis [51]. These data imply that a balance between c-Kit and FasR signalling is instrumental in maintaining proper beta cell mass turnover.

c-Kit signalling pathways in the beta cell. c-Kit regulates beta cell proliferation, survival and function through the Akt–GSK3β pathway, the suppression of FasR-mediated caspase-dependent apoptotic signalling; and by crosstalking with the IR. These interactions occur via (1) direct physical association between two receptors, (2) the common signalling components downstream of c-Kit and IR, or (3) the enhanced autocrine insulin effects on IR function. c-FLIP, cellular FLICE-inhibitory protein; FADD, Fas-associated death domain-containing protein; FasL, Fas ligand; MafA, v-maf musculoaponeurotic fibrosarcoma oncogene family, protein A (avian); NF-κB, nuclear factor κB, S6, ribosomal protein S6 kinase
c-Kit and insulin receptor crosstalk in islets
It has been documented that c-Kit is able to crosstalk with many growth factor and cytokine receptors [26–28, 81]. For example, erythropoietin and SCF have synergistic effects on erythropoiesis. In fact, erythropoietin appears to stimulate c-Kit dimerisation and tyrosine phosphorylation in human hematopoietic cell lines [81]. Recently, treatment with recombinant human erythropoietin was shown to provide protection against the development of diabetes in STZ-induced and db/db mice. This was associated with a significant up regulation of c-Kit and vascular endothelial growth factor-A (VEGF-A) protein expression [82]. It is well established that insulin signalling pathways can participate in crosstalk with other receptors [83, 84]. Insulin, secreted from beta cells in response to glucose stimulation, activates the beta cell insulin receptor (IR) and the downstream signalling transducer, IRS. This, in turn, triggers the PI3K–Akt pathway, which is an important mediator of beta cell proliferation, function and survival. Overexpression of c-KIT leads to increased IR protein expression and tyrosine phosphorylation of IRS1 and -2, and treatment of INS-1 cells with exogenous SCF leads to enhanced co-localisation of c-Kit and IR, as revealed by double immunofluorescence [12]. The effect of c-Kit and IR signalling was suppressed by wortmannin (PI3K inhibitor) or c-Kit siRNA, suggesting an interaction between these two upstream pathways through: (1) physical association between two receptors, (2) common pathway components downstream of c-Kit and IR (e.g. PI3K/Akt or GSK3β [45]), or (3) enhanced insulin biosynthesis and autocrine feedback of insulin effects on IR function, as seen in dose- and time-dependent models, where positive signals were observed in juvenile c-KitβTg islets and negative signals were seen in aged c-KitβTg mice (Fig. 3) [12].
c-Kit induces beta cell VEGF-A secretion thereby modulating islet vascular formation
Pancreatic islets are richly vascularised and feature highly permeable capillaries. In the mature islet, every beta cell is located adjacent to a capillary. This spatial organisation provides an access point for insulin to enter circulation, and proper vascular formation is essential for the normal development and function of the islet [85, 86]. The formation of islet vasculature relies on VEGF-A secretion primarily from beta cells. VEGF-A binding to VEGF receptor 2 on neighbouring endothelial cells promotes endothelial cell proliferation and migration, eventually resulting in islet vessel formation [86]. By deleting VEGF-A, either in the whole pancreas or in beta cells, the fenestration of islet capillaries, required for fine-tuning blood glucose regulation, was severely impaired [86], and led to beta cell dysfunction [87, 88].
The causal link between c-Kit and VEGF-A production has been documented previously in multiple cancer pathologies [89, 90], leading to the question of whether or not c-Kit regulates islet vascular formation via VEGF-A production. In a recent study we provided evidence that VEGF-A production in INS-1 cells was augmented by SCF treatment; an effect mediated by the PI3K-mammalian target of rapamycin (mTOR) pathway [91]. Substantial loss of islet vasculature was discovered in c-Kit Wv/+ mice, which was reversed by beta cell-specific c-KIT overexpression. These data suggest that c-Kit-induced VEGF-A production is critical for normal islet vascularisation. Interestingly, endothelial–beta cell interactions may not always result in a positive outcome, as islet endothelial cells have been shown to contribute to islet destruction following an inflammatory response during HFD treatment [92]. Inflammatory factor expression was increased in islets, which resulted in a decrease in beta cell function and led to hyperglycaemia [92]. These findings indicate that c-Kit can be a therapeutic target that promotes revascularisation during islet replacement procedures. However, c-Kit activity must be tightly regulated in order to control the VEGF-A concentration of the microenvironment, for the purpose of maintaining a normal and stable islet vascular network and proper beta cell function and survival.
c-Kit, stem cell and cell-based therapies for diabetes treatment
A significant limitation of cell-based therapies for diabetes treatment is the lack of donor cell sources. To circumvent this, research has been dedicated to isolating pancreatic islet precursors and applying differentiation protocols to obtain mature functional islets [93]. To accomplish this, the identification of cell types with the potential to differentiate into islets is of critical importance. Multiple studies have attempted to differentiate c-Kit+ cells gathered from different organs into beta cells or beta-like cells. In humans, a population of pancreatic progenitor cells containing multiple stem cell markers, including c-Kit+ cells purified from the early trimester fetal pancreas, were shown to adopt a pancreatic phenotype ex vivo by differentiating into insulin-expressing cells [94]. Human exfoliated deciduous teeth expressing c-Kit were capable of differentiation towards functional pancreatic endocrine cells when exposed to serum-free conditions [95]. Furthermore, another study used human adipose tissue-derived mesenchymal stem cells, initially expressing c-Kit, to induce pancreatic endocrine cell differentiation under stringent in vitro conditions [92]. In mice, PDX-1 and NGN3 overexpression induced formation of immature insulin/C-peptide-producing cells from mouse embryonic stem cell-derived c-Kit+ endoderm populations [96]. In addition, c-Kit+ cells isolated from the salivary gland of adult mice were differentiated into pancreatic endocrine lineages under glucagon-like peptide 1 treatment [97, 98]. Isolated Thy1+ cells from mouse liver, which expressed c-Kit and other stem cell markers, generated islet-like clusters that expressed islet differentiation markers and endocrine hormones under high glucose conditions [99]. Taken together, these studies provide evidence of the plasticity of c-Kit+ cells in non-pancreatic tissues and their potential to become or support beta cells. The major consideration is the functional relevance of these differentiated clusters and their potential for inducing tumorigenesis. Thus, before these c-Kit+ progenitors can be used in a therapeutic setting, a feasible differentiation protocol must be developed to study their functional status and tumorigenic properties. These risks would seemingly be lessened if terminally differentiated cells were used during transplantation.
Summary and perspectives
Data obtained within the past decade have provided insights into the biological role of c-Kit in the pancreas, particularly with respect to beta cell growth and function. c-Kit is detectable not only in the pancreatic ductal region, but also in a subpopulation of beta cells with a high proliferative capacity. c-Kit-expressing cells participate in the reversal of pancreatic damage and diabetes by assisting in pancreatic endocrine regeneration, suggesting that c-Kit may be required for maintaining the pancreatic stem/progenitor cell population. More recent studies have uncovered the mechanism by which manipulation of these c-Kit-expressing cells in vitro can result in their ability to proliferate, differentiate and secrete insulin in response to glucose. In addition, animal studies have provided detailed information on the functional role of c-Kit in vivo. c-Kit influences FasR and IR signalling such that, because of convergence of these pathways, it can affect the dynamic balance of beta cell turnover and appropriate function of beta cells under basal and glucose-stimulated conditions. Furthermore, the connection between c-Kit signalling and VEGF-A production suggests a paracrine regulatory role of pancreatic endocrine cells in the formation of islet vasculature and autocrine signalling that promotes beta cell differentiation and proliferation. These findings are particularly valuable in understanding the underlying mechanisms responsible for beta cell loss and dysfunction, and, to a greater extent, will contribute to new and more physiologically relevant cell replacement therapies for the treatment of diabetes.
Abbreviations
- c-Kit Wv/+ :
-
Heterozygous c-Kit W v mutant mice
- c-KitβTg :
-
Transgenic mice with beta cell-specific c-KIT overexpression
- ESC:
-
Embryonic stem cell
- FasR:
-
Fas receptor
- GSK3β:
-
Glycogen synthase kinase 3β
- GWAS:
-
Genome-wide association studies
- HFD:
-
High-fat diet
- IR:
-
Insulin receptor
- MAPK:
-
Mitogen-activated protein kinase
- NGN3:
-
Neurogenin 3
- PDGFR:
-
Platelet-derived growth factor receptor
- PDX-1:
-
Pancreatic and duodenal homeobox 1
- PI3K:
-
Phosphoinositide 3-kinase
- SCF:
-
Stem cell factor (s- soluble m- membrane)
- SFK:
-
Src family of tyrosine kinases
- siRNA:
-
Small interfering RNA
- SNP:
-
Single nucleotide polymorphism
- SOCS:
-
Suppressor of cytokine signalling
- STZ:
-
Streptozotocin
- VEGF-A:
-
Vascular endothelial growth factor-A
References
American Diabetes Association (2009) Diagnosis and classification of diabetes mellitus. Diabetes Care 32(Suppl 1):S62–S67
Chabot B, Stephenson DA, Chapman VM et al (1988) The proto-oncogene c-kit encoding a transmembrane tyrosine kinase receptor maps to the mouse W locus. Nature 335:88–89
Yarden Y, Kuang WJ, Yang-Feng T et al (1987) Human proto-oncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J 6:3341–3351
Yarden Y, Escobedo JA, Kuang WJ et al (1986) Structure of the receptor for platelet-derived growth factor helps define a family of closely related growth factor receptors. Nature 323:226–232
Yuzawa S, Opatowsky Y, Zhang Z et al (2007) Structural basis for activation of the receptor tyrosine kinase KIT by stem cell factor. Cell 130:323–334
Ropers HH, Craig IW (1989) Report of the committee on the genetic constitution of chromosomes 12 and 13. Cytogenet Cell Genet 51:259–279
Majumdar MK, Feng L, Medlock E et al (1994) Identification and mutation of primary and secondary proteolytic cleavage sites in murine stem cell factor cDNA yields biologically active, cell-associated protein. J Biol Chem 269:1237–1242
Hsu YR, Wu GM, Mendiaz EA et al (1997) The majority of stem cell factor exists as monomer under physiological conditions. Implications for dimerization mediating biological activity. J Biol Chem 272:6406–6415
Langley KE, Bennett LG, Wypych J et al (1993) Soluble stem cell factor in human serum. Blood 81:656–660
Welker P, Grabbe J, Gibbs B et al (1999) Human mast cells produce and differentially express both soluble and membrane-bound stem cell factor. Scand J Immunol 49:495–500
Li J, Goodyer CG, Fellows F, Wang R (2006) Stem cell factor/c-Kit interactions regulate human islet-epithelial cluster proliferation and differentiation. Int J Biochem Cell Biol 38:961–972
Feng ZC, Li J, Turco BA et al (2012) Critical role of c-Kit in beta cell function: increased insulin secretion and protection against diabetes in a mouse model. Diabetologia 55:2214–2225
Reber L, Da Silva CA, Frossard N (2006) Stem cell factor and its receptor c-Kit as targets for inflammatory diseases. Eur J Pharmacol 533:327–340
Blechman JM, Lev S, Barg J et al (1995) The fourth immunoglobulin domain of the stem cell factor receptor couples ligand binding to signal transduction. Cell 80:103–113
Philo JS, Wen J, Wypych J et al (1996) Human stem cell factor dimer forms a complex with two molecules of the extracellular domain of its receptor, Kit. J Biol Chem 271:6895–6902
Chan PM, Ilangumaran S, La Rose J et al (2003) Autoinhibition of the kit receptor tyrosine kinase by the cytosolic juxtamembrane region. Mol Cell Biol 23:3067–3078
Hashimoto K (2002) Necessity of tyrosine 719 and phosphatidylinositol 3′-kinase-mediated signal pathway in constitutive activation and oncogenic potential of c-kit receptor tyrosine kinase with the Asp814Val mutation. Blood 101:1094–1102
Thömmes K, Lennartsson J, Carlberg M, Rönnstrand L (1999) Identification of Tyr-703 and Tyr-936 as the primary association sites for Grb2 and Grb7 in the c-Kit/stem cell factor receptor. Biochem J 341:211–216
Timokhina I, Kissel H, Stella G, Besmer P (1998) Kit signaling through PI 3-kinase and Src kinase pathways: an essential role for Rac1 and JNK activation in mast cell proliferation. EMBO J 17:6250–6262
Bondzi C, Litz J, Dent P, Krystal GW (2000) Src family kinase activity is required for Kit-mediated mitogen-activated protein (MAP) kinase activation, however loss of functional retinoblastoma protein makes MAP kinase activation unnecessary for growth of small cell lung cancer cells. Cell Growth Differ 11:305–314
Sieg DJ, Hauck CR, Ilic D et al (2000) FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol 2:249–256
Gommerman JL, Sittaro D, Klebasz NZ et al (2000) Differential stimulation of c-Kit mutants by membrane-bound and soluble Steel Factor correlates with leukemic potential. Blood 96:3734–3742
Maddens S, Charruyer A, Plo I et al (2002) Kit signaling inhibits the sphingomyelin-ceramide pathway through PLCγ1: implication in stem cell factor radioprotective effect. Blood 100:1294–1301
Agarwal S, Kazi JU, Rönnstrand L (2013) Phosphorylation of the activation loop tyrosine 823 in c-Kit is crucial for cell survival and proliferation. J Biol Chem 288:22460–22468
Lennartsson J, Wernstedt C, Engström U et al (2003) Identification of Tyr900 in the kinase domain of c-Kit as a Src-dependent phosphorylation site mediating interaction with c-Crk. Exp Cell Res 288:110–118
Ye Z-J, Gulcicek E, Stone K et al (2011) Complex interactions in EML cell stimulation by stem cell factor and IL-3. Proc Natl Acad Sci U S A 108:4882–4887
Drube S, Heink S, Walter S et al (2010) The receptor tyrosine kinase c-Kit controls IL-33 receptor signaling in mast cells. Blood 115:3899–3906
Jahn T, Sindhu S, Gooch S et al (2007) Direct interaction between Kit and the interleukin-7 receptor. Blood 110:1840–1847
Kapur R, Zhang L (2001) A novel mechanism of cooperation between c-Kit and erythropoietin receptor. Stem cell factor induces the expression of Stat5 and erythropoietin receptor, resulting in efficient proliferation and survival by erythropoietin. J Biol Chem 276:1099–1106
Masson K, Heiss E, Band H, Rönnstrand L (2006) Direct binding of Cbl to Tyr568 and Tyr936 of the stem cell factor receptor/c-Kit is required for ligand-induced ubiquitination, internalization and degradation. Biochem J 399:59–67
Wollberg P, Lennartsson J, Gottfridsson E et al (2003) The adapter protein APS associates with the multifunctional docking sites Tyr-568 and Tyr-936 in c-Kit. Biochem J 370:1033–1038
Bayle J, Letard S, Frank R et al (2004) Suppressor of cytokine signaling 6 associates with KIT and regulates KIT receptor signaling. J Biol Chem 279:12249–12259
Edling CE, Pedersen M, Carlsson L et al (2007) Haematopoietic progenitor cells utilise conventional PKC to suppress PKB/Akt activity in response to c-Kit stimulation. Br J Haematol 136:260–268
Simon C, Dondi E, Chaix A et al (2008) Lnk adaptor protein down-regulates specific Kit-induced signaling pathways in primary mast cells. Blood 112:4039–4047
Kozlowski M, Larose L, Lee F et al (1998) SHP-1 binds and negatively modulates the c-Kit receptor by interaction with tyrosine 569 in the c-Kit juxtamembrane domain. Mol Cell Biol 18:2089–2099
Bashamboo A, Taylor AH, Samuel K et al (2006) The survival of differentiating embryonic stem cells is dependent on the SCF-KIT pathway. J Cell Sci 119:3039–3046
Ledran MH, Krassowska A, Armstrong L et al (2008) Efficient hematopoietic differentiation of human embryonic stem cells on stromal cells derived from hematopoietic niches. Cell Stem Cell 3:85–98
Okumura N, Tsuji K, Ebihara Y et al (1996) Chemotactic and chemokinetic activities of stem cell factor on murine hematopoietic progenitor cells. Blood 87:4100–4108
Baxter LL, Hou L, Loftus SK, Pavan WJ (2004) Spotlight on spotted mice: a review of white spotting mouse mutants and associated human pigmentation disorders. Pigment Cell Res 17:215–224
Galan JJ, de Felici M, Buch B et al (2006) Association of genetic markers within the KIT and KITLG genes with human male infertility. Hum Reprod 21:3185–3192
Loveland KL, Schlatt S (1997) Stem cell factor and c-kit in the mammalian testis: lessons originating from Mother Nature’s gene knockouts. J Endocrinol 153:337–344
Oberg-Welsh C, Welsh M (1996) Effects of certain growth factors on in vitro maturation of rat fetal islet-like structures. Pancreas 12:334–339
Oberg C, Waltenberger J, Claesson-Welsh L, Welsh M (1994) Expression of protein tyrosine kinases in islet cells: possible role of the Flk-1 receptor for beta-cell maturation from duct cells. Growth Factors 10:115–126
Welsh M, Annerén C, Lindholm C et al (2000) Role of tyrosine kinase signaling for beta-cell replication and survival. Ups J Med Sci 105:7–15
Welsh M (2012) The platelet-derived growth factor (PDGF) family of tyrosine kinase receptors: a Kit to fix the beta cell? Diabetologia 55:2092–2095
Rachdi L, El Ghazi L, Bernex F et al (2001) Expression of the receptor tyrosine kinase KIT in mature beta-cells and in the pancreas in development. Diabetes 50:2021–2028
Yashpal NK, Li J, Wang R (2004) Characterization of c-Kit and nestin expression during islet cell development in the prenatal and postnatal rat pancreas. Dev Dyn 229:813–825
LeBras S, Czernichow P, Scharfmann R (1998) A search for tyrosine kinase receptors expressed in the rat embryonic pancreas. Diabetologia 41:1474–1481
Li J, Quirt J, Do HQ et al (2007) Expression of c-Kit receptor tyrosine kinase and effect on beta-cell development in the human fetal pancreas. Am J Physiol Endocrinol Metab 293:E475–E483
Krishnamurthy M, Ayazi F, Li J et al (2007) c-Kit in early onset of diabetes: a morphological and functional analysis of pancreatic beta-cells in c-KitW-v mutant mice. Endocrinology 148:5520–5530
Feng Z-C, Riopel M, Li J et al (2013) Downregulation of Fas activity rescues early onset of diabetes in c-Kit Wv/+ mice. Am J Physiol Endocrinol Metab 304:E557–E565
Feng Z-C, Donnelly L, Li J et al (2012) Inhibition of Gsk3β activity improves β-cell function in c-Kit Wv/+ male mice. Lab Investig 92:543–555
Wu Y, Li J, Saleem S et al (2010) c-Kit and stem cell factor regulate PANC-1 cell differentiation into insulin- and glucagon-producing cells. Lab Investig 90:1373–1384
Ma F, Chen F, Chi Y et al (2012) Isolation of pancreatic progenitor cells with the surface marker of hematopoietic stem cells. Int J Endocrinol 2012:948683
Bernex F, de Sepulveda P, Kress C et al (1996) Spatial and temporal patterns of c-kit-expressing cells in W lacZ/+ and W lacZ/W lacZ mouse embryos. Development 122:3023–3033
Yasuda A, Sawai H, Takahashi H et al (2006) The stem cell factor/c-kit receptor pathway enhances proliferation and invasion of pancreatic cancer cells. Mol Cancer 5:46
Wang R, Li J, Yashpal N (2004) Phenotypic analysis of c-Kit expression in epithelial monolayers derived from postnatal rat pancreatic islets. J Endocrinol 182:113–122
Mancuso F, Calvitti M, Luca G et al (2010) Acceleration of functional maturation and differentiation of neonatal porcine islet cell monolayers shortly in vitro cocultured with microencapsulated sertoli cells. Stem Cells Int 2010:587213
Peters K, Panienka R, Li J et al (2005) Expression of stem cell markers and transcription factors during the remodeling of the rat pancreas after duct ligation. Virchows Arch 446:56–63
Tiemann K, Panienka R, Klöppel G (2007) Expression of transcription factors and precursor cell markers during regeneration of beta cells in pancreata of rats treated with streptozotocin. Virchows Arch 450:261–266
Gong J, Zhang G, Tian F, Wang Y (2012) Islet-derived stem cells from adult rats participate in the repair of islet damage. J Mol Histol 43:745–750
Hess D, Li L, Martin M et al (2003) Bone marrow-derived stem cells initiate pancreatic regeneration. Nat Biotechnol 21:763–770
Bell GI, Putman DM, Hughes-Large JM, Hess DA (2012) Intrapancreatic delivery of human umbilical cord blood aldehyde dehydrogenase-producing cells promotes islet regeneration. Diabetologia 55:1755–1760
Raimondi S, Lowenfels AB, Morselli-Labate AM et al (2010) Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract Res Clin Gastroenterol 24:349–358
Esposito I, Friess H, Kappeler A et al (2001) Mast cell distribution and activation in chronic pancreatitis. Hum Pathol 32:1174–1183
Raimondi S, Maisonneuve P, Lowenfels AB (2009) Epidemiology of pancreatic cancer: an overview. Nat Rev Gastroenterol Hepatol 6:699–708
Esposito I, Kleeff J, Bischoff SC et al (2002) The stem cell factor-c-kit system and mast cells in human pancreatic cancer. Lab Investig 82:1481–1492
Kanetsky PA, Mitra N, Vardhanabhuti S et al (2009) Common variation in KITLG and at 5q31.3 predisposes to testicular germ cell cancer. Nat Genet 41:811–815
Chen Q-R, Braun R, Hu Y et al (2013) Multi-SNP analysis of GWAS data identifies pathways associated with nonalcoholic fatty liver disease. PLoS One 8:e65982
Willer CJ, Speliotes EK, Loos RJF et al (2009) Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet 41:25–34
Thorleifsson G, Walters GB, Gudbjartsson DF et al (2009) Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat Genet 41:18–24
Doche ME, Bochukova EG, Su H-W et al (2012) Human SH2B1 mutations are associated with maladaptive behaviors and obesity. J Clin Invest 122:4732–4736
Nocka K, Majumder S, Chabot B et al (1989) Expression of c-kit gene products in known cellular targets of W mutations in normal and W mutant mice—evidence for an impaired c-kit kinase in mutant mice. Genes Dev 3:816–826
Sharov AA, Li G-Z, Palkina TN et al (2003) Fas and c-kit are involved in the control of hair follicle melanocyte apoptosis and migration in chemotherapy-induced hair loss. J Invest Dermatol 120:27–35
Sakata S, Sakamaki K, Watanabe K et al (2003) Involvement of death receptor Fas in germ cell degeneration in gonads of Kit-deficient Wv/Wv mutant mice. Cell Death Differ 10:676–686
Moniruzzaman M, Sakamaki K, Akazawa Y, Miyano T (2007) Oocyte growth and follicular development in KIT-deficient Fas-knockout mice. Reproduction 133:117–125
Mori T, Ando K, Tanaka K et al (1997) Fas-mediated apoptosis of the hematopoietic progenitor cells in mice infected with murine cytomegalovirus. Blood 89:3565–3573
Rocha S, Martin AM, Meek DW, Perkins ND (2003) p53 represses cyclin D1 transcription through down regulation of Bcl-3 and inducing increased association of the p52 NF-kappaB subunit with histone deacetylase 1. Mol Cell Biol 23:4713–4727
Bennett M, Macdonald K, Chan SW et al (1998) Cell surface trafficking of Fas: a rapid mechanism of p53-mediated apoptosis. Science 282:290–293
Müller M, Wilder S, Bannasch D et al (1998) p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med 188:2033–2045
Kapur R, Cooper R, Zhang L, Williams DA (2001) Cross-talk between α4β1/α5β1 and c-Kit results in opposing effect on growth and survival of hematopoietic cells via the activation of focal adhesion kinase, mitogen-activated protein kinase, and Akt signaling pathways. Blood 97:1975–1981
Choi D, Schroer SA, Lu SY et al (2010) Erythropoietin protects against diabetes through direct effects on pancreatic beta cells. J Exp Med 207:2831–2842
Ricort JM, Tanti JF, van Obberghen E, Le Marchand-Brustel Y (1997) Cross-talk between the platelet-derived growth factor and the insulin signaling pathways in 3T3-L1 adipocytes. J Biol Chem 272:19814–19818
Li J, DeFea K, Roth RA (1999) Modulation of insulin receptor substrate-1 tyrosine phosphorylation by an Akt/phosphatidylinositol 3-kinase pathway. J Biol Chem 274:9351–9356
Lammert E, Cleaver O, Melton D (2001) Induction of pancreatic differentiation by signals from blood vessels. Science 294:564–567
Lammert E, Gu G, McLaughlin M et al (2003) Role of VEGF-A in vascularization of pancreatic islets. Curr Biol 13:1070–1074
Brissova M, Shostak A, Shiota M et al (2006) Pancreatic islet production of vascular endothelial growth factor–a is essential for islet vascularization, revascularization, and function. Diabetes 55:2974–2985
Iwashita N, Uchida T, Choi JB et al (2007) Impaired insulin secretion in vivo but enhanced insulin secretion from isolated islets in pancreatic beta cell-specific vascular endothelial growth factor-A knock-out mice. Diabetologia 50:380–389
Litz J, Krystal GW (2006) Imatinib inhibits c-Kit-induced hypoxia-inducible factor-1alpha activity and vascular endothelial growth factor expression in small cell lung cancer cells. Mol Cancer Ther 5:1415–1422
Zhang N, Richter A, Suriawinata J et al (2004) Elevated vascular endothelial growth factor production in islets improves islet graft vascularization. Diabetes 53:963–970
Feng ZC, Li J, Silverstein J, Popell A, Yee SP, Wang R (2013) Long-term overexpression of c-Kit negatively impacts beta-cell function and survival via capillary and inflammatory recruitment in the islet. Diabetologia 56(Suppl 1):S222, Abstract
Agudo J, Ayuso E, Jimenez V et al (2012) Vascular endothelial growth factor-mediated islet hypervascularization and inflammation contribute to progressive reduction of β-cell mass. Diabetes 61:2851–2861
Pagliuca FW, Millman JR, Gürtler M et al (2014) Generation of functional human pancreatic β cells in vitro. Cell 159:428–439
Suen PM, Zou C, Zhang YA et al (2008) PDZ-domain containing-2 (PDZD2) is a novel factor that affects the growth and differentiation of human fetal pancreatic progenitor cells. Int J Biochem Cell Biol 40:789–803
Ishkitiev N, Yaegaki K, Kozhuharova A et al (2013) Pancreatic differentiation of human dental pulp CD117+ stem cells. Regen Med 8:597–612
Kubo A, Stull R, Takeuchi M et al (2011) Pdx1 and Ngn3 overexpression enhances pancreatic differentiation of mouse ES cell-derived endoderm population. PLoS One 6:e24058
Matsumoto S, Okumura K, Ogata A et al (2007) Isolation of tissue progenitor cells from duct-ligated salivary glands of swine. Cloning Stem Cells 9:176–190
Baek H, Noh YH, Lee JH et al (2014) Autonomous isolation, long-term culture and differentiation potential of adult salivary gland-derived stem/progenitor cells. J Tissue Eng Regen Med 8:717–727
Yang L, Li S, Hatch H et al (2002) In vitro trans-differentiation of adult hepatic stem cells into pancreatic endocrine hormone-producing cells. Proc Natl Acad Sci U S A 99:8078–8083
Funding
This work was supported by grants from the Canadian Institutes of Health Research (CIHR, Grant No. MOP 89800). ZCF is a recipient of a Canadian Diabetes Association Doctoral Student Research Award.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
All authors were involved in drafting the article and in revising the manuscript. RW was also involved in the conception of the article. All authors approved the final version.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Feng, ZC., Riopel, M., Popell, A. et al. A survival Kit for pancreatic beta cells: stem cell factor and c-Kit receptor tyrosine kinase. Diabetologia 58, 654–665 (2015). https://doi.org/10.1007/s00125-015-3504-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-015-3504-0