
Abstract
Aims/hypothesis
Upregulation of the reactive oxygen species (ROS)-producing enzyme NADPH oxidase (NOX)-1 in islets and beta cells follows acute exposure to inflammatory cytokines and is concomitant with beta cell dysfunction. NOX-1 is a candidate mediator of inflammation-induced beta cell dysfunction. This study aimed to determine whether selective inhibition of NADPH oxidase-1 presents a new strategy to preserve beta cell function.
Methods
Induced beta cell dysfunction was studied in primary human donor islets, isolated mouse islets and murine beta cell lines. Islets and beta cells were stimulated with inflammatory cytokines (TNF-α, IL-1β, IFN-γ). NOX-1 activity was blocked by the selective inhibitor ML171.
Results
Cytokine induction of intracellular ROS was reduced 80% with 1 μmol/l ML171 in murine beta cell lines (p < 0.01). Cytokine-induced apoptosis, measured by caspase-3 activation or quantified fluorescence microscopy, was prevented in islets and beta cell lines up to 100% with ML171 in a concentration-dependent manner (p < 0.05). Functionally, glucose-stimulated insulin secretion was abolished by cytokine exposure but preserved by ML171 in isolated mouse islets and murine beta cell lines. A feed-forward regulation of NOX-1 in islets and beta cell lines was disrupted by ML171.
Conclusions/interpretation
Stimulation of NOX-1 activity is a major component of inflammatory cytokine-induced beta cell dysfunction. Significant protection of beta cells is conferred with selective inhibition of NOX-1. Suppression of NOX-1 activity may present a new therapeutic strategy to preserve and protect beta cell function in diabetes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A loss of functional beta cell mass is an underlying feature of diabetes and oxidative stress is a recognised contributing factor to beta cell failure. Relative to other cells, beta cells are particularly sensitive to sustained elevation in intracellular reactive oxygen species (ROS) and resultant oxidative stress [1–5]. Some serum conditions associated with diabetes (e.g. elevated inflammatory cytokines, increased levels of NEFAs and high plasma glucose concentration) are known to induce cellular ROS [6–13]. Sources of cellular ROS in the beta cell include endoplasmic reticulum stress (reviewed [14]), mitochondria stress (reviewed [15]) and NAPDH oxidase (NOX) activity (reviewed [16, 17]).
NOX enzymes function to generate ROS. Seven members in the NOX family have been identified, NOX-1, -2, -3, -4 and -5 and dual oxidase-1/-2. Recently, expression of catalytic subunits and required associated proteins for NOX-1, NOX-2 and NOX-4 has been reported in beta cells [7, 10, 11, 18–21]. Physiologically, NOX activity in beta cells has been linked with glucose-stimulated insulin secretion (GSIS) [12, 22], although transgenic deletion studies have questioned this direct role [23]. Pathophysiologically, sustained NOX activity may contribute to stimuli-induced beta cell dysfunction associated with diabetes development and progression [16, 17].
Inflammation-induced beta cell dysfunction is a recognised feature of type 1 [24, 25] and type 2 diabetes [26–29]. Inflammatory cytokine stimulation of beta cells and islets induces loss of GSIS, increase in intracellular ROS and induction of apoptosis [21, 30]. These outcomes of proinflammatory cytokine (PIC) stimulation are concomitant with an elevated expression of NOX-1 [21]. Expression of NOX-1 in beta cells involves a feed-forward regulation driven by elevated intracellular ROS and redox signalling [30]. In terms of beta cell dysfunction, feed-forward regulation of NOX-1 following stimuli induction could result in a sustained and chronic increase in intracellular ROS and oxidative stress. Inhibition of NOX-1 activity and/or NOX-1 feed-forward regulation could provide a candidate target to preserve beta cell function in an inflammatory environment.
Lack of selective NOX inhibitors has presented a barrier to interpreting the functional importance of NOX isotypes. Recent high-throughput screening-based campaigns have identified new small molecules that are selective inhibitors of NOX enzymes [31–33]. The inhibitor ML171, a 2-acetylphenothiazine compound, is a nanomolar inhibitor of NOX-1 with greater than 30-fold selectivity over other NOX isotypes. ML171 inhibits NOX-1 activity in human colon cancer cells and its inhibition is overcome by overexpression of NOX-1, thus supporting the target selectivity of ML171 [34].
Here we report on the selective chemical inhibition of NOX-1 in islets and beta cells using ML171. This proof-of-concept study supports the viability for targeted inhibition of NOX-1 as a strategy to protect and preserve beta cells in an inflammatory environment.
Methods
Ethics
Protocols and procedures were reviewed and approved by relevant institutional regulatory committees.
Mouse islet isolation and human islets
Human donor islets were obtained from integrated islet distribution project (http://iidp.coh.org) and cultured in CMRL media (Mediatech, Manassas, VA, USA). Mouse islets were freshly isolated from 8-week-old male C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME, USA) by common bile duct cannulation and collagenase digestion [35]. Islets were hand picked before use.
Islets and cell lines
INS-1 cells (rat) were cultured in RPMI-1640 media (Life Technologies, Grand Island, NY, USA) containing 10% fetal calf serum, 1% penicillin/streptomycin, 10 mmol/l HEPES, 2 mmol/l l-glutamine, 1 mmol/l sodium pyruvate and 0.05 mmol/l 2-mercaptoethanol. βTC-3 cells (mouse) were cultured in DMEM media (Life Technologies) containing 18% fetal calf serum, 1% penicillin/streptomycin, 4 mmol/l l-glutamine, 5.5 mmol/l glucose and 1 mmol/l sodium pyruvate.
Treatment and RT-PCR
Islets or cells lines were treated with a PIC cocktail (5 ng/ml IL-1β, 10 ng/ml TNF-α, 100 ng/ml IFN-γ; R&D Systems, Minneapolis, MN, USA) for 24 h: 2-acetylphenothiazine (ML171) (R&D Systems) was added at the stated concentration 30 min before. Total RNA (RNeasy Mini Kit; Qiagen, Valencia, CA, USA) was transcribed using murine leukaemia virus reverse transcriptase (Life Technologies) and random hexamers (Life Technologies) prior to PCR with Jump Start Taq polymerase (Life Technologies). Primers used with SYBR Green 1 (Molecular Probes, Carlsbad, CA, USA) are shown in electronic supplementary material (ESM) Table 1. RT-PCR was performed in triplicate (CFX96; Bio-Rad, Hercules, CA, USA). Taqman primers (Life Technologies) were also used. Data were normalised to the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and analysed using the \( {2}^{-\varDelta \varDelta {\mathrm{C}}_{\mathrm{t}}} \) method. Mean Ct values were 12.6 and 13.5 for GAPDH, 29.8 and 29.7 Nox-1 control and 24.9 and 25.8 Nox-1 cytokine-stimulated for βTC-3 and INS-1 cells, respectively.
Western blotting
Beta cells (INS-1 or βTC-3) treated with PIC cocktail for 24 h without or with 0.1–10 μmol/l ML171 were lysed (1× PBS, 1% Triton X-100, 1 mmol/l phenylmethylsulfonyl fluoride, 1× Halt Protease Inhibitor Cocktail [Pierce, Rockford, IL, USA], 1 mmol/l NaVO4), incubated on ice for 20 min and centrifuged (20,817 g, 15 min, 4°C). Gel-separated proteins were transferred onto polyvinylidene fluoride (Immobilon-FL; Millipore, Billerica, MA, USA) and blocked with Tris-buffered saline (TBS)/0.05% Tween-20 (Sigma, St. Louis, MO, USA) containing 5% non-fat milk (Bio-Rad) for 1 h at room temperature. Primary antibody incubation was carried out overnight at 4°C. Wash cycles were in TBS/0.05% Tween-20 (four washes, 5 min). Primary antibodies used were 1:250 Anti-NOX-1 or 1:1,000 β-actin (Abcam, Cambridge, MA, USA). The signal was detected using 1:2,000 horseradish peroxidase-conjugated secondary antibody for 1 h (GE Healthcare, Little Chalfont, UK), SuperSignal Chemiluminescent Substrate (Thermo Scientific, Rockford, IL, USA) on a ChemiDoc XRS System (Bio-Rad). Densitometry was determined using Image J version 1.42q (imagej.nih.gov/ij/).
ROS measurement
Ten micromoles per litre of 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate, di(acetoxymethyl ester) (DCF-DA, Life Technologies) was added to treated (1 h) and washed cells for 30 min at 37°C. Post-PBS-wash cells were incubated at 37°C for 1 h. Fluorescence measurement was at excitation wavelength 480 nm and emission wavelength 530 nm (SpectraMax Molecular Devices, Sunnyvale, CA, USA).
GSIS
Treated islets or cells were washed with PBS and placed in 1 ml serum-free Krebs–Ringer buffer (composition in mmol/l: 115 NaCl, 24 NaHCO3, 5 KCl, 1 MgCl2 and 25 HEPES) for 1 h at 37°C. Low (1 mmol/l) or high (16 mmol/l) glucose was added for 30 min at 37°C. Insulin levels in media were measured by ELISA (Mercodia, Winston Salem, NC, USA) according to the manufacturer’s instructions. The following stimuli were used: PIC cocktail for 4 h, 0.5 mmol/l H2O2 for 1 h (Sigma) and 100 μmol/l pyocyanin (Sigma) for 1 h. ML171 (10 μmol/l) (R&D Systems) was added 30 min prior to treatment.
Apoptosis detection
For the caspase-3 assay, pro-caspase-3 cleavage was measured using a caspase-3 Assay kit (BD Pharmingen, Franklin Lakes, NJ, USA) according to the manufacturer’s instructions; fluorescence was measured (SpectraMax) at excitation wavelength 380 nm and emission wavelength 440 nm (INS-1) or 460 nm (βTC-3, mouse).
For fluorescence microscopy, stimulated cells or islets washed in cold PBS were incubated in cold PBS containing 1 μg/ml propidium iodide and 0.1 μmol/l YO-PRO-1 (Life Technologies) for 30 min at 4°C. For cell lines, five random fields per well were analysed and for islets, all islets (>1,000 µm2 in size) were analysed. The densitometric fluorescence value for YO-PRO-1 (green channel) was normalised to background and expressed proportional to cell-occupied area (phase contrast). An apoptotic index was established by relative expression of normalised signal with PIC-cocktail being defined as unity. Images were captured with Axiophot (Zeiss, Jena, Germany) and Axiovision (Zeiss) image analysis.
Statistical analysis
Experiments were performed in triplicate. Student’s t test or one-way ANOVA with Tukey post hoc testing (Prism 4.0; Graph-Pad Software, La Jolla, CA, USA) were used to determine statistical significance (95% CI and p < 0.05).
Results
Inhibition of NOX-1 prevents inflammatory cytokine-induced ROS in beta cells
Elevation of cellular ROS in beta cell lines INS-1 (rat) and βTC-3 (mouse) was evaluated by conversion of fluorescent substrate DCF-DA. Following 1 h stimulation with a PIC cocktail (IL-1β, TNF-α and IFN-γ), detectable ROS were elevated from control levels of 0.05 ± 0.02 and 0.09 ± 0.03 relative fluorescence units (RFU) to 0.25 ± 0.04 (p < 0.05) and 0.23 ± 0.03 (p < 0.01) RFU for INS-1 and βTC-3, respectively (Fig. 1). Addition of NOX-1 inhibitor ML171 (0.1, 1, 10 μmol/l) resulted in a concentration-dependent inhibition of PIC-induced ROS in both INS-1 (Fig. 1a) and βTC-3 (Fig. 1b) beta cells. These data identify NOX-1 activity as a contributing factor to PIC-induced elevation of intracellular ROS in beta cells.

Induction of ROS by inflammatory cytokines in beta cells is mediated by NOX-1. Beta cells were treated with PIC cocktail with or without ML171. Graphs show DCF-DA conversion (RFU) for INS-1 (a) and βTC-3 (b) beta cells. Data are presented as mean ± SEM. *p < 0.05 and **p < 0.01 relative to control (Ctl); † p < 0.05, †† p < 0.01 and ††† p < 0.001 relative to PIC; n = 3
Inhibition of NOX-1 prevents inflammatory cytokine-induced cell death in beta cells
Exogenous exposure to PICs induces beta cell death. Quantification of caspase-3 activation was used as a measure of induced apoptosis. Following cytokine exposure, caspase-3 activity increased from control levels of 430 ± 13.74 RFU to 467 ± 11.86 RFU (p < 0.05) in INS-1 cells and from control levels of 220 ± 7.2 RFU to 255 ± 6.8 RFU (p < 0.01) in βTC-3 cells (Fig. 2). Addition of ML171 (0.1, 1, 10 μmol/l) blocked the PIC-induced activation of caspase-3 up to 100 ± 3.4% (p < 0.05) and 97 ± 3.1% (p < 0.05) in INS-1 (Fig. 2a) and βTC-3 (Fig. 2b) cells, respectively. Induction of apoptosis was additionally studied microscopically. The fluorescent dye YO-PRO-1 (green) was used to detect cells undergoing apoptosis. Treating INS-1 and βTC-3 cells with PIC cocktail significantly increased apoptosis, as shown by an increase in fluorescence (p < 0.001) when compared with control cells (Fig. 3c, h vs Fig. 3b, g). Inclusion of ML171 (1 and 10 μmol/l) with PICs significantly protected both beta cell lines from cell death (p < 0.05 and p < 0.001, respectively). Fluorescence intensity was quantified for control, PIC- and PIC plus ML171-treated cell lines (Fig. 3a, f). Representative images for each cell line and treatment are shown in Fig. 3b–e, g–j.

Protection of beta cell lines from cytokine-induced caspase-3 activation. Beta cells were treated with PIC cocktail with or without ML171. Graphs show pro-caspase-3 cleavage (RFU) for INS-1 (a) and βTC-3 (b) beta cells. Data are presented as mean ± SEM. *p < 0.05 and **p < 0.01 relative to control (Ctl); † p < 0.05 relative to PIC; n = 3

Protection of beta cell lines from cytokine-induced apoptosis. INS-1 cells (a–e) and βTC-3 cells (f–j) were treated with PIC cocktail with or without ML171. Cells were examined microscopically (magnification ×10) following labelling with YO-PRO-1 (green). Graphs for INS-1 (a) and βTC-3 (f) show quantified apoptosis. Representative images of INS-1 and βTC-3 cells, respectively, are shown for untreated cells (b, g), PIC-treated cells (c, h), cells treated with PIC and ML171 at 1 μmol/l (d, i) or cells treated with PIC and M171 at 10 μmol/l (e, j). Data are presented as mean ± SEM. ***p < 0.001 relative to control (Ctl); † p < 0.05 and ††† p < 0.001 relative to PIC; n = 3
Inhibition of NOX-1 prevents Nox-1 gene expression initiated by inflammatory cytokines in beta cells
Expression of the Nox-1 gene in INS-1 and βTC-3 cells was upregulated 23 ± 2.1-fold and 32 ± 2.4-fold, respectively (Fig. 4a, b). Addition of ML171 (0.1, 1, 10 μmol/l) inhibited PIC-induced expression of Nox-1 by 33% and 23% in INS-1 and βTC-3 cells, respectively. Expression of NOX-1 protein increased by 25 ± 0.3% in INS-1 cells and 10 ± 0.2% in βTC-3 cells when beta cells were treated with PICs (Fig. 4c, d). PIC-induced production of NOX-1 protein was significantly inhibited by 1 μmol/l ML171 (100 ± 4.9%, p < 0.05 in INS-1 cells and 100 ± 1.5%, p < 0.01 in βTC-3 cells).

Expression of NOX-1 gene and protein production is inhibited by ML171. Nox-1 gene expression relative to control (untreated) cells in INS-1 cells (a) and βTC-3 cells (b) following stimulation with PIC cocktail without or with ML171. NOX-1 protein expression in INS-1 (c) and βTC-3 cells (d) for untreated cells, PIC-treated or PIC plus 1 μmol/l ML171. Graphs show densitometric analysis of western blots from INS-1 cells (c) and βTC-3 cells (d) relative to PIC-treated cells. Representative blots are shown for NOX-1 (top lane) or β-actin (bottom lane). Data are presented as mean ± SEM. *p < 0.05 relative to control (Ctl); † p < 0.05 and †† p < 0.01 relative to PIC; n = 3
Inhibition of NOX-1 preserves GSIS in beta cells exposed to inflammatory cytokines
Insulin secretion was increased in control INS-1 and βTC-3 beta cell lines when low-glucose medium (1 mmol/l) was exchanged for high-glucose medium (16 mmol/l) (Fig. 5a, b; p < 0.05). After exposure to PICs, GSIS was absent in both INS-1 and βTC-3 cells. After addition of ML171 (1 and 10 μmol/l) to PIC exposure, insulin secretion in response to high-glucose media was significantly increased in both INS-1 (p < 0.05 for both concentrations) and βTC-3 cells (p < 0.05 p < 0.001 for 1 and 10 μmol/l, respectively), restoring the glucose-sensitive insulin response. No significant changes in cellular total insulin (low glucose) or total protein were observed with cytokine treatment (data not shown). These data suggest that NOX-1 inhibition by ML171 protects beta cells from inflammation-induced cell dysfunction. Exogenous exposure of beta cell lines INS-1 or βTC-3 to the ROS-generating agents hydrogen peroxide (H2O2) or pyocyanin induced cell death (Fig. 6a–d) and uncoupling of GSIS (Fig. 6e, f for INS-1 cells; Fig. 6g, h for βTC-3 cells). Inclusion of 10 μmol/l ML171 significantly blocked apoptosis and uncoupling of GSIS by H2O2 or pyocyanin in beta cell lines (p < 0.05, p < 0.01 and p < 0.001). These data suggest that elevated ROS mediates beta cell dysfunction and that protection can be conferred by inhibiting NOX-1.

Beta cell function is preserved by inhibition of NOX-1. GSIS in INS-1 (a) and βTC-3 cells (b) for untreated cells (Ctl) or cells treated with PIC cocktail without or with ML171. Cells were exposed to low-glucose (1 mmol/l, grey bars) or high-glucose medium (16 mmol/l, black bars). Data are presented as mean ± SEM. *p < 0.05 and **p < 0.01 relative to low glucose; n = 3

Inhibition of NOX-1 protects beta cells from ROS-mediated dysfunction. Beta cells were treated with H2O2 (a, c, e, g) or pyocyanin (Pyo) (b, d, f, h) with or without 10 μmol/l ML171. Apoptosis (a–d) and GSIS (e–h) were measured in INS-1 (a, b, e, f) or βTC-3 (c, d, g, h) cells. Cells were exposed to 1 mmol/l glucose (L, grey bars) or 16 mmol/l glucose (H, black bars). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01 and ***p < 0.001 relative to control (Ctl); † p < 0.05, †† p < 0.01 and ††† p < 0.001 relative to H2O2 or pyocyanin; n = 3
Inhibition of NOX-1 preserves function and survival of primary mouse islets exposed to inflammatory cytokines
Stimulation of primary mouse islets with PICs resulted in cell death. Cleavage of a pro-caspase-3 substrate was used to measure cell death in isolated mouse islets. Caspase-3 increased from control levels of 52.7 ± 0.3 RFU to 54 ± 0.3 RFU (p < 0.05) following exposure to PIC (Fig. 7a). Addition of 1 μmol/l and 10 μmol/l ML171 blocked the PIC-induced activation of caspase-3 up to 100 ± 7% and 100 ± 3%, respectively.

Inhibition of NOX-1 protects primary mouse islets from inflammatory cytokines. (a) Pro-caspase-3 cleavage was measured in isolated primary mouse islets treated with PIC cocktail in the absence or presence of ML171. (b) GSIS was assessed in untreated (Ctl) islets or islets treated with PIC with or without 10 μmol/l ML171. Islets were exposed to 1 mmol/l glucose (grey bars) or 16 mmol/l glucose (black bars). (c) Expression of Nox-1 gene is shown in primary mouse islets treated with PIC without or with 1 μmol/l ML171. Data show fold expression relative to untreated (control) islets. Data are presented as mean ± SEM. *p < 0.05 and **p < 0.01 relative to Ctl; † p < 0.05 and †† p < 0.01 relative to PIC; n = 3
Beta cell function was assessed in primary mouse islets by measuring GSIS. Mouse islets exposed to 16 mmol/l glucose vs 1 mmol/l glucose showed a significant increase in insulin secretion (p < 0.01) (Fig. 7b). PIC-treated islets did not exhibit a glucose-stimulated insulin response. The glucose-stimulated insulin response was partially restored when islets were treated with PICs plus ML171. When 10 μmol/l ML171 was included, insulin secretion was significantly increased in response to high-glucose media (p < 0.01).
Nox-1 gene expression was also measured in mouse islets. Islets treated with PICs showed a 44.4 ± 6.9-fold increase in the expression of Nox-1 compared with untreated islets (Fig. 7c). Addition of 1 μmol/l ML171 inhibited PIC-induced Nox-1 expression in mouse islets by 84% (p < 0.05).
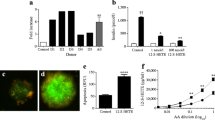
Primary mouse islets were assessed microscopically for PIC-induced apoptosis (Fig. 8a). Following overnight exposure to PIC cocktail, a 40 ± 3% (p < 0.001) increase in apoptosis was observed (Fig. 8a). Inclusion of 10 μmol/l ML171 significantly reduced PIC-induced islet apoptosis by 86 ± 4% (p < 0.001). Representative images for each condition are shown in Fig. 8b–d.

Protection of primary mouse islets from cytokine-induced apoptosis. Apoptosis was measured by YO-PRO-1 (green) labelling in isolated primary mouse islets treated with PIC cocktail without or with 10 μmol/l ML171. (a) Quantified apoptosis from all islets per experiment. Data presented are an apoptotic index where PIC response was set to unity. (b–d) Representative images for untreated islets (b), PIC-treated islets (c) and PIC plus 10 μmol/l ML171-treated islets (d) are shown (magnification ×10). Data are presented as mean ± SEM. ***p < 0.001 relative to control (Ctl); ††† p < 0.001 relative to PIC; n = 3
Inhibition of NOX-1 protects human donor islets exposed to inflammatory cytokines
Primary human donor islets were treated with PIC cocktail, and cell death was measured microscopically using the fluorescent dye YO-PRO-1. Human islets treated with PICs showed a significant increase in apoptosis (44 ± 9%) (p < 0.001) (Fig. 9a). When the islets were treated with PIC plus the NOX-1 inhibitor ML171 (10 μmol/l), the PIC-induced apoptosis was significantly reduced by 30 ± 3% (p < 0.05). Representative images for each condition are shown in Fig. 9b–d. Co-staining of human islets with insulin to identify beta cells is shown in ESM Fig. 1. Induced NOX-1 gene expression was measured in human islets (Fig. 10). Treatment of primary human donor islets with PICs upregulated NOX-1 expression 9.4 ± 1.9-fold. Addition of 1 μmol/l ML171 significantly inhibited PIC-induced expression of NOX-1 by 50% (4.4 ± 1.2-fold; p < 0.05).

Protection of primary human donor islets from cytokine-induced apoptosis. Apoptosis was measured by YO-PRO-1 (green) labelling in primary human donor islets treated with PIC cocktail in the absence or presence of 10 μmol/l ML171. (a) Quantified apoptosis from all islets per experiment. Data presented are an apoptotic index where PIC response was set to unity across donors. (b–d) Representative images for untreated islets (b), PIC-treated islets (c) and PIC plus 10 μmol/l ML171-treated islets (d) are shown (magnification ×10). Data are presented as mean ± SEM. ***p < 0.001 relative to control (Ctl); † p < 0.05 relative to PIC; n = 3

Regulation of NOX-1 gene expression by NOX-1 activity in primary human donor islets. Expression of NOX-1 gene in human donor islets treated with PIC cocktail in the absence or presence of 1 μmol/l ML171. Data show fold expression relative to untreated (control, Ctl) islets. Data are presented as mean ± SEM. † p < 0.05 relative to PIC; n = 3
Discussion
We have previously described NOX-1 activity as a mediator of inflammation-induced beta cell dysfunction [21]. The importance of this pathway to beta cell pathology was reinforced by identification of a feed-forward regulation of NOX-1 in beta cells [30]. In this report we have investigated the consequence of NOX-1 inhibition in terms of conferring protection to islets and beta cell lines exposed to an inflammatory environment.
A chronic, albeit subclinical, inflammatory state is a recognised feature of both type 1 and type 2 diabetes [24–29, 36–38]. Study of islets and beta cell lines exposed to PICs in vitro/ex vivo has shown marked loss of beta cell function and survival when compared with non-cytokine-exposed controls. Acute exposure of beta cells to PICs (6 h) results in measurable cell death and decreased function. Cellular changes in the beta cells that are concomitant with inflammatory cytokine exposure include an elevation of intracellular ROS and upregulation of genes, including those encoding 12-lipoxygenase, monocyte chemoattractant protein-1 and NOX-1. Based on our previous findings showing a selective upregulation of NOX-1 in beta cells exposed to PICs, and association of NOX-1 with PIC-induced islet dysfunction, we have sought a selective inhibitor of NOX-1 to validate the role of NOX-1 in mediating acute inflammation-induced beta cell dysfunction.
Recent high-throughput screening campaigns have successfully identified compound classes with improved selectivity within the NOX family of enzymes. In addition to dual specific NOX-1/4 pyrazolopyrimide dione inhibitors, a selective NOX-1 inhibitor, 2-acetylphenothiazine compound (ML171) was identified in an HT29 cell-based screen. ML171 has an enzyme IC50 of 129–250 nmol/l and a greater than 30-fold specificity for inhibition of NOX-1 over NOX-2 or other NOX isoforms [34].
Homogenous beta cell lines INS-1 and βTC-3 showed a significant upregulation in intracellular ROS following acute exposure to a PIC cocktail of IL-1β, TNF-α and IFN-γ. The same stimulation preferentially upregulates NOX-1 when compared with other NADPH oxidases [21]. Inclusion of the selective NOX-1 inhibitor ML171 was used to determine the importance of NOX-1 activity in PIC-induced beta cell dysfunction. ML171 effectively decreased the elevation of intracellular ROS resulting from PIC exposure. Inhibition due to ML171 was concentration dependent, with protection being maximal at 10 μmol/l. Analogous results were observed for both INS-1 and βTC-3 cells. These data suggest that a significant component of PIC-induced ROS elevation in beta cell lines is NOX-1 dependent since induction of ROS is blocked with the selective NOX-1 inhibitor, ML171.
Association of elevated ROS with beta cell dysfunction is established and equates to the relatively low activity of ROS scavenger systems in beta cells [1–4]. PICs induce cell death and beta cell dysfunction. We determined whether these outcomes involve NOX-1 activation. Induction of cell death in beta cell lines was evident following PIC stimulation, measured both by induction of caspase-3 cleavage or by direct quantitative microscopic analysis. Inclusion of the NOX-1 inhibitor ML171 effectively reduced PIC-induced beta cell death observed in INS-1 and βTC-3 cells. Equally, the function of beta cells, assessed by measuring static GSIS, is uncoupled by PIC exposure and preserved in the presence of ML171. The lower starting concentration of ML171 required to inhibit the effects of PICs on ROS production, caspase-3 or GSIS in beta cells likely reflects the shorter duration of these assays relative to the overnight apoptosis assay. Collectively, these data in homogenous beta cells lines demonstrate that NOX-1 activity is a major cellular event that mediates PIC-induced beta cell dysfunction. Further, inhibition of NOX-1 can preserve function and survival in beta cell lines exposed to inflammatory conditions.
Beta cell dysfunction and islet cell death were observed following exposure of primary islets to cytokines. Addition of ML171 protected the islets from the effects of PICs for the measured variables of function and cell viability. Significant protection was observed in mouse GSIS, cell death and caspase-3 cleavage. Relative to cell lines, the basal caspase-3 signal was reduced in primary islets; this may reflect the lower cell proliferation rate. Importantly, for the translational potential of NOX-1 inhibition, these data in primary islets provide proof-of-concept validation that disruption of NOX-1 activity may be a targetable strategy to preserve islet function under inflammatory conditions.
In terms of systemic inhibition of NOX-1, any potential off-target effects will need to be considered. In a broad-spectrum receptor inhibition screen, ML171 exhibited a very low potency and very low affinity at serotonin and adrenergic receptors [39]. Mice with a global deletion of NOX-1 are viable and do not exhibit any gross physical or behavioural phenotype [40]. Study of these mice has identified a physiological role for NOX-1 in acute response to hypertension induced by exogenous angiotensin II [40, 41], though a chronic upregulation of the endogenous renin–angiotensin system was not observed [42]. Pathophysiological upregulation of NOX-1 expression is associated with certain colon and prostate cancers [43–46]. Thus, while NOX-1 is non-essential, mice with complete deletion of Nox-1 have impaired regulation of hypertension, suggesting a partial inhibition may be tolerated. Inhibition of NOX-1 could additionally be beneficial in certain cancers [43–47]. Further, inhibition of NOX-1 has been proposed as a strategy to suppress thrombotic events [48]. The pyrazolopyrimide dione NOX-1/4 selective inhibitor GK-136901 is reported to be well tolerated in mice, with dosing tested up to 1,000 mg/kg per os [33]. Inhibition of NOX-1 activity by the selective inhibitor ML171 offered the cleanest exploration for the role of NOX-1 in PIC-mediated beta cell dysfunction. Potential off-target effects of the inhibitor, while not reported, cannot be eliminated. Alternative approaches to downregulating NOX-1 will provide helpful validation. Molecular deletion studies offer the challenge of achieving meaningful knockdown on the background of stimuli-induced expression of NOX-1. Our previous studies have identified a feed-forward regulation of NOX-1 in beta cells [30]. Elevated intracellular ROS and subsequent redox signalling drive a primed NOX-1 gene. The importance of this to islet pathology is the implied potential for a reinforcing elevation in intracellular ROS and progression to oxidative stress. Direct elevation of ROS in beta cell lines results in apoptosis. This is inhibited in βTC-3 and INS-1 cells by inclusion of 10 μmol/l ML171. In terms of a therapeutic index for systemic NOX-1 inhibition, partial disruption of the feed-forward regulation of NOX-1 in beta cells has the potential for therapeutic beta cell protection with minimal off-target effects. Providing support for this concept, ML171 disrupted PIC-induced expression of NOX-1 in beta cell lines and primary islets.
An unaddressed clinical challenge of diabetes is the establishment of an effective strategy for preserving existing beta cell mass and/or conferring protection to replenished islets in an existing inflammatory environment. We have identified NOX-1 as an effector of inflammation-induced beta cell dysfunction. Using a selective inhibitor of NOX-1, this study shows NOX-1 to be a major mediator of PIC-induced beta cell dysfunction. Development and translation of NOX-1 inhibitors may provide novel strategies for preserving and protecting beta cell function in diabetes. These applications could include monocomponent or combination therapies with islet regenerative agents, in addition to protection of islets pre-transplantation/xenotransplantation with or without encapsulation.
Abbreviations
- C57BL/6J:
-
C57 black 6
- DCF-DA:
-
6-Carboxy-2′,7′-dichlorodihydrofluorescein diacetate, di(acetoxymethyl ester)
- GADPH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- GSIS:
-
Glucose-stimulated insulin secretion
- NOX:
-
NADPH oxidase
- PIC:
-
Proinflammatory cytokine
- RFU:
-
Relative fluorescence unit
- ROS:
-
Reactive oxygen species
- TBS:
-
Tris-buffered saline
References
Grankvist K, Marklund SL, Taljedal IB (1981) CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochem J 199:393–398
Lenzen S, Drinkgern J, Tiedge M (1996) Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med 20:463–466
Tiedge M, Lortz S, Drinkgern J, Lenzen S (1997) Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 46:1733–1742
Modak MA, Datar SP, Bhonde RR, Ghaskadbi SS (2007) Differential susceptibility of chick and mouse islets to streptozotocin and its co-relation with islet antioxidant status. J Comp Physiol B 177:247–257
Lenzen S (2008) Oxidative stress: the vulnerable beta-cell. Biochem Soc Trans 36:343–347
Janciauskiene S, Ahren B (2000) Fibrillar islet amyloid polypeptide differentially affects oxidative mechanisms and lipoprotein uptake in correlation with cytotoxicity in two insulin-producing cell lines. Biochem Biophys Res Commun 267:619–625
Oliveira HR, Verlengia R, Carvalho CR, Britto LR, Curi R, Carpinelli AR (2003) Pancreatic beta-cells express phagocyte-like NAD(P)H oxidase. Diabetes 52:1457–1463
Cunningham GA, McClenaghan NH, Flatt PR, Newsholme P (2005) L-Alanine induces changes in metabolic and signal transduction gene expression in a clonal rat pancreatic beta-cell line and protects from pro-inflammatory cytokine-induced apoptosis. Clin Sci (Lond) 109:447–455
Inoguchi T, Nawata H (2005) NAD(P)H oxidase activation: a potential target mechanism for diabetic vascular complications, progressive beta-cell dysfunction and metabolic syndrome. Curr Drug Targets 6:495–501
Nakayama M, Inoguchi T, Sonta T et al (2005) Increased expression of NAD(P)H oxidase in islets of animal models of type 2 diabetes and its improvement by an AT1 receptor antagonist. Biochem Biophys Res Commun 332:927–933
Uchizono Y, Takeya R, Iwase M et al (2006) Expression of isoforms of NADPH oxidase components in rat pancreatic islets. Life Sci 80:133–139
Morgan D, Oliveira-Emilio HR, Keane D et al (2007) Glucose, palmitate and pro-inflammatory cytokines modulate production and activity of a phagocyte-like NADPH oxidase in rat pancreatic islets and a clonal beta cell line. Diabetologia 50:359–369
Michalska M, Wolf G, Walther R, Newsholme P (2010) Effects of pharmacological inhibition of NADPH oxidase or iNOS on pro-inflammatory cytokine, palmitic acid or H2O2-induced mouse islet or clonal pancreatic beta-cell dysfunction. Biosci Rep 30:445–453
Volchuk A, Ron D (2010) The endoplasmic reticulum stress response in the pancreatic beta-cell. Diabetes Obes Metab 12(Suppl 2):48–57
Newsholme P, Morgan D, Rebelato E et al (2009) Insights into the critical role of NADPH oxidase(s) in the normal and dysregulated pancreatic beta cell. Diabetologia 52:2489–2498
Weaver JR, Taylor-Fishwick DA (2014) Role of NADPH oxidase in beta cell dysfunction islets of Langerhans. Springer, Dordrecht
Taylor-Fishwick DA (2013) NOX, NOX who is there? The contribution of NADPH oxidase one to beta cell dysfunction. Front Endocrinol (Lausanne) 4:40
Lupi R, del Guerra S, Bugliani M et al (2006) The direct effects of the angiotensin-converting enzyme inhibitors, zofenoprilat and enalaprilat, on isolated human pancreatic islets. Eur J Endocrinol 154:355–361
Shao J, Iwashita N, Ikeda F et al (2006) Beneficial effects of candesartan, an angiotensin II type 1 receptor blocker, on beta-cell function and morphology in db/db mice. Biochem Biophys Res Commun 344:1224–1233
Rebelato E, Mares-Guia TR, Graciano MF et al (2012) Expression of NADPH oxidase in human pancreatic islets. Life Sci 91:244–249
Weaver JR, Holman TR, Imai Y et al (2012) Integration of pro-inflammatory cytokines, 12-lipoxygenase and NOX-1 in pancreatic islet beta cell dysfunction. Mol Cell Endocrinol 358:88–95
Morgan D, Rebelato E, Abdulkader F et al (2009) Association of NAD(P)H oxidase with glucose-induced insulin secretion by pancreatic beta-cells. Endocrinology 150:2197–2201
Li N, Li B, Brun T et al (2012) NADPH oxidase NOX2 defines a new antagonistic role for reactive oxygen species and cAMP/PKA in the regulation of insulin secretion. Diabetes 61:2842–2850
Jorns A, Gunther A, Hedrich HJ, Wedekind D, Tiedge M, Lenzen S (2005) Immune cell infiltration, cytokine expression, and beta-cell apoptosis during the development of type 1 diabetes in the spontaneously diabetic LEW.1AR1/Ztm-iddm rat. Diabetes 54:2041–2052
Eizirik DL, Mandrup-Poulsen T (2001) A choice of death–the signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia 44:2115–2133
Catalan V, Gomez-Ambrosi J, Ramirez B et al (2007) Proinflammatory cytokines in obesity: impact of type 2 diabetes mellitus and gastric bypass. Obes Surg 17:1464–1474
Steinberg GR (2007) Inflammation in obesity is the common link between defects in fatty acid metabolism and insulin resistance. Cell Cycle 6:888–894
Igoillo-Esteve M, Marselli L, Cunha DA et al (2010) Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics CCL2 expression by beta cells in type 2 diabetes. Diabetologia 53:1395–1405
Su SC, Pei D, Hsieh CH, Hsiao FC, Wu CZ, Hung YJ (2011) Circulating pro-inflammatory cytokines and adiponectin in young men with type 2 diabetes. Acta Diabetol 48:113–119
Weaver JR, Taylor-Fishwick DA (2013) Regulation of NOX-1 expression in beta cells: a positive feedback loop involving the Src-kinase signaling pathway. Mol Cell Endocrinol 369:35–41
ten Freyhaus H, Huntgeburth M, Wingler K et al (2006) Novel Nox inhibitor VAS2870 attenuates PDGF-dependent smooth muscle cell chemotaxis, but not proliferation. Cardiovasc Res 71:331–341
Wind S, Beuerlein K, Eucker T et al (2010) Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br J Pharmacol 161:885–898
Laleu B, Gaggini F, Orchard M et al (2010) First in class, potent, and orally bioavailable NADPH oxidase isoform 4 (Nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis. J Med Chem 53:7715–7730
Gianni D, Nicolas N, Zhang H, et al (2010) Optimization and characterization of an inhibitor for NADPH oxidase 1 (NOX-1) Probe Reports from the NIH Molecular Libraries Program. National Center for Biotechnology Information (US), Bethesda (MD)
Yang ZD, Chen M, Wu R, McDuffie M, Nadler JL (2002) The anti-inflammatory compound lisofylline prevents type I diabetes in non-obese diabetic mice. Diabetologia 45:1307–1314
Tilg H, Moschen AR (2008) Inflammatory mechanisms in the regulation of insulin resistance. Mol Med 14:222–231
Al-Maskari M, Al-Shukaili A, Al-Mammari A (2010) Pro-inflammatory cytokines in Omani type 2 diabetic patients presenting anxiety and depression. Iran J Immunol 7:124–129
Kang YS, Song HK, Lee MH, Ko GJ, Cha DR (2010) Plasma concentration of visfatin is a new surrogate marker of systemic inflammation in type 2 diabetic patients. Diabetes Res Clin Pract 89:141–149
Altenhofer S, Radermacher KA, Kleikers PW, Wingler K, Schmidt HH (2014) Evolution of NADPH oxidase inhibitors: selectivity and mechanisms for target engagement. Antioxid Redox Signal. doi:10.1089/ars.2013.5814
Gavazzi G, Banfi B, Deffert C et al (2006) Decreased blood pressure in NOX1-deficient mice. FEBS Lett 580:497–504
Matsuno K, Yamada H, Iwata K et al (2005) Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation 112:2677–2685
Yogi A, Mercure C, Touyz J et al (2008) Renal redox-sensitive signaling, but not blood pressure, is attenuated by Nox1 knockout in angiotensin II-dependent chronic hypertension. Hypertension 51:500–506
O’Leary DP, Bhatt L, Woolley JF et al (2012) TLR-4 signalling accelerates colon cancer cell adhesion via NF-kappaB mediated transcriptional up-regulation of Nox-1. PLoS One 7:e44176
Fukuyama M, Rokutan K, Sano T, Miyake H, Shimada M, Tashiro S (2005) Overexpression of a novel superoxide-producing enzyme, NADPH oxidase 1, in adenoma and well differentiated adenocarcinoma of the human colon. Cancer Lett 221:97–104
Arnold RS, He J, Remo A et al (2007) Nox1 expression determines cellular reactive oxygen and modulates c-fos-induced growth factor, interleukin-8, and Cav-1. Am J Pathol 171:2021–2032
Lim SD, Sun C, Lambeth JD et al (2005) Increased Nox1 and hydrogen peroxide in prostate cancer. Prostate 62:200–207
Sancho P, Fabregat I (2010) NADPH oxidase NOX1 controls autocrine growth of liver tumor cells through up-regulation of the epidermal growth factor receptor pathway. J Biol Chem 285:24815–24824
Walsh TG, Berndt MC, Carrim N, Cowman J, Kenny D, Metharom P (2014) The role of Nox1 and Nox2 in GPVI-dependent platelet activation and thrombus formation. Redox Biol 2:178–186
Acknowledgements
Human donor islets were provided by the Integrated Islet Distribution Program (IIDP, http://iipd.coh.org/).
Funding
Funding for this work was provided by CDMRP, Department of Defense (PR093521, DAT-F), JDRF International and the Leona M. and Harry B. Helmsley Charitable Trust (DAT-F).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
DAT-F directed the work, provided data interpretation and wrote the article. JRW and WJG acquired research data, contributed to the analysis and interpretation of data and helped to draft and/or review the article. All authors approved this manuscript version. DAT-F is responsible for the integrity of the work as a whole.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
(PDF 62 kb)
ESM Fig. 1
(PDF 99 kb)
Rights and permissions
About this article

Cite this article
Weaver, J.R., Grzesik, W. & Taylor-Fishwick, D.A. Inhibition of NADPH oxidase-1 preserves beta cell function. Diabetologia 58, 113–121 (2015). https://doi.org/10.1007/s00125-014-3398-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-014-3398-2