
Abstract
Key Message
Developing genetically resistant soybean cultivars is key in controlling the destructive Sclerotinia Stem Rot (SSR) disease. Here, a GWAS study in Canadian soybeans identified potential marker-trait associations and candidate genes, paving the way for more efficient breeding methods for SSR.
Abstract
Sclerotinia stem rot (SSR), caused by the fungal pathogen Sclerotinia sclerotiorum, is one of the most important diseases leading to significant soybean yield losses in Canada and worldwide. Developing soybean cultivars that are genetically resistant to the disease is the most inexpensive and reliable method to control the disease. However, breeding for resistance is hampered by the highly complex nature of genetic resistance to SSR in soybean. This study sought to understand the genetic basis underlying SSR resistance particularly in soybean grown in Canada. Consequently, a panel of 193 genotypes was assembled based on maturity group and genetic diversity as representative of Canadian soybean cultivars. Plants were inoculated and screened for SSR resistance in controlled environments, where variation for SSR phenotypic response was observed. The panel was also genotyped via genotyping-by-sequencing and the resulting genotypic data were imputed using BEAGLE v5 leading to a catalogue of 417 K SNPs. Through genome-wide association analyses (GWAS) using FarmCPU method with threshold of FDR-adjusted p-values < 0.1, we identified significant SNPs on chromosomes 2 and 9 with allele effects of 16.1 and 14.3, respectively. Further analysis identified three potential candidate genes linked to SSR disease resistance within a 100 Kb window surrounding each of the peak SNPs. Our results will be important in developing molecular markers that can speed up the breeding for SSR resistance in Canadian grown soybean.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Soybean [Glycine max (L.) Merr] is one of the most important oil and protein crops in the world and in Canada due to its highly versatile role in feed and food production and as part of cropping systems (Yoosefzadeh-Najafabadi and Rajcan 2022). The unique agronomic and nutritional characteristics of soybean present a valuable opportunity to address the growing strain on food resources as a result of the rising human population and the environmental transformations taking place as a consequence of climate change (Lal 2013; Ray et al. 2013). Climate change can be considered as a major global issue with far-reaching implications for human existence, including triggering numerous abiotic and biotic stresses that are leading to significant yield loss of strategically important crops such as soybean (Yoosefzadeh-Najafabadi and Rajcan 2022; Yoosefzadeh-Najafabadi et al. 2022). In particular, the effects of biotic stresses on crop production can be particularly severe and widely felt, causing destabilization of food production and security in different world regions.
Soybean production in United States (US) and Canada faces major biotic stresses, primarily crop diseases (Allen et al. 2017; Koenning and Wrather 2010). Among the most significant diseases in soybean, sclerotinia stem rot (SSR), also known as white mould, is known as one of the important diseases that significantly impact the crop’s grain yield and quality (Allen et al. 2017). Sclerotinia stem rot is caused by Sclerotinia sclerotiorum (Lib) de Bary, a necrotrophic fungal pathogen that has been documented to infect more than 408 crop species (Boland and Hall 1994). In soybean, the fungus infects the plant through the flower, where it spreads through the stem, leading to bleaching, wilting, and shredding of the tissues (Bolton et al. 2006). At harvest, additional symptoms can be observed through severe damage to the soybean grain, leading to loss of yield and seed quality (Bolton et al. 2006). Since, its first documentation in the US in 1924, SSR has expanded into the soybean growing regions of the Northern US and Canada (Peltier et al. 2012). In 1994, SSR was ranked the second most yield loss-causing disease for soybeans in Canada and ranked similarly ranked in 1994, 2004, and 2009 in the US (Koenning and Wrather 2010).
Biological, chemical, and cultural control measures can be used by farmers to mitigate the impact of SSR (Peltier et al. 2012). However, these methods can be ineffective when the disease incidence is higher than 50% (Mueller et al. 2002; Zeng et al. 2012). Furthermore, SSR incidence and severity vary from year to year due to its high sensitivity to temperature and humidity, which makes it difficult for growers to predict and effectively deploy control measures (Peltier et al. 2012). Genetic resistance offers the most economical, environmentally sustainable, and reliable solution to control crop diseases, including SSR. There are no reported soybean cultivars with complete SSR resistance to date; however, partial resistance has been reported in different soybean lines (Boudhrioua et al. 2020; Jing et al. 2021). Partial resistance to SSR in soybeans has been reported as a polygenic trait that is controlled by several intrinsic and extrinsic factors, including genotype, environment, and the interaction between genotype and environment (Boudhrioua et al. 2020; Jing et al. 2021).
Several mapping studies have previously identified quantitative trait loci (QTL) that control partial resistance to SSR in soybean through biparental linkage mapping. Over 100 of the reported QTL have been found on 18 out of 20 soybean chromosomes as recorded on SoyBase, the USDA-ARS soybean genetics and genomics database (https://www.soybase.org/ accessed on 15 June, 2023). Although biparental linkage mapping offers useful insights into the genetic architecture of complex traits, it is limited to the genetic diversity in two parents. As a result, only a few of the QTL discovered by the different linkage mapping studies overlap, which make them challenging to use for marker-assisted breeding.
Recent improvements in high throughput sequencing technologies and their associated reductions in genotyping costs have facilitated the ability to employ genome-wide association studies (GWAS) to identify partial resistance to SSR in a diverse and unrelated soybean population. In comparison to the traditional biparental QTL mapping, GWAS provides a higher resolution of marker-trait association (MTA) due to increased recombination possibilities and a lower level of linkage disequilibrium (LD) in an unrelated population (Hong et al. 2022; Korte and Farlow 2013; Yoosefzadeh-Najafabadi et al. 2023).
Several studies have used GWAS and reported over 130 MTAs with SSR resistance in different soybean populations (Boudhrioua et al. 2020; Moellers et al. 2017; Wei et al. 2017). Boudhrioua et al. (2020) screened a population of 127 soybean accessions using 1.5 million single nucleotide polymorphism (SNP) markers through genotyping-by-sequencing (GBS) and whole genome sequencing (WGS) to identify a new major effect MTA on chromosome 1 linked to disease development on the main stem. In the study, genotypes carrying the resistance allele were reported to develop lesions nearly half the length of those carrying the susceptible allele (Boudhrioua et al. 2020). In another study, Wei et al. (2017) utilized two different advanced mapping models to identify three of the same MTA (chromosome 1, 11 and 18) associated with SSR resistance in a population of 420 soybean lines, providing additional confidence in the genetic control being located in part on chromosome 1. A study by Moellers et al. (2017) screened a large panel of 466 soybean accessions and identified 58 significant main effect loci and 24 significant epistatic interactions linked to SSR resistance. Candidate genes reported in the study are known to be involved in various plant immunity-related processes such as cell wall structure, hormone signalling and sugar allocation, which further indicates the complexity of SSR resistance.
Although several previous studies have reported MTAs for SSR in soybean, there is lack of information on genetic control of SSR resistance in soybean primarily grown in Canada. Therefore, in this study we aimed to investigate the genetic basis of partial resistance to SSR and identify potential candidate genes that are strongly involved in controlling the trait particularly in soybean genotypes that are suitable for Canadian environments. For this objective, a Canadian soybean germplasm panel (CSGP) was screened for SSR resistance in controlled environments and genotypically evaluated through GWAS to detect reliable SSR MTAs. The utilization of detected MTAs can potentially facilitate marker-assisted selection (MAS) for large scale screening of breeding populations based on their level of SSR resistance, resulting in minimizing the time required for soybean breeding.
Materials and methods
Plant material
The Canadian soybean germplasm panel (CSGP) is composed of 193 lines that are representative of the genetic diversity in Canadian soybean. Several criteria were applied to collect the CSGP, including genetic variation in response to SSR and adaptation to the different soybean maturity ranging within 2400 to 2900 crop heat units (Bootsma et al. 2007; Brown and Bootsma 2000) or maturity groups 000 to I, according to the United States Department of Agriculture classification. This represents the growing regions of Ontario, Quebec, and Southern Manitoba. Sources of the lines in the CSGP were from the University of Guelph Soybean breeding program, which contributed most to the panel with 119 cultivars and breeding lines, 35 cultivars were sourced from agriculture and agri-food Canada (AAFC) at Ottawa Research and Development Centre, 19 cultivars from CÉROM (Le Centre de recherche sur les grains, Quebec), ten cultivars from northern US (obtained through the United States Department of Agriculture—Agriculture Research Services, Germplasm Resources Information Network: https://www.ars-grin.gov/ accessed on 15 June, 2023), nine diverse ancestral cultivars, and one commercial cultivar from Syngenta. In this study, a maximum of 10 year-old seeds were selected to ensure acceptable germination levels. The panel also included three checks: partially resistant commercial cultivar from AAFC, Maple Donovan; moderately resistant Guelph cultivar OAC Bayfield, and highly susceptible AAFC natto cultivar, Nattosan.
Experimental design
To evaluate for resistance to SSR, the 193 lines were grown in growth room facilities of the Crop Science building at the University of Guelph, Guelph, Ontario, Canada. The experimental design was a randomized complete block design with five replications blocked in time. Prior to the experiment’s setup, planting pots were soaked in 1% Virkon (Aston Pharma, London, United Kingdom) at 10 g/litre, rinsed, and dried overnight to sanitize and avoid contamination. Four seeds per genotype were directly planted in a five-litre premium plastic planter garden pot filled with Moisten Sunshine L4 potting mix (Green Island Distributors, Inc., Riverhead New York, United States). To synchronize panel flowering date, planting was conducted on different days based on previously recorded flowering data. Plants were thinned down to a single plant per pot after ten days. The classic N-P-K 20–20-20 fertilizer formula measured at 50 g per litre of water was used for fertilization and was applied by integrating it into the irrigation system. Swirski mite (Amblyseius swirskii) biological insect control (Biobest, Leamington, Ontario, Canada) was sprinkled on top of each pot before planting and on each plant 15 days post-planting. Growth room environments were set to 25 °C day/21 °C night for temperature, 16 day/8 night for light (light intensity: 265 umol. m2s1, 20 cm above bench, light bank 125 cm from bench), automated drip irrigation twice a day for 5 min, and relative humidity (RH) at 65—70% pre inoculation and 80—85% post-inoculation.
Phenotypic evaluation for SSR
The inoculation method used to phenotype for SSR resistance is the cotton pad method, as described by Bastien et al. (2014). The NB-5 strain of S. sclerotiorum obtained from Dr. Francois Belzile, Laval University, Quebec, Canada was used for inoculation. Cultures were maintained on potato dextrose agar (PDA) (Fisher Scientific International Inc., Hampton, New Hampshire, United States) medium and kept at 4 °C temperature. For inoculation assay preparation, a piece of sclerotiorum was transferred to a new PDA plate and incubated in a dark room between 22 and 25 °C temperature for five days. At this point, a mass of white mycelia would be grown, filling the plate up to the edge. Mycelial plugs of 5 mm diameter were collected from the edge of the plate and transferred to Erlenmeyer flasks containing potato dextrose broth (PDB) (Fisher Scientific International Inc., Hampton, New Hampshire, United States). Two mycelial plugs were added per 800 ml. The inoculation assay was incubated on an orbital shaker (MAXQ 4000, Thermo Fisher Scientific, Waltham, Massachusetts, United States) at 130 RPM and 22 to 25 °C temperature for five days until almost reaching saturation. Inoculation was conducted at the start of flowering. The inoculation process began with homogenizing the suspension in a blender (Sunbeam-Oster Co., Inc., Fort Lauderdale, Florida, United States) for 30 s.
Half a piece of a 100% pure cotton round was soaked in the suspension and applied on the petiole of the flowers on the lowest node. To maintain high relative humidity in the growth room, a misting system was installed above the plant canopy and on the floors of each bench. The system was set up to mist for two minutes every four hours above the plants and continuously running on the floor during the inoculation period. Disease resistance was measured as lesion size on the plant stem at eight days post-inoculation. Measurements were taken in mm using a digital calliper (Mastercraft, Toronto, Ontario, Canada).
Genotyping
Leaf tissue samples were collected from three week-old plants into 10 ml plant-tissue collection tubes and freeze-dried using the Labonco FreeZone® freeze-dry system (Savant Moduly, Kansas City, MO, USA) for 24 h. DNA extraction was conducted using the NucleoSpin® Plant II DNA extraction kit by Macherey–Nagel following the manufacturer’s protocol. The Nanodrop ND 1000 Spectrophotometer (Nanodrop Technologies, Inc., Wilmington, DE, USA) was used to examine contamination and check the DNA quality, while the Qubit 2.0 Fluorometer (Thermo Fisher Scientific Inc., USA) was used to analyse DNA concentration and standardize to 10 ng/ul before sequencing.
Panel genotyping was conducted via the genotyping-by-sequencing (GBS) method described by Elshire et al. (2011). The GBS library was prepared with ApeKI enzymatic digestion system and single end sequencing was conducted at the Plateforme d’analyses génomiques [Institut de Biologie Intégrative et des Systèmes (IBIS)], Université Laval (Quebec, QC, Canada). Up to 470 million single end reads covering 50 bp to 160 bp were produced using six chips on the Ion Proton System (ThermoFisher Scientific Inc. USA). Generated reads were aligned onto the soybean reference genome (William 82, Gmax_275_Wm82.a2. v2) (Schmutz et al. 2010) and SNP calling was conducted using the Fast-GBS pipeline (Torkamaneh et al. 2017). This led to a catalogue of 109 K GBS-derived SNPs on which initial imputation was performed to complete the missing genotypes. A secondary imputation was conducted using 4 million SNPs generated via whole-genome-sequencing of a reference panel consisting of 102 Canadian elite soybean genotypes. Both imputation procedures were performed using BEAGLE v5 (Browning et al. 2018) and resulted in a 417 K SNP catalogue. The full 417 K SNP catalogue was used in the Linkage disequilibrium (LD) analysis, which was estimated and plotted by using the PopLDdecay version 3.42 (Zhang et al. 2019).
Population structure analysis
Kinship and population structure were estimated and incorporated in the GWAS analysis to address false positives and confounding due to genetic relatedness. The kinship matrix was calculated using the VanRaden method (VanRaden 2007), while the population structure was estimated through principal component (PC) analysis using the FastSTRUCTURE software (Raj et al. 2014). To increase the computational efficiency of FastSTRUCTURE, only 16 K SNPs from the catalogue were used for the population structure analysis. For this step, the LD decay was calculated, followed by estimation of the SNPs’ LD, then 16 K SNPs that are outside of the LD decay range were randomly selected for analysis. Three runs were then conducted with K set between 1 and 9 to estimate the optimum number of subpopulations using the chooseK tool in FastSTRUCTURE.
Statistical analysis
The genetic value of each soybean genotype was estimated using the best linear unbiased prediction (BLUP), one of the most used linear mixed models (Robinson 1991). BLUPs were used as the phenotypic response to account for fixed effects and random effects more accurately. Although plants were grown in controlled environments using uniform settings, minor variations in temperature and relative humidity were observed across experiments. These environmental parameters are very important for Sclerotinia sclerotium infection of soybean. BLUPS are known for their efficiency in including information from various sources and accounting for genotype-by-environment interactions (Bernardo 2010). Estimated breeding values (EBVs) can also be used as phenotypic response; however, they are often more suitable for animal GWAS compared to plant GWAS. This is due to the simplicity and more structured pedigrees in animal populations compared to plant populations (Jannink and Walsh 2002; Bernando et al., 2008; Hayes et al. 2009; Gorjanc et al. 2015). For complex traits with low heritability, using EBVs as the trait score for GWAS has been reported to produce very high false-positive rates (Ekine et al. 2014). BLUPs have been previously used for phenotypic response in several previous plant GWAS studies (Sun et al., 2012; Tsai et al. 2020; Yoosefzadeh-Najafabadi et al. 2021; Tagliotti et al. 2021).
The BLUP values were calculated by AllInOne preprocessing R package version 1.9.5 (Yoosefzadeh Najafabadi et al. 2023). Due to growth room space limitations, three experiments in total were conducted at different times. All parameters including individual genotypes as well as each experiment were included in the BLUP model as random effects to control for heterogeneity among the environments and minimize experimental error. BLUP estimation was performed based on the following statistical model (Eq. 1):
where, Y represents disease resistance trait, measured as lesion length (mm) on the inoculated plant stem; G stands for genotype, which is considered as random effect; E represents the three experimental blocks; R stands for replicates within each environmental block; T is the interaction between genotype and environmental block (G × E); and ε is the error of the unknown source of the variation.
Association analysis
The generated BLUPs values were used for GWAS analysis, which was performed using the Fixed and Random Model Circulating Probability Unification (FarmCPU) in the rMVP package version 1.0.8 in R software (Yin et al. 2021). FarmCPU was developed by Liu et al., (2016) to minimize limitations and false discoveries in previous GWAS tools. The algorithm achieves this by using mixed linear model (MLM) as the random effect model and stepwise regression as the fixed-effect model iteratively, thus leveraging advantages offered by both (Liu et al. 2016). In FarmCPU, the random effect model (REM) is used to optimize the SNP selection based on the p-values as follows (Eq. 2):
, where Yi represents the observation on the ith sample, ei is the residual, while Ui is the total genetic effect of the ith sample.
The fixed-effect model (FEM) in FarmCPU is used to simultaneously test the N number of SNPs following the equation (Eq. 3):
where, Yi represents the observation on the ith sample; Mi1, Mi2, …, Mit stands for the genotypes of the t pseudo-quantitative trait nucleotides (QTNs); P1, P2, P3, …, Pt is the respective effect of the pseudo-QTNs; Nij is the genotype of the jth SNPs and ith sample; Kj is the effect of the jth SNPs; and ei is the residual.
Furthermore, the false discovery rate (FDR) was estimated using the Q-value package to set the threshold for the significant MTAs. The FDR provides more power than the Bonferroni correction method in GWAS (Yang et al. 2005; Zablocki et al. 2014). The Q-value package used a list of SNP marker p-values estimated through FarmCPU simultaneous tests and measured the proportion of false positives each time the test was called significant. It then provided the pi0 value, which was used to adjust the default significance threshold in the Rmvp package (Pavlopoulos et al. 2023). This method is more appropriate as the FDR is set locally based on the SNP p-values (Storey et al., 2002; Liu et al., 2018).
Extracting candidate genes underlying detected quantitative trait loci
Following the GWAS process, the flanking region for each of the discovered MTA was established by calculating the LD decay distance, which was performed using randomly selected 17 K SNPs across chromosomes. Potential candidate genes in the region were then obtained from the G. max cv. William 82 reference genome gene models 2.0 in SoyBase (https://www.soybase.org accessed on 15 June, 2023). To select the list of relevant candidate genes, gene ontology (GO) annotation and the GO term enrichment report on the soybase.org database were used to evaluate the biological and molecular function of each potential candidate gene.
Following the GWAS process, the flanking region for each of the discovered MTA was established by calculating the LD decay distance. Potential candidate genes in the region were obtained from the G. max cv. William 82 reference-genome gene models 2.0 in SoyBase (https://www.soybase.org accessed on 15 June, 2023). To select the list of relevant candidate genes, gene ontology (GO) annotation and the GO term enrichment report on the soybase.org database were used to evaluate the biological and molecular function of each potential candidate gene. Finally, the electronic fluorescent pictograph (eFP) browser for soybean (www.bar.utoronto.ca, accessed on 16 May 2023) was used to further investigate additional information on each of the selected candidate genes including tissue and developmental stage-dependent expression, as reported in transcriptomic data from Severin et al. (2010).
Results
Phenotyping evaluations
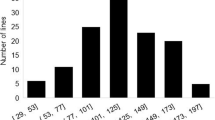
A total of 193 genotypes in the CSGP were evaluated in three independent growth room experiments: GR1, GR2 and GR3. All lines in the panel indicated known signs and symptoms of sclerotinia stem rot disease. Plants developed necrotic, water-soaked lesions starting from the inoculation point on the stem, which suggests the effectiveness of inoculation, as shown in Fig. 1. A wide variation in SSR disease response was observed among the 193 genotypes screened in the controlled environments. Figure 2 illustrates the phenotypic distribution for each genotype in the CSGP. Lesion lengths showed a wide range from 3.7 to 115.6 mm with a mean of 33.4 mm ± 27.4. Disease response groups were categorized into partially resistant: > 25 mm, moderately resistant: > 25–50 mm, and susceptible: > 50 mm. Although the CSGP displayed variation in disease response, phenotypic response distribution was skewed to the right, indicating the majority (111) of genotypes had partial resistance reaction, a few (31) genotypes showing moderate resistance and the remaining (51) genotypes showing a susceptibility response (Figs. 3, 4). The partial resistance check Maple Donovan, and moderate resistance check OAC Bayfield showed shorter lesion lengths than the mean: 17.9 ± 8.2 and 29.5 mm ± 3, respectively, while the susceptible check, Nattosan had longer lesions than the mean: 107.3 mm ± 75.3.

Cotton pad inoculation method (Bastien et al., 2012) and stem lesion due to SSR infection eight days post-inoculation. SSR severity scale illustrated for partial resistance A; moderate resistance B and susceptibility C

Lesion length distribution across the 193 soybean lines. The arrows indicate the position of checks in the distribution: partially resistant Maple Donovan (green arrow), moderately resistant OAC Bayfield (brown arrow), and highly susceptible Nattosan (red arrow)

Population structure A and kinship B plots for the 193 soybean lines. The number of genotypes used in the GWAS analysis are indicated on the x-axis, while individual subgroup membership is indicated on the y-axis. SP 1—SP 6 represents the number of subpopulations in the panel

Quantile–Quantile plot A of estimated–log10(P) from marker—trait association of SSR resistance trait in the CSGP, and Manhattan plot B for GWAS analysis for SSR resistance of the 193 soybean lines. The x-axis indicates the chromosome number and Y-axis indicates − log10(P). The horizontal line represents the significance threshold (FDR = 0.1), with peak SNPs (S02_5352442; C/T on chromosome 2, and S09_45604994; T/C on chromosome 9)
Genotyping
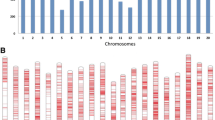
A total of 417 K SNPs was generated from the 193 genotypes and mapped on the 20 chromosomes of soybean. The catalogue was produced after retaining SNPs with a minor allele frequency (MAF) ≥ 0.05 and heterozygosity ≤ 0.1, and filtering out redundant SNPs (LD > 0.99). The average number of SNPs across all 20 chromosomes was 20,865, with a mean density of one SNP per 0.04 cM across the genome. The shortest chromosome was chr 11 with 4,046 SNPs, while the longest was chromosome 18 with 61,236 SNPs as shown in Fig. 5B.

Average linkage disequilibrium decay rate A of the GWAS analysis panel, and the distribution and marker density B of 417 K SNPs
Kinship and population structure
Results from population structure evaluation suggested that the association mapping panel was made up of between three to six subpopulations as shown in Fig. 3A. Based on the results, structure analysis was conducted using K = 6 as the suitable K for the structure profile of the CSGP. Additionally, the kinship matrix between genotypes on the CSGP was estimated to further reduce confounding due to relatedness (Fig. 3B).
GWAS analysis
Different MTAs for SSR were calculated using the FarmCPU algorithm considering the BLUP values as the phenotype and the filtered SNP dataset of 417 K markers as the genotype. Based on the FarmCPU algorithm, two SNPs were found to have a significant association with resistance to SSR (Fig. 4B). As shown in Table 1, the first significant SNP (S02_5352442) was located on chromosome 2 at position 5,352,442 bp, with an allele effect of 16.1. The SNP, S02_5352442 was found in the same QTL region, approximately 25 kb downstream from a previously reported QTL for SSR resistance in soybean by Moellers et al. 2017. The second significant SNP (S09_45604994) was found on chromosome 9 at position 45,604,994 bp, with an allelic effect of 14.1. Studies by Moellers et al. 2017 and Wei et al. 2017 also identified two QTL for SSR resistance on chromosome 9 in two different soybean association mapping panels. Peak SNPs reported in both studies were found around 13,000 Kb downstream from S09_45604994 reported in the current study.
Extraction of candidate genes in identified QTL region
Linkage disequilibrium (LD) estimates, calculated based on the correlation coefficient (r2) of alleles, were used to specify boundaries for potential candidate gene search boundaries within each of the detected MTA region. The average LD (r2) in the panel was estimated to drop to 0.3 per 100 Kb of physical distance, a slow but common LD decay rate for a self-pollinated crop such as soybean. The target region for potential candidate genes was, therefore, selected to be 100 Kb upstream and downstream from each SNP’s peak. Based on GO annotation and GO term enrichment, Glyma.02G059000, Glyma.02G059400, Glyma.02G059700 on chromosome 2, and Glyma.09G232100, Glyma.09G232600, Glyma.09G233400 on chromosome 9 were listed and as strong candidate genes linked to SSR resistance (Table 1).
Discussion
SSR, caused by Sclerotinia sclerotiorum (lib.) de Bary, is one of the most catastrophic, yield limiting diseases of soybean in Canada and worldwide (Willbur et al. 2019). Due to inefficiency and cost of current methods of SSR control, improving plant resistance to the disease through breeding is regarded as the most reliable and economic solution (Willbur et al. 2019). Although several studies have previously reported over a hundred QTL for SSR partial resistance in soybean, only a few of the studies have comprehensively surveyed genotypes that are specific to Canadian environments (www.soybase.org, accessed on 15 June, 2023), where the disease is the third most damaging to soybean production. This study sought to screen a Canadian soybean germplasm panel (CSGP) for SSR resistance, and identify QTL, and potential candidate genes are biologically relevant to SSR resistance that could be leveraged for MAS in Canada.
The results of this study showed significant differences in phenotypic response to S. Sclerotiorum inoculation among the CSGP genotypes. The variation in phenotypic response was consistently evident throughout the experiments. This was expected given that CSGP was assembled to represent the maturity groups of Canadian environments as well as genetic differences in reaction to SSR (Rajcan, unpublished work). All experiments were conducted in controlled environments following optimal growth conditions for soybean. Screening in controlled environments was chosen, because SSR occurrence is known to be extremely dependent on specific environment conditions (20–25 °C temperature and > 80% relative humidity) and the flowering stage of the plants (Mila and Yang 2008). Therefore, controlled environments can allow to distinguish between resistance due to physiological mechanisms and avoidance (e.g. early or late development or canopy arrangement). Furthermore, the cotton pad method, a non-invasive technique (Bastien et al. 2014), was used to mimic the natural infection of S. Sclerotiorum through soybean flowers in the field. According to Bastien et al. (2014), disease response of genotypes in controlled environments predicted results in the field.
The phenotypic data followed a continuous distribution, which illustrates the quantitative nature of the disease. The panel was largely partially resistant (PR) (57%) but also consisted of moderately resistant (MR) (16%) and susceptible (S) genotypes (27%). The check genotypes: Maple Donovan, OAC Bayfield and Natossan, displayed PR, MR, and S phenotypic response respectively as expected. Four out of 10 most PR lines were cultivars from the University of Guelph Soybean breeding program which may accelerate the introgression into its and other breeding pipelines. Furthermore, there was a strong positive correlation across experimental blocks experiments, which suggests consistency and reliability of the phenotypic results. We believe that the MTA results in the current study are enhanced by the robust SNP marker coverage in the employed genetic map. A total of 417 K SNPs were used as were distributed throughout the genome, which is significantly higher than the number of SNPs that is needed for sufficient coverage (Bastien et al. 2014; Hyten et al. 2007). This is also a significantly higher SNP number than used in previous studies that conducted GWAS for SSR resistance in Soybean. Only a study conducted by Boudhrioua et al. (2020) used a higher marker coverage (1.5 M SNPs) than the current study.
By using FarmCPU, two SNPs were found to be significantly associated with resistance to SSR. The first SNP (S02_5352442) was found on position 5,352,442 of chromosome 2. We believe that S02_5352442 may be close to the one discovered by Moellers et al. (2017). The peak SNPs in both studies are located approximately 25 Kb apart. Moellers et al. (2017)’s study was conducted using a much larger and association panel consisting of different soybean genotypes from ours. This provides additional confidence that this QTL may play an important role in partial resistance to SSR in soybean. The second SNP (S09_45604994) was found on position 45,604,994 of chromosome 9. Two separate studies (Moellers et al. 2017; Wei et al. 2017) have also previously found QTL for SSR resistance in soybean on chromosome 9. However, none of the previous QTL overlaps with S09_45604994 in the current study. The lack of overlap could be explained by several factors including variability in screening techniques and environments, using different soybean genotypes in association panels, use of different pathogen isolates for inoculation, and different marker coverage of genotypic data. The lack of overlap could also be explained by the very small effect of identified MTAs in each study. The lack of MTA’s overlap and failure of rediscovery has been reported in several previous studies, including some that have used identical mapping panels and inoculation techniques MTAs (Boudhrioua et al. 2020; Iquira et al. 2015; Moellers et al. 2017; Wei et al. 2017).
Six genes were presented as potential candidate genes for SSR resistance in this study. Candidate gene identification was based on LD region colocation with the peak SNP for SSR resistance and biological function obtained through gene ontology (GO) annotation on Soybase database (www.soybase.org, accessed on 15 June, 2023). As such, three of the candidate genes: Glyma.02G059000, Glyma.02G059400, Glyma.02G059700 were found in the chr2 MTA region, while the other three: Glyma.09G232100, Glyma.09G232600, and Glyma.09G233400 were found in the MTA region on chr 9. The shown candidate genes belong to different gene families that are important for plant defence against pathogens. The defence roles include involvement in cell wall structure (Glyma.09G232100), recognition and/or binding to molecules of pathogen origin, signalling and/or activation of defence responses, etc. as shown in Table 1 (Soybase.org). Some of the selected candidate genes have been reported in previous work (Ali et al. 2022; Khoei et al. 2021; Severin et al. 2010; Wang et al. 2020). Specifically, phylogenic analysis has indicated Glyma.02G059000 belongs to the terpenoid biosynthesis gene family of soybean (Ali et al. 2022). Terpenoids are a highly diverse group compounds that are produced by both plants and animals, where they serve different physiological functions. The terpenoids produced by plants have been reported to play an important role in defence against pathogens, particularly offering protection from fungal diseases (Ali et al. 2022; Devarenne 2009). Glyma.02G059400 gene annotation involvement in the carbohydrate biosynthetic process, jasmonic acid biosynthetic process and response to fungus, wounding, and jasmonic acid stimulus which suggests an essential role in plant defence mechanism. Additionally, previous work from Khoei et al. (2021) to understand the regulation of the defence mechanism against plant-parasitic nematodes (PPN) in soybean highlighted Glyma.02G059400 as one of the major differentially expressed target genes involved in the soybean immune response to PPN (Khoei et al. 2021). Glyma.02g059700 was reported among the candidate genes linked to SSR resistance in soybean reported by Moellers et al. (2017). The candidate gene was found chromosome 2 QTL (Sclero 3-g52) and is known to be involved in threonine kinase signalling, cell wall macromolecule catabolic process, cell surface receptor signalling and other functions of plant defence (Moellers et al. 2017). Furthermore, Glyma.09G232100 on chr 9 has been shown to belong to the Pectin Methylesterase Inhibitors (PMEI) gene family in soybean (Wang et al. 2020). Genes in the PMEI family play a critical role in plant-pathogen interaction, particularly through the cell wall structure and properties (Lionetti et al. 2012; Wang et al. 2020).
Despite our efforts to screen for SSR resistance using an inoculation method that mimics natural infection (Bastien et al. 2014) as well as employing a large SNP catalogue with extensive genome coverage, our study faced a few limitations. Our results show a significant portion of variation that was not captured in our GWAS analysis. This could be due to the highly quantitative nature of SSR resistance, where the trait may be largely governed minor-effect QTL in the used association panel. Furthermore, all our phenotyping experiments were conducted in controlled environments, which can lead to results that are inconsistent with field environments (Hoffman et al. 2002; Kim et al. 2000; Wegulo et al. 1998). Future studies should aim to discover QTL using similarly extensive or more marker coverage coupled with phenotyping larger and more diverse populations in controlled environment and field environment phenotyping could help to capture the remaining variation and discover additional MTAs that can be deployed for MAS in breeding programs to accelerate improvement of SSR resistance in soybean.
Conclusion
Through screening a Canadian Soybean germplasm panel for SSR, this study identified partially resistant soybean lines that will be useful sources for the development of soybean cultivars with improved resistance to SSR. GWAS studies discovered two significant MTAs linked to resistance to SSR in the panel. Neither MTA has been reported previously, nor do we believe that they may represent novel sources of genetic resistance to SSR in soybean. Several candidate genes were found in the MTAs regions, including some that belong to soybean gene families that are known to play key roles in plant defence. Results from this study are expected to provide a useful basis for further investigation on gene functional analysis, identify causal analysis and ultimate development of molecular markers that can be used by breeders to develop SSR-resistant soybean cultivars for Canadian environments.
Data availability
The raw data supporting the conclusions of this article will be available without undue reservation.
References
Ali M, Miao L, Soudy FA, Darwish DBE, Alrdahe SS, Alshehri D, Benedito VA, Tadege M, Wang X, Zhao J (2022) Overexpression of Terpenoid biosynthesis genes modifies root growth and nodulation in soybean (Glycine max). Cells 11:2622
Allen TW, Bradley CA, Sisson AJ, Byamukama E, Chilvers MI, Coker CM, Collins AA, Damicone JP, Dorrance AE, Dufault NS (2017) Soybean yield loss estimates due to diseases in the united states and Ontario, Canada, from 2010 to 2014. Plant Health Progress 18:19–27
Bastien M, Sonah H (2010) Belzile F (2014) Genome wide association mapping of Sclerotinia sclerotiorum resistance in soybean with a genotyping-by-sequencing approach. The Plant Genome 7:0030
Bernardo R (2008) Molecular markers and selection for complex traits in plants: learning from the last 20 years. Crop Sci 48(5):1649–1664
Bernardo R. (2010) Breeding for Quantitative Traits in Plants (2nd Edition). Stemma Press
Boland G, Hall R (1994) Index of plant hosts of sclerotinia sclerotiorum. Can J Plant Path 16:93–108
Bolton MD, Thomma BP, Nelson BD (2006) Sclerotinia sclerotiorum (Lib.) de Bary: biology and molecular traits of a cosmopolitan pathogen. Mol Plant Pathol 7:1–16
Bootsma A, McKenney D, Anderson D, Papadopol P (2007) A re-evaluation of crop heat units in the maritime provinces of Canada. Can J Plant Sci 87:281–287
Boudhrioua C, Bastien M, Torkamaneh D, Belzile F (2020) Genome-wide association mapping of Sclerotinia sclerotiorum resistance in soybean using whole-genome resequencing data. BMC Plant Biol 20:1–9
Brown D, Bootsma A (2000) Crop heat units for corn and other warm-season crops in Ontario. Ministry of Agriculture, Food & Rural Affairs, Agriculture & Rural
Browning BL, Zhou Y, Browning SR (2018) A one-penny imputed genome from next-generation reference panels. Am J Hum Genet 103:338–348
Devarenne TP (2009) Terpenoids: higher. eLS
Ekine CC, Rowe SJ, Bishop SC, De Koning DJ (2014) Why breeding values estimated using familial data should not be used for genome-wide association studies. G3: Genes Genomes, Genet 4:341–347
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379
Gorjanc G, Cleveland MA, Houston RD, Hickey JM (2015) Potential of genotyping-by-sequencing for genomic selection in livestock populations. genetics, selection, evolution, 47(1), 12.Grau C, Radke V, Gillespie F (1982) resistance of soybean cultivars to Sclerotinia sclerotiorum. Plant Dis 66:506–508
Hayes BJ, Bowman PJ, Chamberlain AC, Goddard ME (2009) Invited review: genomic selection in dairy cattle: progress and challenges. J Dairy Sci 92(2):433–443
Hoffman DD, Diers BW, Hartman GL, Nickell CD, Nelson RL, Pedersen WL, Cober ER, Graef GL, Steadman JR, Grau CR, Nelson BD, del Rio LE, Helms T, Anderson T, Poysa V, Rajcan I, Stienstra WC (2002) Selected soybean plant introductions with partial resistance to sclerotinia sclerotiorum. Plant Dis 86:971–980
Hong H, Najafabadi MY, Torkamaneh D, Rajcan I (2022) Identification of quantitative trait loci associated with seed quality traits between Canadian and Ukrainian mega-environments using genome-wide association study. Theor Appl Genet 135:2515–2530
Hyten DL, Choi I-Y, Song Q, Shoemaker RC, Nelson RL, Costa JM, Specht JE, Cregan PB (2007) Highly variable patterns of linkage disequilibrium in multiple soybean populations. Genetics 175:1937–1944
Iquira E, Humira S, François B (2015) Association mapping of QTLs for sclerotinia stem rot resistance in a collection of soybean plant introductions using a genotyping by sequencing (GBS) approach. BMC Plant Biol 15:5
Jannink JL, Walsh B (2002) Association mapping in plant populations. Springer
Jing Y, Teng W, Qiu L, Zheng H, Li W, Han Y, Zhao X (2021) Genetic dissection of soybean partial resistance to sclerotinia stem rot through genome wide association study and high throughout single nucleotide polymorphisms. Genomics 113:1262–1271
Khoei MA, Karimi M, Karamian R, Amini S, Soorni A (2021) Identification of the complex interplay between nematode-related lncrnas and their target genes in glycine max L. Front Plant Sci. https://doi.org/10.3389/fpls.2021.779597
Kim HS, Hartman GL, Manandhar JB, Graef GL, Steadman JR, Diers BW (2000) Reaction of soybean cultivars to sclerotinia stem rot in field, greenhouse, and laboratory evaluations. Crop Sci 40:665–669
Koenning SR, Wrather JA (2010) Suppression of soybean yield potential in the continental united states by plant diseases from 2006 to 2009. Plant Health Progress 11:5
Korte A, Farlow A (2013) The advantages and limitations of trait analysis with GWAS: a review. Plant Methods 9:1–9
Lal R (2013) Food security in a changing climate. Ecohydrol Hydrobiol 13:8–21
Lionetti V, Cervone F, Bellincampi D (2012) Methyl esterification of pectin plays a role during plant–pathogen interactions and affects plant resistance to diseases. J Plant Physiol 169:1623–1630
Liu X, Huang M, Fan B, Buckler ES, Zhang Z (2016) Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet 12:e1005767
Mila AL, Yang XB (2008) Effects of fluctuating soil temperature and water potential on sclerotia germination and apothecial production of sclerotinia sclerotiorum. Plant Dis 92:78–82
Moellers TC, Singh A, Zhang J, Brungardt J, Kabbage M, Mueller DS, Grau CR, Ranjan A, Smith DL, Chowda-Reddy R (2017) Main and epistatic loci studies in soybean for Sclerotinia sclerotiorum resistance reveal multiple modes of resistance in multi-environments. Sci Rep 7:3554
Mueller D, Dorrance A, Derksen R, Ozkan E, Kurle J, Grau C, Gaska J, Hartman G, Bradley C, Pedersen W (2002) Efficacy of fungicides on Sclerotinia sclerotiorum and their potential for control of Sclerotinia stem rot on soybean. Plant Dis 86:26–31
Pavlopoulos GA, Baltoumas FA, Liu S, Selvitopi O, Camargo AP, Nayfach S, Azad A, Roux S, Call L, Ivanova NN, Chen IM, Paez-Espino D, Karatzas E, Acinas SG, Ahlgren N, Attwood G, Baldrian P, Berry T, Bhatnagar JM, Bhaya D, Bidle KD, Blanchard JL, Boyd ES, Bowen JL, Bowman J, Brawley SH, Brodie EL, Brune A, Bryant DA, Buchan A, Cadillo-Quiroz H, Campbell BJ, Cavicchioli R, Chuckran PF, Coleman M, Crowe S, Colman DR, Currie CR, Dangl J, Delherbe N, Denef VJ, Dijkstra P, Distel DD, Eloe-Fadrosh E, Fisher K, Francis C, Garoutte A, Gaudin A, Gerwick L, Godoy-Vitorino F, Guerra P, Guo J, Habteselassie MY, Hallam SJ, Hatzenpichler R, Hentschel U, Hess M, Hirsch AM, Hug LA, Hultman J, Hunt DE, Huntemann M, Inskeep WP, James TY, Jansson J, Johnston ER, Kalyuzhnaya M, Kelly CN, Kelly RM, Klassen JL, Nüsslein K, Kostka JE, Lindow S, Lilleskov E, Lynes M, Mackelprang R, Martin FM, Mason OU, McKay RM, McMahon K, Mead DA, Medina M, Meredith LK, Mock T, Mohn WW, Moran MA, Murray A, Neufeld JD, Neumann R, Norton JM, Partida-Martinez LP, Pietrasiak N, Pelletier D, Reddy TBK, Reese BK, Reichart NJ, Reiss R, Saito MA, Schachtman DP, Seshadri R, Shade A, Sherman D, Simister R, Simon H, Stegen J, Stepanauskas R, Sullivan M, Sumner DY, Teeling H, Thamatrakoln K, Treseder K, Tringe S, Vaishampayan P, Valentine DL, Waldo NB, Waldrop MP, Walsh DA, Ward DM, Wilkins M, Whitman T, Woolet J, Woyke T, Iliopoulos I, Konstantinidis K, Tiedje JM, Pett-Ridge J, Baker D, Visel A, Ouzounis CA, Ovchinnikov S, Buluç A, Kyrpides NC, Families NMP, C, (2023) Unraveling the functional dark matter through global metagenomics. Nature 622:594–602
Peltier AJ, Bradley CA, Chilvers MI, Malvick DK, Mueller DS, Wise KA, Esker PD (2012) Biology, yield loss and control of Sclerotinia stem rot of soybean. J Integr Pest Manage 3:B1–B7
Raj A, Stephens M, Pritchard JK (2014) fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics 197:573–589
Ray DK, Mueller ND, West PC, Foley JA (2013) Yield trends are insufficient to double global crop production by 2050. PLoS ONE 8:e66428
Robinson GK (1991) That BLUP is a good thing: the estimation of random effects. Stat Sci 6:15–32
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, Xu D, Hellsten U, May GD, Yu Y, Sakurai T, Umezawa T, Bhattacharyya MK, Sandhu D, Valliyodan B, Lindquist E, Peto M, Grant D, Shu S, Goodstein D, Barry K, Futrell-Griggs M, Abernathy B, Du J, Tian Z, Zhu L, Gill N, Joshi T, Libault M, Sethuraman A, Zhang X-C, Shinozaki K, Nguyen HT, Wing RA, Cregan P, Specht J, Grimwood J, Rokhsar D, Stacey G, Shoemaker RC, Jackson SA (2010) Genome sequence of the palaeopolyploid soybean. Nature 463:178–183
Severin AJ, Woody JL, Bolon Y-T, Joseph B, Diers BW, Farmer AD, Muehlbauer GJ, Nelson RT, Grant D, Specht JE, Graham MA, Cannon SB, May GD, Vance CP, Shoemaker RC (2010) RNA-seq atlas of glycine max: a guide to the soybean transcriptome. BMC Plant Biol 10:160
Storey JD (2002) A direct approach to false discovery rates. J Royal Stat Soc: Ser B (stat Methodol) 64(3):479–498
Sun C, Wang B, Yan L, Hu K, Liu S, Zhou Y, Chunyun G, Zhenqian Z, Jiana L, Jiefu Z, Song C, Jing W, Chaozhi M, Jinxing T, Jinxiong S, Tingdong F, Yi B (2016) Genome-wide association study provides insight into the genetic control of plant height in rapeseed (Brassica napus L.). Front Plant Sci 7:1102
Tagliotti ME, Deperi SI, Bedogni MC, Huarte MA (2021) Genome-wide association analysis of agronomical and physiological traits linked to drought tolerance in a diverse potatoes (Solanum tuberosum) panel. Plant Breeding 140(4):654–664
Torkamaneh D, Laroche J, Bastien M, Abed A, Belzile F (2017) Fast-GBS: a new pipeline for the efficient and highly accurate calling of SNPs from genotyping-by-sequencing data. BMC Bioinform 18:5
Tsai HY, Janss LL, Andersen JR, Orabi J, Jensen JD, Jahoor A, Jensen J (2020) Genomic prediction and GWAS of yield, quality and disease-related traits in spring barley and winter wheat. Sci Rep 10(1):3347
VanRaden PM (2007) Genomic measures of relationship and inbreeding. INTERBULL Bulletin 37:33–33
Wang J, Ling L, Cai H, Guo C (2020) Gene-wide identification and expression analysis of the PMEI family genes in soybean (Glycine max). 3 Biotech 10:335
Wegulo SN, Yang XB, Martinson CA (1998) Soybean cultivar responses to sclerotinia sclerotiorum in field and controlled environment studies. Plant Dis 82:1264–1270
Wei W, Mesquita ACO, Figueiró AdA, Wu X, Manjunatha S, Wickland DP, Hudson ME, Juliatti FC, Clough SJ (2017) Genome-wide association mapping of resistance to a Brazilian isolate of sclerotinia sclerotiorum in soybean genotypes mostly from Brazil. BMC Genomics 18:1–16
Willbur J, McCaghey M, Kabbage M, Smith DL (2019) An overview of the Sclerotinia sclerotiorum pathosystem in soybean: impact, fungal biology, and current management strategies. Trop Plant Pathol 44:3–11
Yang Q, Cui J, Chazaro I, Cupples LA, Demissie S (2005) Power and type I error rate of false discovery rate approaches in genome-wide association studies. In BMC Genetics 6:1–4
Yin L, Zhang H, Tang Z, Xu J, Yin D, Zhang Z, Yuan X, Zhu M, Zhao S, Li X, Liu X (2021) rMVP: a memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genomics Proteomics Bioinform 19:619–628
Yoosefzadeh Najafabadi M, Heidari A, Rajcan I (2023) Allinone pre-processing: a comprehensive preprocessing framework in plant field phenotyping. SoftwareX 23:101464
Yoosefzadeh-Najafabadi M, Rajcan I (2022) Six decades of soybean breeding in Ontario, Canada: a tradition of innovation. Can J Plant Sci 103:333–352
Yoosefzadeh-Najafabadi M, Torabi S, Tulpan D, Rajcan I, Eskandari M (2021) Genome-wide association studies of soybean yield-related hyperspectral reflectance bands using machine learning-mediated data integration methods. Front Plant Sci 12:777028
Yoosefzadeh-Najafabadi M, Rajcan I, Vazin M (2022) High-throughput plant breeding approaches: Moving along with plant-based food demands for pet food industries. Front Vet Sci 9:991844
Yoosefzadeh-Najafabadi M, Torabi S, Tulpan D, Rajcan I, Eskandari M (2023) Application of SVR-mediated GWAS for identification of durable genetic regions associated with soybean seed quality traits. Plants 12:2659
Zablocki RW, Schork AJ, Levine RA, Andreassen OA, Dale AM, Thompson WK (2014) Covariate-modulated local false discovery rate for genome-wide association studies. Bioinformatics 30(15):2098–2104
Zeng W, Kirk W, Hao J (2012) Field management of Sclerotinia stem rot of soybean using biological control agents. Biol Control 60:141–147
Zhang C, Dong S-S, Xu J-Y, He W-M, Yang T-L (2019) PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 35:1786–1788
Acknowledgements
The authors thanks current and former staff of Dr. Istvan Rajcan’s soybean research team at University of Guelph in terms of field trial management, data collection and analytical supports. The authors also appreciate the generous funding support from in part by Canadian agricultural partnership program (CAP) of agriculture and agri-food Canada (AAFC), Grain Farmers of Ontario and SeCan.
Funding
This project was funded in part by Canadian agricultural partnership program (CAP) of agriculture and agri-food Canada (AAFC), Grain Farmers of Ontario and SeCan. The funding bodies did not play any role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
IR conceptualized, designed, and directed the experiments; DM contributed to conceptualization of the experiments, conducted the experiments, modelled, summarized the results, and wrote the manuscript; MY-N participated in writing and editing the manuscript; MY-N, and IR revised the manuscript and validated the results. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that the research was conducted without any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Communicated by Mikko J. Sillanpää.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mugabe, D., Yoosefzadeh-Najafabadi, M. & Rajcan, I. Genetic diversity and genome-wide association study of partial resistance to Sclerotinia stem rot in a Canadian soybean germplasm panel. Theor Appl Genet 137, 201 (2024). https://doi.org/10.1007/s00122-024-04708-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00122-024-04708-8