
Abstract
Key message
A minor-effect QTL, Qhd.2AS, that affects heading date in wheat was mapped to a genomic interval of 1.70-Mb on 2AS, and gene analysis indicated that the C2H2-type zinc finger protein gene TraesCS2A02G181200 is the best candidate for Qhd.2AS.
Abstract
Heading date (HD) is a complex quantitative trait that determines the regional adaptability of cereal crops, and identifying the underlying genetic elements with minor effects on HD is important for improving wheat production in diverse environments. In this study, a minor QTL for HD that we named Qhd.2AS was detected on the short arm of chromosome 2A by Bulked Segregant Analysis and validated in a recombinant inbred population. Using a segregating population of 4894 individuals, Qhd.2AS was further delimited to an interval of 0.41 cM, corresponding to a genomic region spanning 1.70 Mb (from 138.87 to 140.57 Mb) that contains 16 high-confidence genes based on IWGSC RefSeq v1.0. Analyses of sequence variations and gene transcription indicated that TraesCS2A02G181200, which encodes a C2H2-type zinc finger protein, is the best candidate gene for Qhd.2AS that influences HD. Screening a TILLING mutant library identified two mutants with premature stop codons in TraesCS2A02G181200, both of which exhibited a delay in HD of 2–4 days. Additionally, variations in its putative regulatory sites were widely present in natural accession, and we also identified the allele which was positively selected during wheat breeding. Epistatic analyses indicated that Qhd.2AS-mediated HD variation is independent of VRN-B1 and environmental factors. Phenotypic investigation of homozygous recombinant inbred lines (RILs) and F2:3 families showed that Qhd.2AS has no negative effect on yield-related traits. These results provide important cues for refining HD and therefore improving yield in wheat breeding programs and will deepen our understanding of the genetic regulation of HD in cereal plants.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The timing of flowering marks the transition from vegetative growth to reproductive development, and thus it is essential for plants to optimize seed production and complete a successful life cycle (Andres and Coupland 2012). In cereal crops, heading date (HD; flowering time) is an important and complex agronomic trait that determines regional adaptability under various climatic conditions, thereby affecting crop production. Plants have evolved multiple pathways that integrate intrinsic genetic signals and external environmental stimuli to initiate flowering at the appropriate time (Jung and Muller 2009). Hence, identification and cloning of QTLs/genes that control HD will deepen our understanding of the genetic control of this trait and provide useful genetic resources for the engineering and breeding of novel wheat varieties with high yield and broad adaptability.
Bread wheat (Triticum aestivum L.) is one of the world’s most important staple crops because it produces ~ 20% of the daily calories consumed by humans (Dubcovsky and Dvorak 2007). Wheat is an allohexaploid species (2n = 6x = 42) that was formed through two rounds of polyploidization, and its genetic diversity partially contributed to its spread from the Fertile Crescent area to diverse environments around the world (Dubcovsky and Dvorak 2007; Salamini et al. 2002). In the past several decades, many studies have been performed to dissect the genetic control of HD and elucidate its interactions with environmental factors (Distelfeld et al. 2009a; Shi et al. 2019). These studies have identified the vernalization pathway, photoperiod response, and earliness per se as three main components that shape heading date in wheat.
Many plant species require prolonged exposure to low temperatures in winter to trigger the initiation of flowering; such a requirement is known as vernalization (Chouard 1960). Depending on the requirement of vernalization, wheat is classified into either winter or spring growth habits, under which the genetic regulation is mainly mediated by Vernalization (VRN) genes, including VRN1, VRN2, VRN3, and VRN-D4 (Kippes et al. 2015; Yan et al. 2003, 2004b, 2006). VRN1, a gene that encodes an APETALA1-like MADS-box transcription factor, was isolated from the diploid wheat species Triticum monococcum L. and acts as a flowering promoter (Yan et al. 2003). VRN1 expression is repressed in non-vernalized plants, but a cold treatment dramatically induces transcription in the shoot apex and leaves (Chen and Dubcovsky 2012; Loukoianov et al. 2005; Oliver et al. 2009). Genetic analysis showed that allelic variations in the promoter and intronic region of VRN1 are associated with different growth habits in wheat (Fu et al. 2005; Yan et al. 2004a). Further studies demonstrated that conserved regulatory elements such as the CArG-box are crucial for expression of VRN1 by mediating its binding with other flowering regulators (e.g., VRT2) (Kane et al. 2007; Xie et al. 2021a; Zhang et al. 2012). The VRN2 locus contains two tightly-linked genes, ZCCT1 and ZCCT2, which encode proteins with zinc-finger and CCT domains that function as flowering repressors (Yan et al. 2004b). An investigation into natural allelic variations in ZCCT1 and ZCCT2 indicated that both genes function in delaying HD, and mutations in either one of the two genes cannot completely disrupt the VRN2 function (Distelfeld et al. 2009b). Genetic analysis revealed that mutations in the conserved CCT domains are associated with the non-functional vrn2 allele, suggesting that the CCT domain is essential for VRN2-mediated flowering repression; this hypothesis was further supported by biochemical evidence showing that mutations in the CCT domain reduced the ability of VRN2 to interact with NUCLEAR FACTOR-Y proteins (Distelfeld et al. 2009b; Li et al. 2011). The expression levels of VRN2 remain high in non-vernalized winter wheat to repress flowering by inhibiting the transcription of VRN1. After vernalization, the level of VRN2-specific transcripts gradually decreased, thus releasing the repression of VRN3 (Chen and Dubcovsky 2012; Dubcovsky et al. 2006). In wheat and barley, VRN3 is the closest homolog to Arabidopsis FLOWERING LOCUS T (AtFT) (Yan et al. 2006). AtFT is specifically expressed in leaves and the protein is transported to the shoot apical meristem where it interacts with the bZIP transcription factor FD to activate downstream floral genes (Putterill and Varkonyi-Gasic 2016). Similarly, wheat FT1 (VRN3) physically interacts with FLOWRING LOCUS D like 2 (FDL2) and 14-3-3C proteins to form a florigen activation complex that binds directly to the promoter of VRN1 (Li and Dubcovsky 2008; Li et al. 2015). VRN-D4 was mapped to the short arm of chromosome 5D, and cloning of this locus identified an insertional segment from chromosome 5AL that contained a copy of VRN-A1 with distinctive mutations in its regulatory regions (Kippes et al. 2014, 2015).
Photoperiod is another environmental factor that affects HD in wheat. As a long-day (LD) plant, short-day conditions (SDs) significantly delay heading in most wheat varieties. However, some varieties exhibit a short delay in heading date under SDs; this characteristic has been referred to as photoperiod insensitivity (PI), which confers a wide adaptation on wheat to regions with hotter and drier environments, because the plants can complete their life cycles before experiencing adverse stresses (Beales et al. 2007; Wilhelm et al. 2009). The photoperiod response in wheat is mainly mediated by three homoeologous genes, Ppd-A1, Ppd-B1, and Ppd-D1, in chromosome group 2; Among these genes, many allelic variations have been identified that are associated with PI (Bentley et al. 2013; Diaz et al. 2012; Seki et al. 2011). The semi-dominant Ppd-D1a allele is the major source of PI in wheat; this allele has a 2089 bp deletion in its promoter compared to the wild-type allele Ppd-D1b (Beales et al. 2007). In addition, the Ppd-A1a allele has a 1085 bp deletion and the Ppd-B1a allele has a 308 bp insertion in the 5ʹ upstream regions that interrupt a highly conserved regulatory site that affects PI in wheat (Nishida et al. 2012).
When fully-vernalized wheat cultivars were grown under inductive LD conditions, a large difference in heading date was still present in breeding populations, and the underlying genetic factors were categorized as earliness per se genes (Worland et al. 1994). Such genes are difficult to precisely map and identify under natural conditions due to their minor genetic effects and the complex inheritance of HD in hexaploid wheat (Bullrich et al. 2002; Chen et al. 2015). The major Earliness per se loci have been mapped to chromosomes 1A (Eps-Am1, QEps.dms-1A), 1D (Eps-D1), and 4A (QEps.dms-4A); some of these genes display significant epistatic interactions with vernalization and the photoperiod response (Appendino and Slafer 2003; Bullrich et al. 2002; Chen et al. 2015). Further studies have suggested that TaELF3-D1 and TaELF3-A1, which are both orthologous to the Arabidopsis circadian clock gene Early Flowering 3 (ELF3), are the most likely causal genes responsible for Eps-D1 and Eps-Am1, respectively.
Although extensive studies have identified many major genes involved in the genetic regulation of HD in wheat, the dramatic effects that these genes have on HD and other agronomic traits (e.g., plant height and yield) make them unsuitable for fine-tuning the improvement of wheat cultivars. Genes with minor effects on HD are more desirable targets for improving regional adaptability through modern breeding. In this study, we report the identification and fine-mapping of a minor QTL for heading date, Qhd.2AS, that had no negative effect on yield. Sequence variation, gene expression, and mutant analyses suggested that TraesCS2A02G181200, a locus that encodes a C2H2-type zinc finger protein, is the most likely causal gene for Qhd.2AS. These results provide basic information and novel genetic resources for marker-assisted selection in wheat breeding.
Materials and methods
Plant materials and phenotypic investigations
The early heading 1 (eh1) wheat mutant was induced by gamma ray treatment of seeds of the winter wheat inbred line ‘Zhongyuan 9’ (ZY9). The M3 plants were crossed with an elite winter cultivar ‘Lunxuan 987’ (LX987) to develop 400 F6-generation recombinant inbred lines (RILs) by single-seed descent. The F6 RILs were planted in three consecutive years for phenotypic assessments. The RILs and the parental lines were sown in 1.5 m row plots with 20 seeds in each row. All field trials were designed in randomized complete blocks with three replications at the Zhongpuchang Experimental Station of the Institute of Crop Sciences, Chinese Academy of Agricultural Sciences (Beijing, China). The secondary mapping population was derived from RILs that were homozygous for the recessive Vrn-B1v allele but heterozygous for Qhd.2AS. Herein we used Vrn-B1a and Vrn-B1v to refer to the dominant and recessive alleles at VRN-B1 locus, as previously described (Eagles et al. 2010). For individual plants, HD was set as the time when at least half of the spikes had emerged from the leaf sheath. When two-thirds of the individual plants headed in a RIL, the number of days from sowing to heading were recorded. Plant height (PH) was measured from the bottom of the stem to the top of the spike in the main stem at maturity. Thousand grain weight (TGW) was determined as the grain weight divided by the number of grains in each plant and then multiplying by 1000. For kernel-dimension-related traits, 100–200 plump seeds from each plant were measured with an SC-A1 automatic seeds tester (Wseen Technology, China). PH and TWG were measured in 2016, 2017, and 2018; the kernel-dimension-related traits were measured in 2017 and 2018.
SNP-array-based bulked segregant analysis (BSA)
Based on the HD collected over the 3 years, extremely early- or late-heading RILs were selected for DNA extraction using the PVP 40 protocol (Guillemaut and Mardchal-Drouard 1992). The quality and quantity of genomic DNA was determined using a NanoDrop ND-2000 spectrophotometer (Thermo Scientific) and agarose gel electrophoresis. The BSA was conducted as previously described (Li et al. 2020). Briefly, each bulk was constructed by mixing an equal amount of DNA from 18 to 22 RILs. A total of eight bulks, including three early-heading, three late-heading, and two parental bulks, were constructed and genotyped with the Affymetrix® Axiom® Wheat 660 K SNP array. High-quality Single Nucleotide Polymorphisms (SNPs) were selected between the two parental lines based on the threshold of Dash QC value > 0.82, Call-Rate > 0.94, and missing data rate < 0.25. The homozygous SNPs present in the parents were identified in the extremely-heading bulk pairs for SNP-trait association analysis. The allele frequency in each early- or late-heading bulk was calculated as the number of early or late SNPs (eh1 or LX987 allele) divided by the total number of homozygous SNPs. SNP density was determined by counting the number of SNPs within 1 mega-base (Mb) on chromosomes 2A and 5B.
Development of molecular markers and genotyping
Based on the Wheat 660 K SNP assay and our previous BSR-seq analysis, high-quality SNPs were converted to Kompetitive Allele Specific PCR (KASP) markers using the online primer design pipeline (https://galaxy.triticeaetoolbox.org/). Each of the two allele-specific forward primers were labeled with a FAM (5ʹ-GAAGGTGACCAAGTTCATGCT-3ʹ) or a HEX (5ʹ-GAAGGTCGGAGTCAACGGATT-3ʹ) tail at the 3ʹ end. All primers were synthesized by Sangon Biotechnology (Shanghai, China) and the names and sequences are given in Table S6. A single PCR reaction contained 2.5 μl KASP master mixture (LGC Genomics, Middlesex, UK), 2.4 μl template DNA (60 ng μl−1), 0.04 μl MgCl2, and 0.06 μl of a primer mixture that consisted of 12 μl of each forward primer (100 μM), 30 μl of the reverse primer (100 μM), and 46 μl of ddH2O. PCR cycling was performed in the CFX 96 Real-Time System (Bio-Rad, USA) using the following conditions: pre-denaturation at 95 °C for 15 min, followed by 10 touchdown cycles (95 °C for 20 s, touchdown at 65 °C initially and decreasing by 0.8 °C per cycle for 30 s), with an additional 32 cycles of 95 °C for 10 s; and 57 °C for 60 s. After the amplification reactions, fluorescence was detected using the FLUOstar Omega reader (BMG LABTECH, Germany), and signal discrimination was performed using Klustercaller software (v2.22.0.5, LGC Genomics).
Genetic mapping of Qhd.2AS
Based on the genotypes obtained from the KASP assays in the RILs, a genetic linkage map of chromosome 2A was constructed using the MAP function of IciMapping 4.1 (Meng et al. 2015). The re-combination frequency was converted to centi-Morgans (cMs) using the Kosambi mapping function (Kosambi 1943). QTLs for HD were detected in the BIP module using an inclusive composite interval mapping (ICIM) algorithm. Significant QTLs were identified at a logarithm of odds difference (LOD) threshold of 2.5. The phenotypic variance explained (PVE) of significant QTLs was calculated by ICIM analysis. The degree of dominance was calculated using the mean values of homozygous and heterozygous lines as previously described (Li et al. 2020). For fine mapping of Qhd.2AS, we constructed a segregating population derived from heterozygous RILs. Two molecular markers from the initial mapping that map to flanking loci were used to screen recombinant events for Qhd.2AS. The recombinant plants were self-pollinated and their progeny were genotyped to identify homozygous individuals. The HDs of homozygous recombinant progeny plants were used to compare with lines homozygous for LX987 or the eh1 allele. Based on the genotypes and phenotypes of these recombinant plants, the Qhd.2AS locus was precisely mapped to a limited region.
Variation annotation and expression analysis
We previously re-sequenced the whole genomes of ZY9, LX987, and the eh1 mutant (Li et al. 2021; Xie et al. 2021b). SNPs and Insertion/deletion polymorphisms (Indels) in the candidate mapping region were identified and filtered based on a sequencing depth of > 20 and mapping quality > 5. High-quality SNPs and Indels were annotated using ANNOVAR software (Wang et al. 2010) according to the reference genome annotation (IWGSC RefSeq annotation v1.0). Predication of cis-regulatory elements were performed in PlantCARE (Rombauts et al. 1999). For gene expression analysis, fresh leaves and young spikes were sampled at the booting stage. Total RNA was isolated using a PrimeScript RT reagent Kit with genomic DNA Eraser (TaKaRa, China). First-strand cDNA synthesis was performed with the Goldenstar® RT6 cDNA Synthesis Kit (Tsingke Biotechnology, China). Quantitative real-time PCR (qRT-PCR) was performed using the ChamQ SYBR qPCR Master Mix (Vazyme, China) in the CFX 96 Real-Time System (Bio-Rad, USA). In each reaction, the Actin gene was used as an endogenous control to normalize the relative expression using the 2−△△CT method (Livak and Schmittgen 2001). All samples were evaluated with at least three biological and three technical replicates.
Screening for TILLING mutants
We generated an in-house mutant library including over 2000 mutant lines induced by 1.0% EMS in the genetic background of the winter wheat cultivar ‘Jing411.’ Mutants with stop-gain or frameshift mutations were identified. The mutations in E0039 and E0683 were further validated by Sanger sequencing with gene-specific primers (Table S6). The HDs of these mutant lines were investigated during the 2020–2021 growing season. Photos were taken on the same day when the wild-type ‘Jing411’ plants had finished heading.
Haplotype analysis
For Qhd.2AS, four tightly linked KASP markers (Table S6) were used to genotype 262 wheat accessions. The frequency of each haplotype was calculated in three categories of plants with different growth habits, and in modern cultivars and landraces. For the candidate gene TraesCS2A02G181200, SNPs in the gene and the 3 kb upstream sequence were obtained from the Wheat Union Database, which contains variations identified by whole genome re-sequencing from 677 hexaploid wheat accessions (Wang et al. 2020). SNPs with a missing data rate of > 0.4 were filtered out. Linkage disequilibrium (LD) analyses of selected SNPs and identification of haplotypes were performed with Haploview software (Barrett et al. 2005). The geographic distribution of the SNP-140361773 was analyzed and visualized using the HapMap function in the SnpHub web server (Wang et al. 2020).
Statistical analysis and visualization
Multivariate analysis of variance for HD was computed using a general linear model in IBM SPSS v22.0 software (http://www.spss.com). Phenotypic differences between homozygous RILs carrying different alleles were evaluated by Student’s t-test. Data visualization was conducted using GraphPad Prism (R) version 8 (https://www.graphpad.com).
Results
SNP-array-based BSA of heading date in a RIL population
We previously identified the early-heading wheat mutant eh1 from gamma-ray irradiation of the inbred line ‘Zhongyuan 9’ (ZY9). A total of 400 recombinant inbred lines (RILs) were developed by crossing eh1 with an elite winter cultivar, ‘Lunxuan 987’ (LX987), which exhibited 4 to 8 days later heading than eh1 but headed 4 to 6 days earlier than ZY9 across three environments (Li et al. 2020). Using a BSR-Seq approach, we previously identified a large fragment deletion in the major flowering gene VRN-B1 that are mainly responsible for the HD variation in the RILs (Li et al. 2020). Additionally, a continuous distribution in HD was observed in the RILs with the homozygous VRN-B1 genotype. The HDs of RILs homozygous for the dominant and recessive VRN-B1 alleles exhibited similar differences (7–10 days) in multiple environments (Fig. S1), suggesting that additional minor-effect QTLs for HD are present in the RILs. In an initial attempt to test this hypothesis, we performed Wheat 660 K array-based BSA with three independent replicates and identified six chromosomes (2A, 2B, 3A, 3B, 5B, and 6A) that might be associated with the variation in HD (Li et al. 2020). In BSA, multiple bulks can be selected independently and compared with parallel bulks to reduce the probability of false positives (Zou et al. 2016). Thus, we re-analyzed the SNP distribution of extremely early and late heading bulks by mixing the three replicates together. Notably, we observed that most SNPs were identified in chromosomes 2A, 2B, 3B, and 5B in the three bulk pairs, which correlated with the SNP distribution between the two parental lines (Fig. 1a). In each early- or late-heading bulk, the HD-associated markers are more likely to be selected together and present a relatively higher frequency of early- or late-heading alleles in the corresponding bulks (Giovannoni et al. 1991; Michelmore et al. 1991). Therefore, we investigated the allele frequency of homozygous SNPs in the candidate chromosomes from each bulk. In the three early-heading bulks, the frequency of the eh1 alleles on chromosomes 2A and 5B ranged from 0.59 to 0.76. Similarly, the LX987 allele in the late-heading bulks showed a relatively higher frequency than the eh1 allele on 2A and 5B, while both the eh1 and LX987 alleles had comparable frequencies on chromosomes 2B and 3B in all bulks (Fig. 1b). These results suggest that chromosomes 2A and 5B are more likely to be associated with heading date variations in the RILs. To further explore the genomic regions that are potentially associated with HD, we scanned the SNPs within 1 Mb windows on the candidate chromosomes. The SNP distributions showed a high density on the short arm of 2A, corresponding to the physical position of 50–150 Mb (Fig. 1c), while the SNP density peaks were on the long arm of 5B (Fig. 1d). In addition, we noted that more SNPs were enriched in the terminal region of 5B compared with the candidate region in 2A, suggesting a strong association with heading date on chromosome 5B. This is in agreement with our previous study that the vernalization gene VRN-B1 is a major HD regulator in the RILs (Li et al. 2020).

Bulked segregant analysis (BSA) of heading date in RILs. a Distribution of homozygous SNPs from three DNA bulk pairs with contrasting HD. Histograms with different colors represent the number of homozygous SNPs identified from different mixed bulks, and the line chart indicates the number of SNPs identified from LX987 and eh1. b Comparison of allele frequencies in each of the early- and late-heading bulks. Green and blue histograms show the LX987 and eh1 alleles, respectively. The dotted red line indicates the allele frequency at 50%. c, d Distributions of SNP densities on chromosomes 2A and 5B. The x-axis represents physical positions on the chromosomes and y-axis shows the number of SNPs. Red dots refer to the numbers of SNPs in a 1 Mb interval. The triangle indicates the physical position of the VRN-B1 locus (Color figure online)
Fine mapping of the HD gene on chromosome 2A
To confirm the potential heading date QTL on chromosome 2A, we developed 22 KASP markers based on SNPs between the two parental lines that were detected by BSR-Seq or in the Wheat660K SNP array. A linkage map was constructed by genotyping the RIL population with these markers, which spanned a genetic distance of 217.4 cM (82.35–760.67 Mb on IWGSC RefSeq v 1.0) with an average density of 9.8 cM per locus. Combining the HD data collected from three consecutive years with the linkage map, we delimited a stable QTL for HD on the short arm of chromosome 2A to a 6.31 Mb interval (135.19–141.50 Mb) that was flanked by KASP markers flk1 and flk3, which we refer to as Qhd.2AS (Fig. 2a). The LOD score of Qhd.2AS ranged from 3.4 to 4.2 and explained 6.8–9.2% of the phenotypic variance over the three years. These results suggested that Qhd.2AS is a minor HD QTL as compared to VRV-B1 that explained a higher proportion of phenotypic variance in the RIL population (Li et al. 2020). To precisely map Qhd.2AS, we selected six RILs that were heterozygous for this locus but homozygous for the recessive Vrn-B1v allele, and used 2112 progeny plants for gene mapping. In this segregating population, the plants differed in HD by 10 days (Fig. S2a), and Qhd.2AS was associated with the variation in HD by statistical analysis of the mean values of HD in homozygous and heterozygous plants (p < 0.01, Fig. S2b). The degree of dominance was calculated to be 0.19, indicating that Qhd.2AS has a minor dominant effect. No recombinant events were identified by screening this population with the flanking markers flk1 and flk3. Furthermore, we generated a large segregating population consisting of 4894 individuals derived from the lines that were heterozygous for Qhd.2AS. Ultimately, Qhd.2AS was mapped to a 0.41 cM interval flanked by marker loci flk2 and flk3 (Fig. 2b). We further developed three KASP markers in the candidate region and identified five key recombination events (R#1–R#5). The recombinant plants were self-pollinated and the homozygous progeny plants were used for phenotype investigation. The homozygous recombinant lines R#3 and R#5 headed significantly earlier than that of lines R#1, R#2, and R#4 (p < 0.05, Fig. 2c). Combining this data with the genotypes in these recombinant lines we were able to delimit Qhd.2AS to a physical interval of 1.7 Mb flanked by marker loci fm71 and fm76. This chromosomal region includes 16 high-confidence genes according to the Chinese Spring reference genome v1.1 annotation (Fig. 2d).

Validation and fine mapping of Qhd.2AS. a Genetic linkage map and QTL analysis of chromosome 2A. The linkage map was constructed using genotypic data from 400 RILs genotyped with 22 KASP markers. HD data was collected in three consecutive years. The LOD threshold value was set to 2.5. The phenotypic variation in HD explained by Qhd.2AS ranged from 6.8% to 9.2%. b A refined map of the Qhd.2AS locus with four additional molecular markers loci. The map was constructed by genotyping 4,894 plants that were segregating for Qhd.2AS. c Fine mapping of Qhd.2AS with five key recombinant events. The HDs of homozygous progeny from the recombinant plants were investigated in the 2020–2021 growing season. n, the number of plants used for phenotypic evaluation. Different letters indicate a significant difference at p < 0.05. d Schematic diagram of the 16 high-confidence candidate genes in the 1.70 Mb Qhd.2AS candidate region (138.87–140.57 Mb; IWGSC RefSeq v1.1) (Color figure online)
Identification of the Qhd.2AS candidate gene by DNA sequencing and gene expression analysis
To identify the causal gene for Qhd.2AS, we analyzed the sequence variations and expression patterns of the genes in the mapped region between the two parental lines. For sequence analysis, we investigated the whole genome re-sequencing and RNA-seq data from eh1 and LX987, and found that three candidate genes (TraesCS2A01G180300, TraesCS2A01G180400, and TraesCS2A01G181600) contained 5, 2, and 1 missense mutations, and additional three genes (TraesCS2A01G180200, TraesCS2A01G180500, and TraesCS2A01G181400) showed 2, 2, and 1 Indels in the intron region, respectively. Notably, we identified two SNPs and one Indel located in the region upstream of TraesCS2A02G181200 (Table 1), which is predicted to be close to a conserved cis-regulatory element CAAT-box (Table S1), suggesting that this locus might be functionally important for regulating the expression of downstream genes. Furthermore, we investigated the expression levels of all candidate genes in young spikes of eh1 and LX987 plants from our previous RNA-seq data (Li et al. 2020). Among the 16 candidate genes, only TraesCS2A02G181200 expression was detected in both samples and was up-regulated in eh1 (Table 2). Transcription of eight genes were not detected in either eh1 or LX987 plants, and three genes were found to be expressed at low levels in eh1 but not in LX987 (Table 2). We also analyzed the expression levels of these 16 genes in published RNA-Seq database that included 318 leaf and 100 spike samples (Borrill et al. 2016), and found that 8 of these 16 genes have undetectable transcript levels in leaves or spikes (Fig. 3a). These genes are less likely to affect HDs since they are not expressed in the leaves and spikes. We further confirmed the up-regulation of TraesCS2A02G181200 expression in eh1 by real-time quantitative PCR (Fig. 3b), and the SNPs and Indel that differed between the two parental lines by Sanger sequencing (Fig. 3c). Taken together, the gene expression and sequence variation analysis indicated that TraesCS2A02G181200 is more likely to be the causal gene for Qhd.2AS; however, we cannot rule out TraesCS2A02G180200 and TraesCS2A02G180500 as candidate genes due to the intronic variations and the fact that both are expressed in LX987 and eh1 plants.

Verification of sequence variations and expression analysis of candidate genes. a Spike and leaf expression patterns for the 16 candidate genes identified in the 1.70 Mb Qhd.2AS candidate gene region on chromosome 2AS. The expression data includes 100 spike and 318 leaf samples in ExpVIP (http://www.wheat-expression.com/). Darker blue indicates a relatively high level of expression. b Expression analysis of the candidate gene TraesCS2A02G181200 in leaves and spikes as determined by qRT-PCR. The flag leaves and young spikes were sampled at the booting stage from LX987 and eh1 in the field. The expression of TraesCS2A02G181200 in each parental line was determined with three biological replicates from different individual plants, and each biological replicate was assayed with three technical replicates. **p < 0.01. c Verification of two SNPs and one Indel in the promoter region of TraesCS2A02G181200 by Sanger sequencing. The primers used for PCR amplification are given in Table S6. Numbers indicate the physical positions of the three variable sites in IWGSC RefSeq v1.1. Red dotted rectangles represent the positions of the variable sites in the promoter sequence of TraesCS2A02G181200 (Color figure online)
Validation of the candidate gene in the TILLING mutants
We previously generated an in-house mutant library in the background of the winter cultivar ‘Jing411.’ A comparison of the promoter sequences between ‘Jing411’ and the parental lines showed that the Jing411’ contained the same allele as LX987 in TraesCS2A02G180200 (Table S2). Screening the mutant library identified five mutants with frameshift or stop-gain mutations in the three candidate genes (Table S3). Functional prediction suggested that all these mutations had highly disruptive effects on protein function (Table S3). Phenotypic analysis suggested that two of the mutants, E0313 and E0984, both in TraesCS2A02G180200, and one mutant, E0415, in TraesCS2A02G180500 had similar HDs to ‘Jing411.’ In contrast, the HDs of the two mutants in TraesCS2A02G181200, E0039 and E0683, were two and four days later than ‘Jing411,’ respectively (Table S3, Fig. 4a). This suggested that the functional mutations in TraesCS2A02G181200 caused delayed HDs compared to the wild-type ‘Jing411.’ We further confirmed the stop-gain mutation in E0039 and the frameshift mutation in E0683 by Sanger sequencing. The premature termination mutation in E0039 and the frameshift mutation in E0683 truncated two and one of the predicted C2H2-type zinc finger motifs at the C terminus, respectively (Fig. 4b). Taken together, the sequence variations present in the regulatory region, the upregulated expression in eh1 plants, and the delayed HD in the two TILLING mutants together provide evidence that the C2H2-type zinc finger protein gene TraesCS2A02G181200 is the most likely candidate to be the Qhd.2AS causal gene.

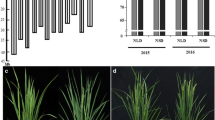
Functional validation of TraesCS2A02G181200 in the TILLING mutants. a Phenotypes of the two mutant lines (E0039 and E0683) grown in the field. Photos were taken on the same day that the wild-type ‘Jing411’ plants had finished heading. b Schematic diagram of gene structure and validation of gene mutations in the TILLING mutants. Green rectangles in the schematic diagram represent the four conserved C2H2-type zinc finger motifs in the protein encoded by TraesCS2A02G181200. Black triangles indicate the positions of the mutations. The stop-gain mutation in E0039 and the frameshift mutation in E0683 were validated by Sanger sequencing. Mutation sites are included in the red dotted rectangles (Color figure online)
Haplotype analysis
To analyze the natural variation present in the causal gene, we identified 15 SNPs in the promoter and gene sequence of TraesCS2A02G181200 in 677 wheat accessions from the Wheat Union Database (Wang et al. 2020). Ten of the 15 SNPs showed linkage disequilibrium (LD) in a 4-kb block (Fig. 5a). We identified four haplotypes (Hap-A, Hap-B, Hap-C, and Hap-D) in this block with a frequency of 0.48, 0.35, 0.14, and 0.03, respectively (Fig. 5b). Notably, two tightly linked SNPs (SNP-140361773 and SNP-140361804) in the LD block were also identified between the parental lines LX987 and eh1 (Fig. 5b, Table 2). We then characterized the geographical distribution of the representative SNP-140361773. The results showed that the G allele from eh1 was predominantly distributed around the world except for part of the wheat production region in Asia (Fig. 5c). Similarly, an investigation of the tightly linked SNP-140361804, which was also identified between the two parental lines, showed that it exhibited a comparable geographical distribution with the SNP-140361773 (Fig. S3a). Additionally, the A allele of SNP-140361773 and the C allele of SNP-140361804 were mainly identified in modern cultivars but was present at a low frequency in the landraces (Fig. S3b). This indicates that the LX987 allele in TraesCS2A02G181200 gene was positively selected in wheat breeding. Additionally, a comparison of the genotypes at the two SNPs and one Indel variation sites identified the LX987 allele as the reference haplotype (Table S2). Moreover, we compared the promoter sequences of TraesCS2A02G181200 with its homeologues in the B and D sub-genome, and the relatives in diploid and tetraploid wheat (Fig. S4). Interestingly, all investigated sequences displayed the eh1 genotypes at SNP-140361773 and Indel-140361786 loci; while only the wild Emmer and Durum wheat had the same allele as LX987 in the SNP-140361804 (Table S2). These results suggested that the eh1 haplotype was the ancestral haplotype.

Haplotype analysis of the candidate gene TraesCS2A02G181200. a Linkage disequilibrium (LD) block analysis of the 15 SNPs identified in the 3 kb promoter region and gene sequences from 677 wheat accessions. Numbers in red squares represent the r2 value of two selected SNPs. b Four haplotypes identified in the LD block based on 10 out of the 15 SNPs. Numbers on the top and right indicate the physical position of the SNPs in IWGSC RefSeq v1.1 and the haplotype frequency, respectively. c Geographical distribution of the representative SNP-140361773. The larger pie chart represents more wheat accessions. The proportions of the G (eh1) and A (LX987) alleles are indicated by different colors (Color figure online)
To explore the genetic diversity of Qhd.2AS, we also used four closely linked KASP markers around this locus to genotype 262 accessions in the Chinese wheat mini-core collection (MCC) (Wang et al. 2012), which includes diverse landraces and elite modern cultivars with different growth habits (Fig. S5a). Notably, these KASP markers exhibited high genotyping efficiency (80.4–97.7%) in the MCC panel, which indicates that the obtained genotypes are reliable and sufficient to perform haplotype analysis (Table S4). The MCC panel accessions were classified into four haplotypes (Hap-1, Hap-2, Hap-3, and Hap-4) based on these diagnostic markers. The frequencies of the individual haplotypes varied considerably, from 2.6 to 57.3% (Table S4), suggesting that Qhd.2AS had experienced some selective pressure in the MCC panel. We characterized the frequency distributions of these haplotypes in accession with different growth habits. For Hap-1, Hap-2, and Hap-3, we observed decreasing frequency distributions in winter, weak-winter, and spring wheat accessions, while the frequency of Hap-4 increased in spring wheat compared to the winter accessions (Fig. S5b). Additionally, we noted that, despite the comparable frequency of Hap-1 in the landraces and modern cultivars, Hap-2 and Hap-3 showed higher frequencies in the modern cultivars compared to the landraces, while Hap-4 exhibited the highest frequency in the landraces (Fig. S5c). These results suggest that Qhd.2AS has undergone divergent selection in wheat with different growth habits and during the process of selection in modern breeding programs.
Epistatic analysis of Qhd.2AS and VRN-B1
To assess the epistatic effect of VRN-B1 on Qhd.2AS, we compared the HDs of homozygous Qhd.2AS RILs in the dominant or recessive VRN-B1 background. Intriguingly, the HDs of homozygous Qhd.2ASeh1 RILs showed 2–3 days earlier than homozygous Qhd.2ASLX987 lines under both the dominant Vrn-B1a and recessive Vrn-B1v genotypes (p < 0.05, Fig. S6). Moreover, multivariate analysis of variance using VRN-B1, Qhd.2AS, and Years as factors revealed that all three factors had significant effects on HD, and there were significant interactions between VRN-B1 and Years (p < 0.001). However, no significant interactions were observed between Qhd.2AS and VRN-B1 (p = 0.300), or between Qhd.2AS and Years (p = 0.461, Table S5). These results suggested that the genetic regulation of Qhd.2AS on HD is independent of VRN-B1 and environmental conditions.
Evaluation of Qhd.2AS on important agronomic traits
To investigate the effect of Qhd.2AS on important agronomic traits, we compared PH, TGW, and five kernel-dimension-related traits (kernel length, width, area, perimeter, and diameter) between RILs homozygous for the alleles Qhd.2ASLX987 and Qhd.2ASeh1. For PH and TGW, we observed no significant differences between the Qhd.2ASLX987 and Qhd.2ASeh1 RILs in 2016 and 2017, but slightly higher values in the Qhd.2ASLX987 lines compared to the Qhd.2ASeh1 lines in 2018 (p < 0.05, Fig. 6a, b). For the five kernel-dimension-related traits measured in 2017 and 2018, the mean values in the two types of RILs showed no significant differences (Fig. 6c–g). In addition, we further evaluated these agronomic traits in a secondary population segregating for the Qhd.2AS locus. Despite the slightly higher mean values of PH and kernel area, and a relatively lower mean value of kernel perimeter in the Qhd.2ASeh1 lines, the other four agronomic traits showed comparable mean values and all seven traits displayed no significant differences between the two types of homozygous plants (Fig. S7). These results indicate that Qhd.2AS is not significantly associated with important agronomic traits.

Evaluation of the effects of Qhd.2AS on agronomic traits in the RILs. Each trait was investigated using at least five randomly selected RILs. For each RIL, three representative plants were used. Error bars represent the standard errors of the means. ns, not significant. *p < 0.05
Discussion
Heading date (HD) in wheat is a key factor that contributes to its global environmental adaptability and yield stability under diverse climatic conditions (Kamran et al. 2014). Dissecting the genetic control of HD is necessary to understand this wide adaptability and stable yield of wheat grown in distinct environments. In the past several decades, studies have identified a number of genes that show high impacts on HD and growth habit in wheat (Distelfeld et al. 2009a; Shi et al. 2019). However, these genes are often unsuitable for deployment in wheat breeding programs due to their dramatic effects on HD and the trade-off effect with yield-related traits; thus, genes with minor effects on HD are more desirable targets for fine-tuning the improvement of crop varieties (Cai et al. 2021; Wu et al. 2018). Using a BSR-Seq approach, we previously identified allelic variations in the vernalization gene VRN-B1 that were mainly responsible for HD variations in the RIL population (Li et al. 2020). However, a continuous distribution in HD was still observed in the RILs carrying homozygous VRN-B1 genotypes. By combining BSA and QTL analysis, we identified an environmentally stable locus, Qhd.2AS, on the short arm of chromosome 2A. The LOD scores and explained phenotypic variance in the RILs indicated that Qhd.2AS is a minor-effect HD gene compared to VRN-B1 (Li et al. 2020). Minor HD genes are useful for adjusting regional and seasonal adaptation in crop varieties; however, they have been poorly characterized using traditional mapping strategies due to their small effects which are often unstable and difficult to score across environments (Chen et al. 2014; Wu et al. 2013). An investigation of the DTH2 gene highlighted the significance of minor HD genes in the historical expansion of cultivated rice to long-day growth conditions (Wu et al. 2013). In our study, the homozygous Qhd.2ASeh1 plants headed approximately 3 days earlier than that of plants carrying the Qhd.2ASLX987 allele, suggesting that the Qhd.2AS locus can be useful for minor adjustment of HD in wheat breeding.
The photoperiod gene Ppd-A1 on chromosome 2A is a key regulator of HD in wheat (Wilhelm et al. 2009). We previously identified a 131-bp deletion at the 3’ end of Ppd-A1 in eh1 but not in LX987; however, this deletion was not associated with HD variation in the RIL population (Li et al. 2020). Additionally, the physical position of Ppd-A1 is located outside of the candidate region for Qhd.2AS, suggesting that Qhd.2AS and Ppd-A1 are not allelic. Recently, several studies have identified numerous QTLs and/or marker-trait associations for HD on wheat chromosomes 1A, 2A, 2B, 3A, 4B, 5B, 7A, 7B, and 7D; many of these QTLs are co-located with known flowering genes such as Ppd-1, Vrn-A1, Vrn-B1, and Vrn-B3 (Avila et al. 2021; Bogard et al. 2020; Chen et al. 2020; Gupta et al. 2020; Liu et al. 2017; Sesiz and Özkan 2021). Chen et al. (2020) reported a QTL that controls HD located close to the centromere region on chromosome 2A, and Bogard et al. (2020) identified a HD QTL on chromosome 2A from Australian wheat cultivars and breeding lines. More recently, a minor HD QTL on chromosome 2AS was identified around the marker locus AX111681607 (106.77 Mb position in IWGSC RefSeq v1.0) (Wu et al. 2022). The physical positions of these loci differed from that of Qhd.2AS identified in this study. Therefore, Qhd.2AS could be a novel HD gene.
We identified a gene that encodes a C2H2-type zinc finger protein that could be the causal gene for Qhd.2AS. Using a large segregating population excluding the VRN-B1 locus, we eventually delimited Qhd.2AS to a genomic interval of 1.70-Mb (138.87–140.57 Mb, IWGSC RefSeq v1.0) that contained 16 annotated high-confidence genes. Our results provide strong evidence to support the notion that the TraesCS2A02G181200 gene, which encodes a C2H2-type zinc finger protein, is the causal gene of Qhd.2AS. First, we analyzed whole genome re-sequencing and RNA-seq data from the two parental lines and identified seven genes that showed sequence variations in the mapped region including eight missense SNPs in the coding sequence of three genes, five Indels in the intronic regions of three genes, and two SNPs and one Indel in the putative regulatory site of TraesCS2A02G181200 (Table 1). Many studies have demonstrated that mutations in the coding sequence, intronic regions, and regulatory sites of flowering genes are important to their function because they affect protein activity or gene expression. For example, a single nucleotide polymorphism changed Ala180 in the dominant Vrn-A1a allele to Vall80, and the mutated protein had a decreased ability to bind with TaHOX1 (Li et al. 2013). Additionally, single nucleotide polymorphisms in the first intron of Vrn-A1 and in the promoter region of Vrn-D1 are associated with different vernalization responses, possibly by mediating the binding activity of other transcription factors (Kippes et al. 2018; Zhang et al. 2012). Therefore, the seven genes that show sequence variations between the two parental lines are potential candidate genes that affect heading time. Furthermore, from our RNA-seq data and an analysis of large-scale leaf or spike samples from the public RNA-seq database, we found that three of the seven candidate genes could be detected in leaves or spikes of wheat, and only the TraesCS2A02G181200 gene was up-regulated in eh1 (Table 2, Fig. 3a, b). The effect of gene expression changes on flowering time mainly depends on their functions in the regulation pathways. For example, the down-regulation of flowering promoter genes such as VRN1 and VRN3 could delay flowering time, while down-regulation of the repressor gene VRN2 accelerates flowering in winter wheat (Yan et al. 2003, 2004b, 2006). We inferred that the three candidate genes could be involved in the regulation of HD since the leaves and spikes are the main tissues where many flowering genes are often expressed (Yan et al. 2003, 2004b, 2006). Finally, we identified five functional TILLING mutations for the three candidate genes (Table S3). Plants carrying the mutations in TraesCS2A02G180200 and TraesCS2A02G180500 exhibited similar HDs to the wild-type ‘Jing411,’ while plants carrying the two mutations in TraesCS2A02G181200 headed 2–4 days later than ‘Jing411’ under field conditions (Fig. 4b, Table S3). Collectively, the sequence variations present in putative regulatory sites, the up-regulated gene expression in eh1, and mutant analysis suggest that TraesCS2A02G181200 is the most likely causal gene for Qhd.2AS. The TraesCS2A02G181200 gene encodes a putative transcription factor with four C2H2-type zinc finger motifs (Fig. 4b). Proteins with zinc finger domains have been widely characterized in plants and are known to participate in multiple biological processes including plant growth, development, and abiotic stress tolerance (Xie et al. 2019; Yan et al. 2014). In Arabidopsis, the nuclear-localized C2H2-type zinc finger protein SUF4 represses flowering by activating the expression of FLC, a well-characterized flowering repressor in the vernalization pathway (Kim et al. 2006). Additionally, the C2H2-type zinc finger protein LATE inhibits flowering in the photoperiod response by down-regulating flowering promoter genes such as CO and FT (Weingartner et al. 2011). These results suggest a potential role for TraesCS2A02G181200 in the regulation of HD in wheat.
HD is sensitive to environmental factors such as temperature and day length. Genes involved in the vernalization pathway and photoperiod response are often integrated with environmental stimuli to determine wheat growth and development (Distelfeld et al. 2009a; Shi et al. 2019). In some winter wheat varieties, a period of short-day treatment can replace the vernalization requirement; this process was found to be associated with down-regulated expression of the flowering repressor VRN2 (Dubcovsky et al. 2006). The vernalization gene VRN1 and photoperiod gene Ppd1 were shown to have epistatic interactions and impacts on the development of winter wheat through a temperature-dependent manner (Dixon et al. 2019; Eagles et al. 2010). The Earliness per se locus also exhibited an additive interaction effect with the vernalization gene Vrn-B1 on HD (Chen et al. 2015). In agreement with previous studies, our multivariate analysis of variance revealed a significant interaction between VRN-B1 and the environment on HD. However, the Qhd.2AS-mediated HD variation in the RILs was independent of VRN-B1 and environmental factors (Table S5), which indicates a distinctive role for Qhd.2AS in the regulation of HD. Growth habit in wheat is mainly determined by major allelic variations in the VRN1 gene (Yan et al. 2003, 2004a). Studies have shown that different haplotype combinations of exonic variations in VRN-A1 are associated with spring or winter growth habits in domesticated wheat (Muterko and Salina 2018). Results of our haplotype analysis suggested that the predominant Hap-4 haplotype of Qhd.2AS has been positively selected for in spring wheat accessions (Fig. S5b). Additionally, the G allele (eh1 genotype) of SNP-140361773 in TraesCS2A02G181200 is widely distributed in wheat cultivars grown around the world (Fig. 5c), suggesting a potential value of this gene in enabling wheat to adapt to different growth conditions. In addition to their effects on HD, many flowering-related genes are often associated with other agronomically important traits in wheat. Ppd-1 has an inhibitory effect on paired spikelet formation by regulating the expression of FT1 (Boden et al. 2015). Knock-out mutants of FT-D1 had increases in spikelet number, plant height, and spike length (Chen et al. 2022). Similarly, deletions in the FT-B1 gene not only delayed flowering time, but also increased the spikelet number and tiller number in wheat (Dixon et al. 2018; Finnegan et al. 2018). We investigated the effects of Qhd.2AS on major yield-related components including plant height, 1000-grain weight, and five kernel-dimension-related traits. Notably, we did not observe significant associations between Qhd.2AS and these agronomic traits, suggesting that the Qhd.2AS is a suitable potential target to balance the trade-off effect between HD and yield components in wheat.
Although the early-heading phenotype in eh1 plants resulted from a gamma-ray induced mutation, we inferred that the Qhd.2AS-mediated variation in HD was not associated with the induced mutation in eh1. Firstly, the HD of eh1 plants was more than 10 days earlier than the HD in wild-type ZY9, while Qhd.2AS only explained a small proportion of the phenotypic variance in the RIL population. Moreover, there is a significant difference in HD between LX987 and ZY9 (Li et al. 2020), suggesting that variations in the genetic background might be associated with HD in RILs. Additionally, we identified 16 SNPs and 6 Indels from the genome re-sequencing data in the candidate region for Qhd.2AS between ZY9 and eh1, and found most variations were located in the intergenic regions and none of them were annotated in the exonic or intronic sequences (Table S7). Lastly, sequence variations in the putative regulatory sites of TraesCS2A02G181200 were widely present in natural wheat accessions (Fig. 5). It is a common phenomenon that the same mutant allele leads to distinct levels of phenotypic variation in different genetic backgrounds. In rice, two cultivars carrying the same deficient mutant allele PDIL1-1 exhibited significant differences in flour characteristics, plant height, and grain yield, suggesting that certain genetic backgrounds can counteract the phenotypic changes that result from gene mutations (Hori et al. 2022). Similarly, the early heading effect caused by the induced mutation in eh1 could be neutralized by the genetic background of LX987; it is therefore difficult to pinpoint the mutated locus that affects HD. Further studies are needed to explore the effects of the induced mutation in a homogeneous genetic background.
Conclusion
We identified an environmentally stable quantitative trait locus named Qhd.2AS that conferred minor effects on HD in a wheat RIL population. Qhd.2AS was validated in a segregating population and the physical location was delimited to a genomic interval of 1.70 Mb. This region contained 16 high-confidence genes based on IWGSC RefSeq v1.1. Phenotypic investigations showed that Qhd.2AS has no negative effects on yield-related traits. Combining sequence variation analysis, gene expression, and phenotypic validation in mutants with truncated proteins, we concluded that the TraesCS2A02G181200 locus that encodes a C2H2-type zinc finger protein is the best candidate to be the causal gene for Qhd.2AS. These results will facilitate a better understanding of the genetic regulation of HD and provide a novel target gene for marker-assisted selection in wheat breeding.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Andres F, Coupland G (2012) The genetic basis of flowering responses to seasonal cues. Nat Rev Genet 13:627–639
Appendino ML, Slafer GA (2003) Earliness per se and its dependence upon temperature in diploid wheat lines differing in the major gene Eps-Am 1 alleles. J Agric Sci 141:149–154
Avila CM, Requena-Ramirez MD, Rodriguez-Suarez C, Flores F, Sillero JC, Atienza SG (2021) Genome-wide association analysis for stem cross section properties, height and heading date in a collection of Spanish durum wheat landraces. Plants 10:1123
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263–265
Beales J, Turner A, GriYths S, Snape JW, Laurie DA (2007) A Pseudo-Response Regulator is misexpressed in the photoperiod insensitive Ppd-D1a mutant of wheat (Triticum aestivum L.). Theor Appl Genet 115:721–733
Bentley AR, Horsnell R, Werner CP, Turner AS, Rose GA, Bedard C, Howell P, Wilhelm EP, Mackay IJ, Howells RM, Greenland A, Laurie DA, Gosman N (2013) Short, natural, and extended photoperiod response in BC2F4 lines of bread wheat with different photoperiod-1 (Ppd-1) alleles. J Exp Bot 64:1783–1793
Boden SA, Cavanagh C, Cullis BR, Ramm K, Greenwood J, Jean Finnegan E, Trevaskis B, Swain SM (2015) Ppd-1 is a key regulator of inflorescence architecture and paired spikelet development in wheat. Nat Plants 1:1–6
Bogard M, Biddulph B, Zheng B, Hayden M, Kuchel H, Mullan D, Allard V, Gouis JL, Chapman SC (2020) Linking genetic maps and simulation to optimize breeding for wheat flowering time in current and future climates. Crop Sci 60:678–699
Borrill P, Ramirez-Gonzalez R, Uauy C (2016) expVIP: a customizable RNA-seq data analysis and visualization platform. Plant Physiol 170:2172–2186
Bullrich L, Appendino L, Tranquilli G, Lewis S, Dubcovsky J (2002) Mapping of a thermo-sensitive earliness per se gene on Triticum monococcum chromosome 1Am. Theor Appl Genet 105:585–593
Cai M, Zhu S, Wu M, Zheng X, Wang J, Zhou L, Zheng T, Cui S, Zhou S, Li C, Zhang H, Chai J, Zhang X, Jin X, Cheng Z, Zhang X, Lei C, Ren Y, Lin Q, Guo X, Zhao L, Wang J, Zhao Z, Jiang L, Wang H, Wan J (2021) DHD4, a CONSTANS-like family transcription factor, delays heading date by affecting the formation of the FAC complex in rice. Mol Plant 14:330–343
Chen A, Dubcovsky J (2012) Wheat TILLING mutants show that the vernalization gene VRN1 down-regulates the flowering repressor VRN2 in leaves but is not essential for flowering. PLoS Genet 8:e1003134
Chen JY, Guo L, Ma H, Chen YY, Zhang HW, Ying JZ, Zhuang JY (2014) Fine mapping of qHd1, a minor heading date QTL with pleiotropism for yield traits in rice (Oryza sativa L.). Theor Appl Genet 127:2515–2524
Chen H, Iqbal M, Perez-Lara E, Yang R-C, Pozniak C, Spaner D (2015) Earliness per se quantitative trait loci and their interaction with Vrn-B1 locus in a spring wheat population. Mol Breed 35:182
Chen Z, Cheng X, Chai L, Wang Z, Du D, Wang Z, Bian R, Zhao A, Xin M, Guo W, Hu Z, Peng H, Yao Y, Sun Q, Ni Z (2020) Pleiotropic QTL influencing spikelet number and heading date in common wheat (Triticum aestivum L.). Theor Appl Genet 133:1825–1838
Chen Z, Ke W, He F, Chai L, Cheng X, Xu H, Wang X, Du D, Zhao Y, Chen X, Xing J, Xin M, Guo W, Hu Z, Su Z, Liu J, Peng H, Yao Y, Sun Q, Ni Z (2022) A single nucleotide deletion in the third exon of FT-D1 increases the spikelet number and delays heading date in wheat (Triticum aestivum L.). Plant Biotechnol J 20:920
Chouard P (1960) Vernalization and its relations to dormancy. Annu Rev Plant Physiol 11:191–238
Diaz A, Zikhali M, Turner AS, Isaac P, Laurie DA (2012) Copy number variation affecting the Photoperiod-B1 and Vernalization-A1 genes is associated with altered flowering time in wheat (Triticum aestivum). PLoS ONE 7:e33234
Distelfeld A, Li C, Dubcovsky J (2009a) Regulation of flowering in temperate cereals. Curr Opin Plant Biol 12:178–184
Distelfeld A, Tranquilli G, Li C, Yan L, Dubcovsky J (2009b) Genetic and molecular characterization of the VRN2 loci in tetraploid wheat. Plant Physiol 149:245–257
Dixon LE, Farre A, Finnegan EJ, Orford S, Griffiths S, Boden SA (2018) Developmental responses of bread wheat to changes in ambient temperature following deletion of a locus that includes FLOWERING LOCUS T1. Plant Cell Environ 41:1715–1725
Dixon LE, Karsai I, Kiss T, Adamski NM, Liu Z, Ding Y, Allard V, Boden SA, Griffiths S (2019) VERNALIZATION1 controls developmental responses of winter wheat under high ambient temperatures. Development. https://doi.org/10.1242/dev.172684
Dubcovsky J, Dvorak J (2007) Genome plasticity a key factor in the success of polyploid wheat under domestication. Science 316:1862–1865
Dubcovsky J, Loukoianov A, Fu D, Valarik M, Sanchez A, Yan L (2006) Effect of photoperiod on the regulation of wheat vernalization genes VRN1 and VRN2. Plant Mol Biol 60:469–480
Eagles H, Cane K, Kuchel H, Hollamby G, Vallance N, Eastwood R, Gororo N, Martin P (2010) Photoperiod and vernalization gene effects in southern Australian wheat. Crop Pasture Sci 61:721–730
Finnegan EJ, Ford B, Wallace X, Pettolino F, Griffin PT, Schmitz RJ, Zhang P, Barrero JM, Hayden MJ, Boden SA, Cavanagh CA, Swain SM, Trevaskis B (2018) Zebularine treatment is associated with deletion of FT-B1 leading to an increase in spikelet number in bread wheat. Plant Cell Environ 41:1346–1360
Fu D, Szucs P, Yan L, Helguera M, Skinner JS, von Zitzewitz J, Hayes PM, Dubcovsky J (2005) Large deletions within the first intron in VRN-1 are associated with spring growth habit in barley and wheat. Mol Genet Genomics 273:54–65
Giovannoni JJ, Wing RA, Ganal MW, Tanksley SD (1991) Isolation of molecular markers from specific chromosomal intervals using DNA pools from existing mapping populations. Nucleic Acids Res 19:6553–6568
Guillemaut P, Mardchal-Drouard L (1992) Isolation of plant DNA: a fast, inexpensive, and reliable method. Plant Mol Biol Rep 10:60–65
Gupta P, Kabbaj H, El Hassouni K, Maccaferri M, Sanchez-Garcia M, Tuberosa R, Bassi FM (2020) Genomic regions associated with the control of flowering time in durum wheat. Plants 9:1628
Hori K, Okunishi T, Nakamura K, Iijima K, Hagimoto M, Hayakawa K, Shu K, Ikka T, Yamashita H, Yamasaki M, Takeuchi Y, Koyama S, Tsujii Y, Kayano T, Ishii T, Kumamaru T, Kawagoe Y, Yamamoto T (2022) Genetic background negates improvements in rice flour characteristics and food processing properties caused by a mutant allele of the PDIL1-1 seed storage protein gene. Rice 15:13
Jung C, Muller AE (2009) Flowering time control and applications in plant breeding. Trends Plant Sci 14:563–573
Kamran A, Iqbal M, Spaner D (2014) Flowering time in wheat (Triticum aestivum L.): a key factor for global adaptability. Euphytica 197:1–26
Kane N, Agharbaoui Z, Diallo A, Adam H, Tominaga Y, Ouellet F, Sarhan F (2007) TaVRT2 represses transcription of the wheat vernalization gene TaVRN1. Plant J 51:670–680
Kim S, Choi K, Park C, Hwang HJ, Lee I (2006) SUPPRESSOR OF FRIGIDA4, encoding a C2H2-type zinc finger protein, represses flowering by transcriptional activation of Arabidopsis FLOWERING LOCUS C. Plant Cell 18:2985–2998
Kippes N, Zhu J, Chen A, Vanzetti L, Lukaszewski A, Nishida H, Kato K, Dvorak J, Dubcovsky J (2014) Fine mapping and epistatic interactions of the vernalization gene VRN-D4 in hexaploid wheat. Mol Genet Genomics 289:47–62
Kippes N, Debernardi JM, Vasquez-Gross HA, Akpinar BA, Budak H, Kato K, Chao S, Akhunov E, Dubcovsky J (2015) Identification of the VERNALIZATION 4 gene reveals the origin of spring growth habit in ancient wheats from South Asia. Proc Natl Acad Sci USA 112:E5401-5410
Kippes N, Guedira M, Lin L, Alvarez MA, Brown-Guedira GL, Dubcovsky J (2018) Single nucleotide polymorphisms in a regulatory site of VRN-A1 first intron are associated with differences in vernalization requirement in winter wheat. Mol Genet Genomics 293:1231–1243
Kosambi D (1943) The estimation of map distances from recombination values. Ann Eugenics 12:172–175
Li C, Dubcovsky J (2008) Wheat FT protein regulates VRN1 transcription through interactions with FDL2. Plant J 55:543–554
Li C, Distelfeld A, Comis A, Dubcovsky J (2011) Wheat flowering repressor VRN2 and promoter CO2 compete for interactions with NUCLEAR FACTOR-Y complexes. Plant J 67:763–773
Li G, Yu M, Fang T, Cao S, Carver BF, Yan L (2013) Vernalization requirement duration in winter wheat is controlled by TaVRN-A1 at the protein level. Plant J 76:742–753
Li C, Lin H, Dubcovsky J (2015) Factorial combinations of protein interactions generate a multiplicity of florigen activation complexes in wheat and barley. Plant J 84:70–82
Li Y, Xiong H, Guo H, Zhou C, Xie Y, Zhao L, Gu J, Zhao S, Ding Y, Liu L (2020) Identification of the vernalization gene VRN-B1 responsible for heading date variation by QTL mapping using a RIL population in wheat. BMC Plant Biol 20:331
Li Y, Xiong H, Zhang J, Guo H, Zhou C, Xie Y, Zhao L, Gu J, Zhao S, Ding Y, Fang Z, Liu L (2021) Genome-wide and exome-capturing sequencing of a gamma-ray-induced mutant reveals biased variations in common wheat. Front Plant Sci 12:793496
Liu Y, Lin Y, Gao S, Li Z, Ma J, Deng M, Chen G, Wei Y, Zheng Y (2017) A genome-wide association study of 23 agronomic traits in Chinese wheat landraces. Plant J 91:861–873
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-△△CT Method. Methods 25:402–408
Loukoianov A, Yan L, Blechl A, Sanchez A, Dubcovsky J (2005) Regulation of VRN-1 vernalization genes in normal and transgenic polyploid wheat. Plant Physiol 138:2364–2373
Meng L, Li H, Zhang L, Wang J (2015) QTL IciMapping: integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J 3:269–283
Michelmore RW, Paran I, Kesseli RV (1991) Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc Natl Acad Sci USA 88:9828–9832
Muterko A, Salina E (2018) Origin and distribution of the VRN-A1 exon 4 and exon 7 haplotypes in domesticated wheat species. Agronomy 8:156
Nishida H, Yoshida T, Kawakami K, Fujita M, Long B, Akashi Y, Laurie DA, Kato K (2012) Structural variation in the 5ʹ upstream region of photoperiod-insensitive alleles Ppd-A1a and Ppd-B1a identified in hexaploid wheat (Triticum aestivum L.), and their effect on heading time. Mol Breed 31:27–37
Oliver S, Finnegan E, Dennis E, Peacock W, Trevaskis B (2009) Vernalization-induced flowering in cereals is associated with changes in histone methylation at the VERNALIZATION1 gene. Proc Natl Acad Sci USA 106:8386–8391
Putterill J, Varkonyi-Gasic E (2016) FT and florigen long-distance flowering control in plants. Curr Opin Plant Biol 33:77–82
Rombauts S, Dehais P, Van Montagu M, Rouze P (1999) PlantCARE, a plant cis-acting regulatory element database. Nucleic Acids Res 27:295–296
Salamini F, Ozkan H, Brandolini A, Schafer-Pregl R, Martin W (2002) Genetics and geography of wild cereal domestication in the near east. Nat Rev Genet 3:429–441
Seki M, Chono M, Matsunaka H, Fujita M, Oda S, Kubo K, Kiribuchi-Otobe C, Kojima H, Nishida H, Kato K (2011) Distribution of photoperiod-insensitive alleles Ppd-B1a and Ppd-D1a and their effect on heading time in Japanese wheat cultivars. Breed Sci 61:405–412
Sesiz U, Özkan H (2021) A new genetic linkage map in einkorn wheat (Triticum monococcum) detects two major QTLs for heading date in chromosome 2A and 5A, probably corresponding to the photoperiod and vernalization genes. Plant Breed 141:12–25
Shi C, Zhao L, Zhang X, Lv G, Pan Y, Chen F (2019) Gene regulatory network and abundant genetic variation play critical roles in heading stage of polyploidy wheat. BMC Plant Biol 19:6
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:e164
Wang L, Ge H, Hao C, Dong Y, Zhang X (2012) Identifying loci influencing 1,000-kernel weight in wheat by microsatellite screening for evidence of selection during breeding. PLoS ONE 7:e29432
Wang W, Wang Z, Li X, Ni Z, Hu Z, Xin M, Peng H, Yao Y, Sun Q, Guo W (2020) SnpHub: an easy-to-set-up web server framework for exploring large-scale genomic variation data in the post-genomic era with applications in wheat. Gigascience. https://doi.org/10.1093/gigascience/giaa060
Weingartner M, Subert C, Sauer N (2011) LATE, a C2H2 zinc-finger protein that acts as floral repressor. Plant J 68:681–692
Wilhelm E, Turner A, Laurie D (2009) Photoperiod insensitive Ppd-A1a mutations in tetraploid wheat (Triticum durum Desf.). Theor Appl Genet 118:285–294
Worland AJ, Appendino ML, Sayers EJ (1994) The distribution in European winter wheats of genes that influence ecoclimatic adaptability whilst determining photoperiodic insensitivity and plant height. Euphytica 80:219–228
Wu W, Zheng XM, Lu G, Zhong Z, Gao H, Chen L, Wu C, Wang HJ, Wang Q, Zhou K, Wang JL, Wu F, Zhang X, Guo X, Cheng Z, Lei C, Lin Q, Jiang L, Wang H, Ge S, Wan J (2013) Association of functional nucleotide polymorphisms at DTH2 with the northward expansion of rice cultivation in Asia. Proc Natl Acad Sci USA 110:2775–2780
Wu W, Zhang Y, Zhang M, Zhan X, Shen X, Yu P, Chen D, Liu Q, Sinumporn S, Hussain K, Cheng S, Cao L (2018) The rice CONSTANS-like protein OsCOL15 suppresses flowering by promoting Ghd7 and repressing RID1. Biochem Biophys Res Commun 495:1349–1355
Wu J, Qiao L, Liu Y, Fu B, Nagarajan R, Rauf Y, Jia H, Yan L (2022) Rapid identification and deployment of major genes for flowering time and awn traits in common wheat. Front Plant Sci 13:992811
Xie M, Sun J, Gong D, Kong Y (2019) The roles of Arabidopsis C1-2i subclass of C2H2-type zinc-finger transcription factors. Genes 10:653
Xie L, Zhang Y, Wang K, Luo X, Xu D, Tian X, Li L, Ye X, Xia X, Li W, Yan L, Cao S (2021a) TaVrt2, an SVP-like gene, cooperates with TaVrn1 to regulate vernalization-induced flowering in wheat. New Phytol 231:834–848
Xie Y, Zeng W, Wang C, Xu D, Guo H, Xiong H, Fang H, Zhao L, Gu J, Zhao S, Ding Y, Liu L (2021b) Fine mapping of qd1, a dominant gene that regulates stem elongation in bread wheat. Front Genet 12:793572
Yan L, Loukoianov A, Tranquilli G, Helguera M, Fahima T, Dubcovsky J (2003) Positional cloning of the wheat vernalization gene VRN1. Proc Natl Acad Sci USA 100:6263–6268
Yan L, Helguera M, Kato K, Fukuyama S, Sherman J, Dubcovsky J (2004a) Allelic variation at the VRN-1 promoter region in polyploid wheat. Theor Appl Genet 109:1677–1686
Yan L, Loukoianov A, Blechl A, Tranquilli G, Ramakrishna W, SanMiguel P, Bennetzen JL, Echenique V, Dubcovsky J (2004b) The wheat VRN2 gene is a flowering repressor down-regulated by vernalization. Science 303:1640–1644
Yan L, Fu D, Li C, Blechl A, Tranquilli G, Bonafede M, Sanchez A, Valarik M, Yasuda S, Dubcovsky J (2006) The wheat and barley vernalization gene VRN3 is an orthologue of FT. Proc Natl Acad Sci USA 103:19581–19586
Yan A, Wu M, Zhao Y, Zhang A, Liu B, Schiefelbein J, Gan Y (2014) Involvement of C2H2 zinc finger proteins in the regulation of epidermal cell fate determination in Arabidopsis. J Integr Plant Biol 56:1112–1117
Zhang J, Wang Y, Wu S, Yang J, Liu H, Zhou Y (2012) A single nucleotide polymorphism at the Vrn-D1 promoter region in common wheat is associated with vernalization response. Theor Appl Genet 125:1697–1704
Zou C, Wang P, Xu Y (2016) Bulked sample analysis in genetics, genomics and crop improvement. Plant Biotechnol J 14:1941–1955
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 32172040), the Crop Varietal Improvement and Insect Pests Control by Nuclear Radiation, the China Agriculture Research System of MOF and MARA (Grant No. CARS-03), and the Agricultural Science and Technology Innovation Program (Grant No. CAAS-ZDRW202002).
Author information
Authors and Affiliations
Contributions
Conceptualization, LL and ZF; formal analysis YL, HX, HG, CZ, and MF; methodology: YL, HX, YX, LZ, SZ, and YD; resources: HG, JG, CW, and AI; Writing—original draft: YL and HX; Writing—review & editing: LL; supervision, LL; funding acquisition: LL. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Communicated by Maria von Korff.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Fig. S1
Distribution of HDs in the RILs that were homozygous for the dominant and recessive VRN-B1 alleles (TIF 7046 KB)
Fig. S2
a Frequency distribution of HDs in the secondary segregating population for Qhd.2AS. b Comparison of HDs in plants with different Qhd.2AS genotypes (TIF 7131 KB)
Fig. S3
a Geographical distribution of the representative SNP-140361804. b Frequency distribution of the SNP-140361773 and SNP-140361804 in wheat landraces and modern cultivars (TIF 14063 KB)
Fig. S4
Multiple sequence alignment of the promoter sequences of TraesCS2A02G181200 and its homeologues (TIF 39149 KB)
Fig. S5
Haplotype analysis of Qhd.2AS in the MCC panel. a Scatter plot of KASP marker assays for genotyping the MCC panel with four closely-linked markers. b Frequency distributions of haplotypes Hap-1-4 in wheat accessions with different growth habits and c in landraces and modern cultivars (TIF 6863 KB)
Fig. S6
Interaction graphs of VRN-B1 and Qhd.2AS in the RIL population. Error bars represent the standard errors of the means. **p < 0.01, *p < 0.05 (TIF 7171 KB)
Fig. S7
Evaluation of the effect of Qhd.2AS on important agronomic traits in the secondary segregating population (TIF 2945 KB)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, Y., Xiong, H., Guo, H. et al. Fine mapping and genetic analysis identified a C2H2-type zinc finger as a candidate gene for heading date regulation in wheat. Theor Appl Genet 136, 140 (2023). https://doi.org/10.1007/s00122-023-04363-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00122-023-04363-5