
Abstract
Key message
Five soybean target lines with recombinase sites at suitable genomic positions were obtained and tested for site-specific gene stacking.
Abstract
For introgression of new transgenic traits to field cultivars, adding new DNA to an existing transgene locus would reduce the number of segregating loci to reassemble back into a breeding line. We described previously an in planta transgene stacking system using the Bxb1 integrase to direct new DNA into a genomic target, but for this system to operate, the target locus must have a preexisting recombination site for Bxb1-mediated integration. Here, we describe 5 soybean target lines from the screening of 118 Agrobacterium-mediated transgenic plants that were positive for gus expression. Each of the 5 target lines has a single copy of the transgenic DNA with precise DNA sequences of the recombinase recognition sites, located at least 1 kb away from the nearest coding region, not close to the centromere, and showed good expression of the reporter gene. We tested Bxb1 integrase-mediated integration of a gfp-containing plasmid into each of these lines and showed precise site-specific integration in bombarded calluses. For plant regeneration, we used embryonic axes of mature soybean seeds to conduct a new set of biolistic transformation with a DsRed-containing plasmid. Three integration events were regenerated into whole plants, demonstrating the principle that target lines can serve as foundation lines for the stacking of DNA to predefined locations in the soybean genome.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Soybean is a major crop and an important source of vegetable protein and oil for human and farm animals. To improve quality and yield, genetic engineering has been a useful approach to augment traditional soybean breeding. By 2019, biotech soybean was the largest planted genetically modified (GM) crop with ~ 92 million hectares representing ~ 74% of the global soybean acreage (ISAAA 2019). Soybean engineered with multiple traits is also becoming more prevalent. As more useful genes are discovered, future crop improvement could be expected to experience the incremental incorporation of new transgenes. How new transgenic traits are added to a plant genome could significantly affect the speed and labor for developing field cultivars. A new transgene is introduced typically into a single cultivar that goes through the deregulation process for that single integration ‘event’ before it is bred out to the numerous location-specific varieties. If a new transgene integrates into a different location, it would create a new genetic locus that must be assorted back into a breeding line. Stacked traits can be obtained by crossbreeding and can be manageable when there are few loci to content with (Bengyella et al. 2018). If more transgenic traits were added, assorting all of them into a single genome could require considerable work. Even without linkage drag, for diploids or polyploids that segregate as diploids, the probability of obtaining homozygosity from a hemizygous transformant is (¼)n, where n is the number of loci. Introgressing a new trait into a different cultivar would also need to introgress other non-transgenic traits as well. It would not be far-fetched to imagine a probability of 1 in a million, from (¼)10, comprising for example, of just 3 transgenic loci along with 7 non-transgenic traits. Naturally, a breeder can assort the many traits through repeated backcrosses, but at the expense of more time and labor.
One way to keep transgenes from inflating the number of segregating loci is to package them in vitro into a single vector and screen for single site integration of the entire transgene package. This ‘in vitro gene stacking’ approach has merit when all of the genes are already available, but if a crop has already been previously engineered with certain traits, adding another new transgene could mean a ‘do over’ of all previously introduced traits. This might not be too great a burden from an engineering point of view, but from a legal perspective, it could trigger the need to go through the deregulation process again for previously introduced traits, since they would then be considered a new integration event.
A second approach calls for introducing the new trait directly into already established transgenic cultivars. Even if the new DNA integrates into a new location, it would still be in the same genome and would bypass the need for introgression. However, unlike the deregulation of a single integration event bred out to numerous field cultivars, transformation into different commercial cultivars with same DNA would produce independent integration events that would require individual deregulation. Regenerating plants from culture is also difficult with most commercial cultivars, especially when a large number of independently transformed plants are needed for field efficacy testing, and efficient transformation protocols would need to be developed for the many locally adapted cultivars.
A third approach is ‘in planta gene stacking’, the insertion of new DNA next to previously placed transgenes. Host-mediated homologous recombination induced by site-specific nucleases, such as zinc finger nuclease, TALEN, meganuclease and CRISPR/Cas9, can produce accurate genomic DNA strand breaks to allow for homology directed repair from the donor transgene fragment. Zinc finger nuclease-induced recombination has been reported in maize to stack an herbicide resistance gene, aad1, next to a preexisting herbicide resistance gene, PAT, with frequencies up to 5% (Ainley et al. 2013), and in soybean to integrate 4 marker genes into the FAD2-1a locus to produce 3 targeted events out of 1,290 selected shoots from immature embryos (Bonawitz et al. 2019). Meganuclease-mediated targeting has also been reported in cotton for integrating two herbicide resistance genes next to preexisting transgenes at up to 2% frequency (D'Halluin et al. 2013).
In addition to site-specific nucleases, site-specific recombinases can also direct the integration of new DNA, and examples in major crop plants include the Cre recombinase-directed cassette exchange of multiple genes in rice (Pathak and Srivastava 2020) and the FLP recombinase-mediated cassette exchange of DNA in soybean (Li et al. 2009). In the latter case, 3 different FRT sites enabled a second round of gene stacking (Li et al. 2010). However, further gene stacking rounds with this approach could be difficult since new recombinase recognition sites would be needed. As shown by the analogous Cre–lox system, few lox mutant sequences within the 8-bp lox spacer region were functional without high cross-reactivity with other lox alleles (Albert et al. 1995). The system we described for in planta gene stacking uses the Mycobacteriophage Bxb1 site-specific integration system (Hou et al. 2014), in which the 500 amino acid Bxb1 integrase (recombinase) can recombine an attP (phage attachment site, minimal 39 bp) and an attB (bacterial attachment site, minimal 34 bp) to generate attL (attachment site left) and attR (attachment site right) (Ghosh et al. 2003; Kim et al. 2003). After site-specific integration, the Coliphage P1 Cre–lox recombination system, in which the 343 aa Cre protein recombines direct-oriented 34-bp lox sites, can be used to delete DNA no longer necessary after successful integration. Gene stacking can proceed to the next round, as each integrating molecule brings in a new recombination target (attB or attP) for the next round of integration.
The one disadvantage of using a recombinase-mediated system is that the plant genome must already have a genomic target site, for example, an attP or an attB sequence to permit the integration of an incoming molecule through Bxb1 integrase-catalyzed site-specific recombination. Although a target site could be engineered into the plant genome by site-specific nucleases, we could not predict suitable chromosome locations for transgene expression. Additionally, we wanted to insure that our plant target lines would not be restricted from commercial use due to site-specific nuclease patents. Hence, in our empirical search of suitable target lines, this study describes the screening of a collection of Agrobacterium-mediated random insertions in the soybean genome, resulting in 5 soybean target lines each having a single precise copy of the transgenic DNA that is at least one kb away from the nearest gene coding region, is not close to the centromere and showed good expression of the gus reporter gene. In biolistic transformation on calluses, we tested the Bxb1 integrase-mediated site-specific integration of a gfp-containing plasmid into each of the 5 target lines and found precise site-specific integration among bombarded calluses based on PCR detection of the correct junctions. Those clones also expressed gfp as well as the gus reporter genes. For easier regeneration, we switched to using embryonic axes of mature soybean seeds for biolistic transformation and three integration events were regenerated from embryonic axes into whole plants. These data demonstrate that soybean target lines can serve as foundation lines for the in planta gene stacking of new DNA to predefined chromosome locations.
Materials and methods
DNA constructs
Phusion High-fidelity DNA polymerase (NEB, Beijing, China) was used for vector construction. Construction of target construct pSOY036B (Fig. 1a), integration vectors pJL210H (Fig. 1b) and pJL210A (Fig. 1g), and Bxb1 integrase vector pYQ78 with a 45-bp NLS sequences at the C terminus of the Bxb1 coding region (Fig. 1c) are shown in Fig. S7–S10. bar (bialaphos resistance) was controlled by the soybean GmScreamM2 promoter (Pubi) from the Glycine max ubiquitin conjugation enzyme gene (LOC547652) (Zhang et al. 2015), and the cauliflower mosaic virus (CaMV) 35S RNA terminator; gus (plus version) was controlled by the soybean GmActin promoter (Pact) (Zhang and Finer 2014) and the rice polyubiquitin gene RUBQ1 terminator (Kuroda et al. 2010). Recombination sites RS2, attP and lox were synthetic DNA designed to flank bar and gus. gfp or DsRed was controlled by the sugarcase bacilliform badnavirus promoter (Tzafrir et al. 1998) and octopine synthase terminator, hpt by soybean ubiquitin gene promoter (Zhang et al. 2015) and CaMV 35S RNA terminator. Arabidopsis ahas was controlled by its own Arabidopsis promoter and CaMV 35S RNA terminator.

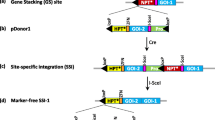
Design and detection of site-specific integration. a Structure of pSOY036B-transferred T-DNA in soybean target lines, with bar, gus, lox, attP and RS2 (symbols in inset legend) between T-DNA left (LB) and right borders (RB); b integrating plasmid pJL210H containing gfp, hpt, attB and lox sites; c Bxb1-integrase expression construct pYQ78 with Bxb1 coding region linked to a C-terminal NLS; d pJL210H type I integration structure from recombination between the chromosome attP site and the hpt distal attB site; e pJL210H type II integration structure from recombination with the hpt proximal attB site; f representative calluses from target line S20 bombarded with pJL210H and screened by PCR for integration junctions with primer sets shown in (d) and (e). Lane N, pJL210H negative control; Lane P, pJL210H with pSOY036B in vitro recombination control. Controls were mixed with S20 genomic DNA at 1 copy per genome. Lane M: DNA size markers in kb; g integrating plasmid pJL210A containing DsRed, ahas, attB and lox sites; h pJL210A type I integration from recombination between the chromosome attP site and the ahas distal attB site; i pJL210A type II integration from recombination with the ahas distal attB; note that primers d and h correspond to the same promoter and terminator, respectively, to drive gfp and DsRed; j PCR data from site-specific integration of pJL210A into target line S20 leading to T0 integrant plants. Lane N, pJL210A mixed with S20 genomic DNA at 1 copy per genome; lane P, pJL210A recombined with pSOY036B in vitro diluted to 1 copy per genome; k Southern blots of 3 integrant plants along with parental line S20 and WT. gus and DsRed DNA probes indicated in (a) and (g) were hybridized to genomic DNA cleaved by SacI, XhoI or MluI. Genomic DNA from induced callus of T0 S20.i1 hemizygous leaf and leaf DNA from T1 hemizygous S20.i2 and S20.i8 plants. Magenta lines represent PCR products from primers in italic lettering, blue lines are DNA fragments from endonuclease cleavages, fragment sizes in kb. Promoters/terminators not shown; genes transcribe in direction of the arrows (color figure online)
Agrobacterium-mediated transformation
Procedures described by Flores et al. (2008) were used on soybean cultivar ‘Dongnong 50’ from Northeast Agricultural University, China, that originated from a Canadian natto soybean [Glycine max (Linn.) Merr.] ‘Electron.’ Sterilized mature seeds were germinated for a day in B5 medium, and cotyledonary nodes were vertically cut after removing remaining axial shoots or buds. Explants were then wounded with a scalpel and dipped into the A. tumefaciens strain EHA105 (pSOY036B). After co-cultivation for 4 days, explants were transferred for shoot induction plates without selection for a week, then to 10 mg L−1 glufosinate plates for 2 weeks and finally to 5 mg L−1 glufosinate plates for 4 to 6 weeks. Elongated shoots ~ 3 cm were transferred for rooting without further selection for about two weeks.
Biolistic-mediated transformation
Soybean target line seeds were sterilized by chlorine gas for 16 h made by pipetting 3.5 mL 12 N HCl into 100 mL 10% sodium hypochlorite solution. Sterilized seeds were sown into germination medium (Murashige and Skoog medium with 3% w/v sucrose, filter-sterilized 1 mg L−1 6-BA and 8 g L−1 Agar, pH 5.8), with 16 h light at 25 °C. After a week, seedling stems were crosscut into thin slices and placed onto callus-inducing medium (Murashige and Skoog medium with 3% w/v sucrose, filter-sterilized 2 mg L−1 6-BA, 0.5 mg L−1 NAA and 8 g L−1 Agar, pH 5.8). Light-colored emerald calluses formed in about 16 days were transferred to fresh callus induction medium. Calluses on fresh callus-inducing medium for 3 days were placed onto osmotic medium (induced medium plus 46.6 g L−1 mannitol and 46.6 g L−1 sorbitol) for 4 h in the dark. Particle bombardment was as described for rice (Li et al. 2016) using the Biolistic Particle Delivery PDS-1000/He System (Bio-Rad, USA). After bombardment, calluses were kept on osmotic medium for 18 h in the dark and GFP fluorescence was monitored under fluorescence microscopy. Calluses were then transferred onto callus-inducing medium plus 100 mg L−1 hygromycin B for one month with light at 25 °C. A callus that grew on hygromycin B-containing plates and showed GFP activity was counted as a single independent ‘event’.
Embryonic axes were prepared by soaking mature seeds in ddH2O for 16 h and placed on bombardment medium (Murashige and Skoog medium with 3% w/v sucrose and 8 g/L Agar, pH 5.8) for biolistic transformation as described for calluses. The subculturing for soybean axes was as described (Rech et al. 2008), except that 100 nM imazapyr was used to screen hygromycin-resistant shoots.
Southern analysis
Soybean genomic DNA was prepared from fresh leaves by incubating in CTAB extract buffer (2% (w/v) CTAB, 100 mM Tris–HCl, 20 mM EDTA, 1.4 M NaCl and 2% (w/v) PVP, pH 8.0) for 30 min, 65 °C. DNA was precipitated by isopropanol and dissolved in ddH2O before extraction by NucleoBond AX 100 Columns (Genopure Plasmid Midi Kit, Roche, Germany). DNA concentration was measured by NanoDrop 2000 (Thermo scientific, USA). Genomic DNA (30 μg) was cleaved with SacI-HF® (NEB, China) in a 50 μl reaction per sample at 37 °C for 12 h, and the products were electrophoresed in a 0.8% agarose gel. Cleaved genomic DNA was transferred to a Hybond-N+ nylon membrane (GE healthcare, UK) by vacuum transfer (Bio-Rad model 785 vacuum blotter, USA). After cross-linking genomic DNA under UV light (UVP CL-1000 Ultraviolet Crosslinker, USA), the membrane was incubated with gus or bar DNA labeled by [α−32P] dCTP (Amersham Rediprime II DNA labeling system, GE, UK) and washed to remove non-specific binding. The hybrid membrane was exposed onto a phosphor screen for 12 h and scanned by Typhoon FLA 9500 (IP: 635 nm, PMT: 500 V, Pixel size 200 μm). For analysis of regenerated integrant plants, the gus or DsRed probe was labeled with DIG-dUTP (DIG labeling system, High Prime DNA Labeling and Detection Starter Kit II, Roche, Germany).
PCR analyses
LA taq (TAKARA, China) was used for PCR of genomic DNA. For left end junctions, a set of primers: j (jS15; jS20, jS32; jS78; jS80) specific to target line genome sequence was used with primer b; for right end junctions, another set of primers l (lS15, lS20; lS32, lS78, lS80) was used with primer k (within gus, Fig. S2). Other primers are described in the text and figures except for primers n and o used for ahas, and primers p and q for Bxb1 integrase gene. All primers are listed in Table S2.
T-DNA insertions in genomic DNA were determined by TAIL-PCR (Liu and Chen 2007) and inverse PCR (Boulin and Bessereau 2007). For TAIL-PCR, first-round primers were chosen from any two random primers AD plus specific primer SP1 based on pSOY036B sequence. First-round PCR products were diluted 1/800 into a 20 ul reaction for the second-round PCR with primers SP2 and AD2. Second-round PCR products were diluted 1/200 into a 20 ul reaction for the third-round PCR with primers SP3 and AD2. For inverse PCR, genomic DNA was cleaved with BamHI at 37 °C for 12 h followed by enzyme inactivation at 75 °C for 15 min and then self-ligated by adding T4 ligase and T4 ligase buffer to a 100 μL reaction per sample at 16 °C for 12 h. High-fidelity DNA polymerase (Phusion, NEB) with primers F1 and R1 was used for the first-round PCR, and products were diluted 3/4000 in a 20 μL reaction for the second-round PCR with primers F2 and R2. TAIL-PCR and inverse PCR final products were purified from 1% agarose gels for sequencing (Sangon Biotech, Shanghai, China). Genomic sequences detected were blasted on database: https://phytozome.jgi.doe.gov/pz/portal.htm; https://www.soybase.org/; Glycine max Wm82.a2.v1 served as reference genome. Vector sequences of PCR products were aligned with pSOY036B sequence by online software ClustalW2 (https://www.ebi.ac.uk/Tools/msa/clustalw2/).
GUS, GFP and DsRed activity
Leaf and root explants or seeds were immersed in GUS staining solution (39 mM KH2PO4, 61 mM K2HPO4·3H2O, 1 mg/mL X-Gluc, 0.5 mM K3[Fe(CN)6], 0.5 mM K4Fe(CN)6·3H2O, 0.1%(v/v) Triton X-100) at 37 °C for 16 h. For GUS enzyme activity, 50 mg soybean leaf tissues were ground and added to 200 μL protein extract buffer (77.4 mM Na2HPO4, 22.6 mM NaH2PO4, 1 mM EDTA, 1% (v/v)TritionX-100, 1 mM PMSF); protein concentration was determined by Pierce BCA Protein Assay Kit (Thermo, USA). β-Glucuronidase activity was assayed through spectrophotometric method as described (Jefferson et al. 1987). DsRed fluorescence was detected using a Mithras2 LB943 Monochromator and Filter Multimode Microplate Reader (Berthold, Germany). Excitation and emission wavelengths were 561 nm and 587 nm, slit widths 6 nm and 12 nm, respectively (Nishizawa et al. 2006). One-way ANOVA was conducted by SPSS 21 (SPSS Inc., USA). Statistical significance was determined by Tukey’s Studentized range (HSD) test.
Fluorescence GFP or DsRed of callus, leaf or stem were visualized using a DMI6000B microscope (Leica, Wetzlar, Germany). For GFP observation, wavelength for excitation filter was from 440 to 520 nm, and 510 LP was chosen for the barrier filter. For DsRed observation, wavelength for excitation filter ranged from 525 to 565 nm, and the wavelength of the barrier filter was from 572 to 648 nm.
Results
Creation of soybean target lines for gene integration
Soybean cotyledon nodes were transformed with Agrobacterium harboring the target construct pSOY036B that contains two genes: the selectable marker bar controlled by the soybean ubiquitin promoter (Zhang et al. 2015)/CaMV 35S RNA terminator and the reporter gene gus by the soybean actin gene promoter (Zhang and Finer 2014)/rice ubiquitin terminator (Kuroda et al. 2010). These two genes were flanked by a set of directly oriented lox recombination sites for subsequent deletion (Fig. 1a). Downstream of gus lies an attP site that serves as a receptor for new DNA, followed by a third lox site in the opposite orientation for the purpose of removing unneeded DNA on the right border side after site-specific integration.
Transformation was attempted on ~ 36,000 soybean cotyledon node explants, and 368 regenerated plants were selected as putative transformants based on growth on glufosinate-containing media. Positive GUS staining was found in 118 regenerated plants, and all but two were found by PCR to have the T-DNA left border (LB) proximal recombination sites based on the 0.9-kb PCR product a-b (Fig. 1a, Fig. S1a). A Southern blot was conducted on those 116 plants with genomic DNA cleaved by SacI and probed with gus or bar DNA. The bar probe was expected to hybridize to a > 1.8-kb left border fragment, while gus probe would detect a > 6-kb right border fragment (Fig. 1a). Of the 116 plants, 35 showed the expected pattern for a single copy insertion of the T-DNA since each probe hybridized to a single band of the expected size (Fig. S1b, c). Although plants with multiple but genetically unlinked hybridizing bands might be convertible to single copy lines in future generations, we did not pursue this possibility.
TAIL-PCR was used to determine the locations of the 35 putative single copy transgenic clones, but was successful with only 13 clones (Fig. S1d). Of those, 11 were discarded because in 3 clones TAIL-PCR results could not be verified by high-fidelity PCR using primers corresponding to the genome sequence; in 4 clones, DNA sequencing found co-integration of the vector-associated aadA (spectinomycin resistance) gene outside of the T-DNA; in 1 clone, the insertion was within repetitive sequences that did not reveal an exact location; in 1 clone, the insertion was into a host gene; and in 2 clones, the insertions were less than 1 kb (673 bp and 173 bp) from a host gene. This left only two suitable clones, S20 and S80, for consideration as target lines.
For the remaining 22 clones where we failed to obtain useful information from TAIL-PCR, we tried inverse PCR following cleavage by BamHI, but were successful in obtaining genome information from only 5 of them (Fig. S1e). However, high-fidelity PCR with primers based on the genome sequence failed to verify the right and/or left junction in 2 of those, hence leaving only 3 clones, S15, S32 and S78, for further investigation.
For all 5 clones (S15, S20, S32, S78 and S80) the DNA sequences of the left and right junctions showed that the relevant RS2, lox and attP recombination sites were correct (Fig. S3, S4). We did not sequence the DNA between the two directly oriented lox sites since bar and gus represent DNA that would be excised by Cre–lox recombination after successful stacking of new DNA. However, bar had to be functional since it was the selection marker for transformation. As for gus, its expression was examined and the soybean actin promoter-driven gus expressed well in the 5 target lines as shown by GUS activity staining in T2 flowers, leaves, roots and seed, as well as by GUS enzyme activity in the 4th trifoliate of the T2 lines (Fig. 2c and d). It is interesting that GUS activity differed among the putative target lines, as GUS enzyme activity of homozygous lines S20 and S80 was up to 4.7- and 2.3-fold higher, respectively, than that of homozygous S78 (Fig. 2d), which showed similar activity as the hemizygous lines S15 and S32. This could suggest that different chromosome locations exert different effects on transgene expression.

Target site location and GUS activity. a Map location of T-DNA insertions in the soybean genome with direction of insertion indicated by the RS site (magenta triangle). All chromosome arranged with base 1 on top; b schematic representation of target site structures. Shown in blue lettering are chromosome positions at left and right ends, with if any, deletions (△) and insertions. Numbers above left (LB) and right (RB) T-DNA borders, and left and right vector sequences between flanking LB or RB to RS2 show the number of base pairs (bp); c GUS staining of different organs (flowers, 2nd trifoliates, roots, seeds) of T2 target lines. S20, S78 and S80 (magenta lettering) were homozygous lines, S15 and S32 hemizygous lines (blue lettering). Relative GUS staining indicated by + (detected) or – (not detected) additional + correspond to stronger blue staining. d β-Glucuronidase activity in extracts of 4th trifoliates of the T2 homozygous target lines (S20, S78 and S80) and T2 hemizygous target lines (S15 and S32) detected from 0 to 180 min. Values are mean ± SD from two independent experiments, each using 3–4 plants (color figure online)
Location of target sites
Line S15 is inserted into the short arm of chromosome 16 between physical map coordinates 4,414,388 to 4,414,393. The DNA between 4,414,389 and 4,414,392 is missing, meaning that line S15 has a 4-bp genome deletion at the site of the insertion (Fig. 2a and b; Fig. S3, S4; Table S1). The T-DNA LB (left border)-containing fragment should be 526 bp comprising of 25-bp LB sequence plus 501-bp vector sequence before the start of the RS2 sequence. In S15, the left end lacks the entire 25-bp T-DNA LB plus 2 bp of adjacent vector sequence (no LB, 499 bp). The T-DNA RB (right border)-containing fragment should be 283 bp comprising of 25-bp RB sequence plus 258-bp vector sequence before the start of the RS2 sequence. In S15, the right end lacks 24 bp of the 25-bp T-DNA RB (1 bp remaining). As in the direction shown in Fig. 2b, nearest coding region to the left and right is 13.1-kb (stop codon) and 14.1-kb (start codon), respectively.
S20 is inserted into long arm of chromosome 15 between position 8,355,701 and 8,355,726. The genomic DNA between 8,355,702 and 8,355,725 is not found, representing a 24-bp deletion at the insertion site (Fig. 2a and b; Fig. S3, S4; Table S1). The left end lacks 23 bp of the 25-bp T-DNA LB (2 bp remaining) with one bp insertion, and the right end has the entire 25-bp T-DNA RB. As in the direction shown in Fig. 2b, nearest coding region to the left and right is 1.5-kb (start codon) and 1.5-kb (stop codon), respectively.
S32 is inserted into short arm of chromosome 2 between position 9,329,711 and 9,329,900. A 188-bp deletion between 9,329,712 and 9,329,899 is found in the genomic DNA, (Fig. 2a and b; Fig. S3, S4; Table S1). The left end lacks 9 bp of the 25-bp T-DNA LB but has a 3-bp insertion; the right end has the entire of the 25-bp T-DNA RB but with an 18-bp insertion. The RS2 region at the LB end has a duplication of RS2 site, but this duplicated RS2 has lost 10 bp (Fig. S3). As in the direction shown in Fig. 2b, nearest coding region to the left is 21.2-kb (start codon) and to the right is 39.0-kb (stop codon).
S78 is inserted into the short arm of chromosome 2 between position 9,310,241 and 9,310,243, representing only a single-bp deletion of position 9,310,242. The left end lacks 17 bp of the 25-bp T-DNA LB, and the right end lost the 16-bp T-DNA RB (Fig. 2a and b; Fig. S3, S4; Table S1). As in the direction shown in Fig. 2b, nearest coding region to the left and right is 58.7-kb (stop codon) and 1.8-kb (start codon), respectively.
S80 is inserted into the long arm of chromosome 11, between position 5,408,016 and 5,408,042. The 25 bp of DNA between 5,408,017 and 5,408,041 is missing (Fig. 2a and b; Fig. S3, S4; Table S1). Both left and right end lost the entire 25-bp T-DNA LB and RB, and 216 bp of adjacent vector sequence on the left end, as well as 19-bp of vector sequence of the right end are missing. As in the direction shown in Fig. 2b, nearest coding region to the left is 16.6-kb (start codon) and to the right is 4.7-kb (stop codon).
Site-specific integration into target lines
To test for Bxb1 integrase-catalyzed integration of new DNA into a target site, biolistic transformation was performed on calluses derived from T2 seedlings of the 5 target lines. As the PCR products representing the native genome could be amplified only from a WT chromosome (primers j + l, Fig. S2a), we found that the calluses of S20, S78 and S80 were homozygous while those of S15 and S32 were hemizygous for the target site.
Integrating vector pJL210H containing gfp and hpt (Fig. 1b) was bombarded into calluses along with the Bxb1 integrase-expressing vector pYQ78 (Fig. 1c). Bxb1 recombinase produced by pYQ78 would catalyze site-specific recombination between the genomic attP and the hpt distal attB to produce the type I integration pattern (Fig. 1d) or the type II integration structure from recombination between the genomic attP and the hpt proximal attB (Fig. 1e). From the five target lines, a total of 2,430 calluses were bombarded and cultured on hygromycin-containing medium. From 981 calluses that grew on the selection medium, 202 were GFP-positive based on fluorescence microscopy observations (Fig. 3a). As integration of pJL210H into random locations could also result in expression of hpt and gfp, this 8.3% transformation efficiency based on GFP activity could include site-specific gene integration, random gene insertion or both types within the same cell.

Site-specific integrant from bombardment of calluses and embryonic axes. a Putative site-specific events selected on hygromycin-containing plates after particle bombardment with pJL210H and pYQ78, and screened for GFP fluorescence; b bombardment of soybean embryonic axes from target line S20. Apical meristems co-bombarded with pJL210A and pYQ78. Clones selected on imazapyr-resistant medium that showed DsRed signals were transferred to soil in the greenhouse after rooting; c GUS and (d) DsRed activity measured in extracts of 4th trifoliates of the T1 hemizygous integrated lines (S20.i2 and S20.i8) and callus of S20.i1, detected from 180 min. Values are mean ± SD n = 3 to 4 and statistically significant differences labeled by letters at p < 0.05
PCR primers c + d and e + f were used to detect the type I integration junctions (Fig. 1d, f), and primers c + g and h + f were used for the type II junctions (Fig. 1e, f). Pooling the PCR data from all 5 target lines, 118 of 202 GFP-positive calluses showed site-specific integration, although 22 of them were deemed incomplete integration since only a single integration junction was detected (Table 1). For the other 96 calluses, left and right integration junctions were detected with 20 type I integration, 23 type II integration and 53 with both type I and type II integration structures (Table 1).
From a total of 2430 calluses, the combined type I plus type II site-specific integration efficiency was about 4%. Based on preselected GFP + calluses, the average efficiency from the five lines was 47.5%. Interestingly, integration efficiency correlated with target site availability, as the integration efficiencies of the homozygous lines S20, S78 and S80 were at least twice as high at 59.6%, 52.4% and 54.5%, respectively, as those of the hemizygous target lines S15 and S32 at 24.2% and 22%, respectively (Table 1).
Having both types of integration could indicate a mixture of different cells within the same callus or different integration at homolog chromosomes in the same cell in the homozygous lines S20, S78 and S80. For the homozygous lines, the detection of only one integration pattern could also be from site-specific integration on both homolog chromosomes. Although calluses with both type I and type II structures could be useful for gene stacking once the type II cells or alleles are separated out, obtaining clones with the type I-only structure was preferred. As shown in Table 1, 20 calluses from 4 target lines were found with the type I-only structure or a ~ 10% efficiency of type I-only integration from GFP-positive calluses. However, this did not exclude the possibility of additional copies of pJL210H integrated elsewhere in the genome.
Representative type I and type II integration events were analyzed at the sequence level. The products amplified by primers c + d and e + f (Fig. 1d) were sequenced for 3 type I calluses each from line S20, S78 and S80, while products by primers c + g and h + f (Fig. 1e) were sequenced for 3 type II calluses from line S20. In every case, the sequence was as expected with precise recombination junctions for both type I and type II integration events (Fig. S5). These data showed that the Bxb1 prokaryotic site-specific integration system operated precisely in the soybean genome.
Site-specific integrant plants
To test obtaining fertile soybean plants from Bxb1-mediated site-specific integration, we used embryonic axes of mature seeds from a T3 homozygous S20 plant for biolistic transformation. This plant line was chosen because it was the first plant characterized which provided sufficient seeds for collection of embryonic axes. Another integration vector, pJL210A, with reporter gene DsRed and the selection gene ahas (imazapyr resistance) was co-transformed with pYQ78 (Fig. 1c). Seventy-five shoots regenerated after microparticle bombardment on apical meristems of 428 embryonic axes (~ 20 axes per plate). Eight shoots (S20.i1-.i8) that were DsRed-positive by fluorescence microscopy were regenerated into plantlets (Fig. 3b). PCR screening of these T0 plantlets with primers c + d and e + f detected 3 plants (S20.i1, S20.i2, S20.i8) with type I integration pattern (Fig. 1h, j). However, for S20.i1, primers c + i and h + f also detected the type II integration structure (Fig. 1i, j), and primers c + f amplified the original target site (Fig. 1a, S6b). This shows that the T0 S20.i1 genomic DNA had 3 types of structures: no integration (S20), type I integration and type II integration. The simplest interpretation would be that the DNA was derived from a plant that was chimeric, which is possible from organogenesis regeneration and that S20.i1 is hemizygous for type I integration in some cells, while hemizygous for type II integration in other cells, or that S20.i1 has cells with both type I and II integration, as well as other cells that are parental S20.
For S20.i1, its T1 seeds failed to germinate, but T1 plants were obtained from plants S20.i2 and S20.i8. Southern blot analysis was conducted on the genomic DNA from S20.i1 (T0), S20.i2 (T1) and S20.i8 (T1). With the homozygous line S20, a gus probe on SacI cleaved DNA should detect a 7.1-kb (> 6 kb; Fig. 1a) border band based on the nearest chromosome SacI site. For type I integration, an 8-kb internal band should hybridize (Fig. 1h). All three plants showed both the 7.1-kb and 8-kb bands to indicate hemizygous integration (Fig. 1k). For the type II integration structure, the gus probe should detect an internal 5-kb SacI band (Fig. 1i), and a faint 5-kb SacI band was observed in S20.i1. With XhoI cleaved DNA, the gus probe should detect a 8.4-kb (> 6.5 kb; Fig. 1a) border band from S20 based on the nearest chromosome XhoI site and internal fragments of 7 kb or 11.3 kb for type I or type II integration, respectively. Figure 1k shows detection of the 8.4- and 7-kb bands from all three plants consistent with hemizygous type I integration, but the 11.3-kb type II integration-specific band was not detected.
The DsRed probe, which does not hybridize to S20, should detect the same 8-kb SacI internal fragment as the gus probe, as well as a 7.2-kb MluI internal fragment for a type I integration structure (Fig. 1h); and both bands were detected in all three integrant plants (Fig. 1k). For the type II integration structure, the DsRed probe should hybridize to a 9.5-kb (> 8.4 kb) SacI and an 11.4-kb (> 9.6 kb) MluI fragment. As shown in Fig. 1k, these bands were not found in S20.i1 or S20.i8, but there were high molecular weight bands found in S20.i1 and S20.i2, although at limiting mobility they might not correspond to the bands expected from type II integration. Given that the gus probe did not detect type II integration in S20.i2, the simplest interpretation is the DsRed probe hybridized to additional copies of pJL210A elsewhere in the genome. Hence, in our 3 regenerated plants, S20.i1 failed to produce T1 progeny, S20.i2 probably contains additional random integration, and only S20.i8 can be considered a clean heritable type I integration event. The possibility does exist however, that the additional random integration events in S20.i2 could be segregated out in subsequent generations, as well as the Bxb1 expression plasmid, as Bxb1 integrase DNA was detected in all integration lines (Fig. S6b).
The detection of a faint 5-kb band in SacI cleaved S20.il raises a possibility that the plant has a minor amount of cells with type II integration, but is otherwise mostly hemizygous for type I integration. However, there might be an alternative explanation. It should be noted that both type I and type II integration structures would have a functional attB (Fig. 1d, e), a substrate for recombination with an available attP. In the case of using lines homozygous for the target site, as in calluses S20, S78 and S80 or the seeds from a T3 homozygous S20 plant, one chromosome would still have an attP if site-specific integration had not occurred in both homologous chromosomes. In that case, it should be possible for attP x attB to recombine the two homologous chromosomes to cause an exchange of chromosome arms (Fig. 4). In that case, gus should hybridize to a 5 kb from one chromosome (Fig. 4f) which might account for the faint 5-kb gus hybridizing band in Fig. 1k. However, the gus probe should also detect 10-kb band from the other chromosome and that was not found (Figs. 1k, 4f), although hybridization of the 10-kb range was detected by the DsRed probe on SacI cleaved S20.i1 DNA (Fig. 1k).

Predicted structure from hemizygous site-specific integration followed by chromosome arm translocation. a Type I integration of pJL210H in one chromosome recombines with a target site of the homologous chromosome to produce the two chromosome structures shown in (b). c Type II integration in one chromosome recombines with a target site of the homologous chromosome to produce the two chromosome structures shown in (d). e Type I integration of pJL210A in one chromosome recombines with a target site of the homologous chromosome to produce the two chromosome structures shown in (f). The gus probe would detect a 5-kb SacI fragment in a Southern blot after chromosome arm exchange consistent with Fig. 1k. g Structure after Cre–lox-mediated excision from structure shown in (e) and (f)
To see whether integration of the DsRed next to gus would affect each other’s expression, GUS activity and DsRed fluorescence were tested on leaf tissue of the T1 hemizygous lines S20.i2 and S20.i8 and for callus tissue from the T0 S20.i1 plant. All three samples showed GUS activity similar to the parental target line S20 (Fig. 3c). For DsRed fluorescence, signal strength was higher in S20.i1 and S20.i2 than in S20.i8 (Fig. 3d). Since S20.i2 and S20.i8 were from comparable leaf tissue of the same T1 generation, the 2 to 3 higher DsRed expression in S20.i2 might be related to additional DsRed plasmid in the genome.
Discussion
The 5 target sites for site-specific gene stacking in soybean were screened from Agrobacterium-mediated random integration events. As target lines could serve as foundation lines for subsequent introduction of transgenes, it was important that the introduced DNA was inserted as an intact molecule and with precise recombination sites. DNA sequence data showed that each target site has precise recognition sites for the Bxb1 integrase and Cre recombinase. The DNA from the RS2 site to the T-DNA borders was dispensable, and indeed, many target lines showed partial deletions of these sequences. The lox-flanked reporter and selection genes were also not important as they were destined to be removed by subsequent Cre-mediated deletion after gus expression provided an indication of whether the genome location would afford expression of new transgenes. Indeed, gus was expressed at different levels among different target sites. To lessen the probability of affecting nearby gene expression, we discarded all clones that had inserted into an open reading frame or with close proximity to an open reading frame and < 1 kb was arbitrary chosen even though cis effects might still be farther away. Finally, since future gene stacking events would be expected to introgress out to field cultivars, we discarded those clones located near a centromere where chromosome recombination would be suppressed.
Since the site-specific integration step requires a circular molecule substrate, biolistic transformation was used to introduce an integration plasmid into target lines. Site-specific integration was shown for all 5 target lines based on PCR analysis of callus tissue. Embryonic axes of one target line, S20, was tested in biolistic transformation which led to regeneration of three transgenic soybean plants with the preferred type I-only site-specific integration structure. However, plant S20.i1 failed to produce T1 progeny. With S20.i2 and S20.i8, both would require segregating out Bxb1 integrase DNA and for S20.i2 also additional copies of the integrating plasmid. With similar experiments in tobacco (Hou et al. 2014) and rice (Li et al. 2022), we did not find a high rate of co-integration of the integrase-expressing plasmid, but in cotton, 3 of 8 integrant plants also harbored Bxb1 integrase gene (Li et al. 2022). Possibly, for soybean and cotton, where obtaining regenerated plants are difficult, we might have used a higher amount of the integrase plasmid DNA to insure site-specific integration. From our experiments, site-specific integration efficiencies among rice, cotton and soybean, were rather similar. From preselected GPF + calluses, the integration efficiencies for the preferred type I-only structure were 11.4% for rice, 6.3% for cotton and 9.9% for soybean.
These frequencies, however, were based on PCR detection of integration junctions, and for plants with homozygous target sites, this does not necessarily indicate a contiguous structure. As depicted in Fig. 4, a Bxb1-mediated chromosome arm exchange after site-specific integration is a possibility (Figs. 1k and 4). We do not have clear evidence that a chromosome arm exchange took place, but if it did, the Southern detection of faint bands do not indicate a high-frequency occurrence. Nonetheless, in future gene stacking attempts, it might be prudent to use only hemizygous seeds to avoid this chromosome arm exchange possibility. The most important consideration, however, is whether having a chromosome arm exchange would affect the recovery of the desired outcome, and the answer is no. After Cre–lox-mediated excision, the final structure would be the same (Fig. 4g).
As for transgene expression, integration of a new transgene did not affect the expression of the previously inserted gus transgene, which was not the case for rice and cotton (Li et al. 2022). Whether the higher DsRed expression in S20.i2 versus S20.i8 is due to a copy effect is not yet certain, as it would await segregating out the extra copy of the DsRed plasmid. In summary, we have documented Bxb1-mediated site-specific integration in soybean, including the recovery of S20.i8, a plant with a clean and heritable type I integration event.
Data availability
The data supporting the findings of this study are available upon request from Ruyu Li.
References
Ainley WM, Sastry-Dent L, Welter ME, Murray MG, Zeitler B, Amora R, Corbin DR, Miles RR, Arnold NL, Strange TL, Simpson MA, Cao Z, Carroll C, Pawelczak KS, Blue R, West K, Rowland LM, Perkins D, Samuel P, Dewes CM, Shen L, Sriram S, Evans SL, Rebar EJ, Zhang L, Gregory PD, Urnov FD, Webb SR, Petolino JF (2013) Trait stacking via targeted genome editing. Plant Biotechnol J 11(9):1126–1134. https://doi.org/10.1111/pbi.12107
Albert H, Dale EC, Lee E, Ow DW (1995) Site-specific integration of DNA into wild-type and mutant lox sites placed in the plant genome. Plant J 7(4):649–659. https://doi.org/10.1046/j.1365-313x.1995.7040649.x
Bengyella L, Yekwa EL, Iftikhar S, Nawaz K, Jose RC, Fonmboh DJ, Tambo E, Roy P (2018) Global challenges faced by engineered Bacillus thuringiensis Cry genes in soybean (Glycine max L.) in the twenty-first century. 3 Biotech 8(11):464. https://doi.org/10.1007/s13205-018-1484-8
Bonawitz ND, Ainley WM, Itaya A, Chennareddy SR, Cicak T, Effinger K, Jiang K, Mall TK, Marri PR, Samuel JP, Sardesai N, Simpson M, Folkerts O, Sarria R, Webb SR, Gonzalez DO, Simmonds DH, Pareddy DR (2019) Zinc finger nuclease-mediated targeting of multiple transgenes to an endogenous soybean genomic locus via non-homologous end joining. Plant Biotechnol J 17(4):750–761. https://doi.org/10.1111/pbi.13012
Boulin T, Bessereau JL (2007) Mos1-mediated insertional mutagenesis in Caenorhabditis elegans. Nat Protoc 2(5):1276–1287. https://doi.org/10.1038/nprot.2007.192
D’Halluin K, Vanderstraeten C, Van Hulle J, Rosolowska J, Van Den Brande I, Pennewaert A, D’Hont K, Bossut M, Jantz D, Ruiter R, Broadhvest J (2013) Targeted molecular trait stacking in cotton through targeted double-strand break induction. Plant Biotechnol J 11(8):933–941. https://doi.org/10.1111/pbi.12085
Flores T, Karpova O, Su X, Zeng P, Bilyeu K, Sleper DA, Kristin B, Nguyen HT, Zhang Z (2008) Silencing of GmFAD3 gene by siRNA leads to low α-linolenic acids (18:3) of fad3-mutant phenotype in soybean [Glycine max (Merr.)]. Transgenic Res 17:839–850. https://doi.org/10.1007/s11248-008-9167-6
Ghosh P, Kim AI, Hatfull GF (2003) The orientation of Mycobacteriophage Bxb1 integration is solely dependent on the central dinucleotide of attP and attB. Mol Cell 12(5):1101–1111. https://doi.org/10.1016/S1097-2765(03)00444-1
Hou L, Yau YY, Wei J, Han Z, Dong Z, Ow DW (2014) An open-source system for in planta gene stacking by Bxb1 and Cre recombinases. Mol Plant 7(12):1756–1765. https://doi.org/10.1093/mp/ssu107
ISAAA (2019) Global status of commercialized biotech/GM crops in 2019: biotech crops drive socio-economic development and sustainable environment in the new frontier. ISAAA Brief No.55. https://www.isaaa.org/resources/publications/briefs/55
Jefferson RA, Kavanagh TA, Bevan MW (1987) GUS fusions: β-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. Embo J 6(13):3901–3907. https://doi.org/10.1002/j.1460-2075.1987.tb02730.x
Kim AI, Ghosh P, Aaron MA, Bibb LA, Jain S, Hatfull GF (2003) Mycobacteriophage Bxb1 integrates into the Mycobacterium smegmatis groEL1 gene. Mol Microbiol 50(2):463–473. https://doi.org/10.1046/j.1365-2958.2003.03723.x
Kuroda M, Kimizu M, Mikami C (2010) A simple set of plasmids for the production of transgenic plants. Biosci Biotechnol Biochem 74(11):2348–2351. https://doi.org/10.1271/bbb.100465
Li Z, Xing A, Moon BP, McCardell RP, Mills K, Falco SC (2009) Site-specific integration of transgenes in soybean via recombinase-mediated DNA cassette exchange. Plant Physiol 151(3):1087–1095. https://doi.org/10.1104/pp.109.137612
Li Z, Moon BP, Xing A, Liu Z, McCardell RP, Damude HG, Falco SC (2010) Stacking multiple transgenes at a selected genomic site via repeated recombinase-mediated dna cassette exchanges. Plant Physiol 154(2):622–631. https://doi.org/10.1104/pp.110.160093
Li R, Han Z, Hou L, Kaur G, Yin Q, Ow DW (2016) Method for biolistic site-specific integration in plants catalyzed by Bxb1 integrase. Methods Mol Biol 1469:15–30. https://doi.org/10.1007/978-1-4939-4931-1_2
Liu Y, Chen Y (2007) High-efficiency thermal asymmetric interlaced PCR for amplification of unknown flanking sequences. Biotechniques 43(5):649–656. https://doi.org/10.2144/000112601
Li Y, Li R, Han Z, Wang H, Zhou S, Li Y, Wang Y, Qi J, Ow DW (2022) Recombinase-mediated gene stacking in cotton. Plant Physiol kiac005. https://doi.org/10.1093/plphys/kiac005
Nishizawa K, Kita Y, Kitayama M, Ishimoto M (2006) A red fluorescent protein, DsRed2, as a visual reporter for transient expression and stable transformation in soybean. Plant Cell Rep 25(12):1355–1361. https://doi.org/10.1007/s00299-006-0210-x
Pathak B, Srivastava V (2020) Recombinase-mediated integration of a multigene cassette in rice leads to stable expression and inheritance of the stacked locus. Plant Direct 4(7):e00236. https://doi.org/10.1002/pld3.236
Rech EL, Vianna GR, Aragao FJ (2008) High-efficiency transformation by biolistics of soybean, common bean and cotton transgenic plants. Nat Protoc 3(3):410–418. https://doi.org/10.1038/nprot.2008.9
Tzafrir I, Torbert KA, Lockhart BEL, Somers DA, Olszewski NE (1998) The sugarcane bacilliform badnavirus promoter is active in both monocots and dicots. Plant Mol Biol 38(3):347–356. https://doi.org/10.1023/A:1006075415686
Zhang Z, Finer JJ (2014) Soybean actin, heat shock protein, and ribosomal protein promoters direct tissue-specific transgene expression in transgenic soybean. In Vitro Cell Dev Biol-Plant 51(1):9–18. https://doi.org/10.1007/s11627-014-9656-7
Zhang N, McHale LK, Finer JJ (2015) Isolation and characterization of “GmScream” promoters that regulate highly expressing soybean (Glycine max Merr.) genes. Plant Sci 241:189–198. https://doi.org/10.1016/j.plantsci.2015.10.010
Acknowledgements
We thank Kan Wang for providing pTF101.1, Yongqing Li for the pZH116-AHAS and Qian Yin for pYQ78.
Funding
This work was supported by the National Key R&D Project (Grant No. 2016YFD0101904) from the Ministry of Science and Technology of China, the Key Research Program of Frontier Sciences CAS, China (Grant No. QYZDY-SSW-SMC010).
Author information
Authors and Affiliations
Contributions
L.J., Z.H., D.C. and X.Z. conducted the experiments; L.J., R.L., Z.H. and D.W.O. designed the experiments; L.J. and D.W.O. wrote the paper.
Corresponding authors
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Ethical approval
The authors declare that the experiments comply with the current laws of the country in which they were performed.
Additional information
Communicated by Ian D. Godwin.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Jiang, L., Li, R., Han, Z. et al. Target lines for recombinase-mediated gene stacking in soybean. Theor Appl Genet 135, 1163–1175 (2022). https://doi.org/10.1007/s00122-021-04015-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-021-04015-6