
Abstract
Key message
The genetic architecture of resistance to Cercospora janseana was examined, and a single resistance locus was identified. A SNP marker was identified and validated for utilization in U.S. breeding germplasm
Abstract
Cercospora janseana (Racib.) is a fungal pathogen that causes narrow brown leaf spot (NBLS) in rice. Although NBLS is a major disease in the southern United States and variation in resistance among U.S. rice germplasm exists, little is known about the genetic architecture underlying the trait. In this study, a recombinant inbred line population was evaluated for NBLS resistance under natural disease infestation in the field across three years. A single, large-effect QTL, CRSP-2.1, was identified that explained 81.4% of the phenotypic variation. The QTL was defined to a 532 kb physical interval and 13 single nucleotide polymorphisms (SNPs) were identified across the region to characterize the haplotype diversity present in U.S. rice germplasm. A panel of 387 U.S. rice germplasm was genotyped with the 13 haplotype SNPs and phenotyped over two years for NBLS resistance. Fourteen haplotypes were identified, with six haplotypes accounting for 94% of the panel. The susceptible haplotype from the RIL population was the only susceptible haplotype observed in the U.S. germplasm. A single SNP was identified that distinguished the susceptible haplotype from all resistant haplotypes, explaining 52.7% of the phenotypic variation for NBLS resistance. Pedigree analysis and haplotype characterization of historical germplasm demonstrated that the susceptible haplotype was introduced into Southern U.S. germplasm through the California line L-202 into the Louisiana variety Cypress. Cypress was extensively used as a parent over the last 25 years, resulting in the susceptible CRSP-2.1 allele increasing in frequency from zero to 44% in the modern U.S. germplasm panel.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cercospora janseana (Racib.) is a fungal pathogen that causes narrow brown leaf spot (NBLS) in rice (Oryza sativa L.). Narrow brown leaf spot can infect leaf sheaths, leaf blades, internodes, and sheaths, and the most recognizable lesions are linear, reddish-brown spots that appear on the leaves and sheaths around heading. The pathogen was first reported in 1900 under the name Napicladium janseanum Racib and first mentioned in North America under the name Cercospora oryzae Miyake in 1906. The pathogen was later renamed C. janseana (Racib.) O. Const. (Farr and Rossman 2020)). Under favorable conditions, NBLS can be a major disease in southern U.S. rice production, causing severe leaf necrosis and premature ripening of seeds, reduced yield, and an overall decrease in milling quality (Groth and Hollier 2010).
C. janseana is one of many Cercospora spp. that affect a variety of crops. Some common crops infected by Cercospora spp. include soybean (C. kikuchii, C. cf. flagellaris, C. cf. sigesbeckiae, and C. sojina) (Mian et al. 2008; Soares et al. 2015; Albu et al. 2016; Borges et al. 2018), maize (C. zeae-maydis and C. zeina) (Crous et al. 2006), sugar beet (C. beticola) (Khan et al. 2008), peanut (C. arachidicola) (Woodward et al. 2010), carrot (C. carotae) (Westerveld et al. 2008), and celery (C. api) (Bounds and Hausbeck 2008). The advances made in understanding the genetics of NBLS within rice can potentially have an impact in these other crops.
Narrow brown leaf spot is predominantly a late-season disease in rice, and as inoculum builds up during the growing season, there is increased infection in later-planted rice (Mani et al. 2016, 2017). This accumulation of inoculum is especially detrimental to two common agricultural practices along the U.S. Gulf Coast: second cropping (ratooning) rice and/or crawfish production. Second cropping rice is the practice of producing a rice crop from the regrowth after the first crop is harvested and is typically harvested three months after the first crop is harvested. Crawfish production is a form of aquaculture in which crawfish are stocked into the flooded rice field and ultimately live off the rice stubble that remains after harvest. Rice varieties that are susceptible to NBLS do not hold up in the crawfish habitat and are not good forage sources in the pond ecosystem. Since these production practices take place late in the season, they are more prone to severe NBLS infection, which can lead to significant economic impacts for farmers.
Until its prevalence and severity increased in the early 2000 s, NBLS had not been considered a significant disease in U.S. rice production. A major outbreak in 2006 was attributed to more than 50% of the total acreage being comprised of susceptible varieties, Cheniere and CL131. In addition to these two varieties, several other varieties planted in 2006 were moderately susceptible. Another contributing factor to the epidemic was a higher presence of inoculum due to a large portion of the 2005 crop being ratooned and used for crawfish production (Groth 2011). The presence of NBLS is now routine, and the degree of infection varies from year to year. In 2019, Arkansas reported an increased incidence and severity of the disease which had previously not been a prominent issue, raising even more concern about the disease (Hardke et al. 2019).
The increased prevalence and severity of NBLS has made it a primary breeding target and a key research focus for rice pathologists in the southern United States. Among U.S. rice varieties and germplasm, there is significant variation for NBLS resistance, with some varieties exhibiting resistance. However, there have only been two studies that examined the inheritance of resistance to NBLS. These studies were conducted before molecular markers, and the resistance genes were never localized (Ryker and Jodon 1940; Ryker and Chilton 1942). The objective of this study was to explore the genetic architecture of NBLS resistance in a recombinant inbred line (RIL) population, map detectable large-effect quantitative trait loci (QTL), and deploy a diagnostic DNA marker for breeding applications in U.S. rice germplasm.
Materials and methods
Plant material
The “MY2” RIL population (n = 286) was previously developed as part of the RiceCap project (Oard et al. 2010). The population was developed from a cross between two released U.S. varieties, Cypress and LaGrue. Cypress is moderately susceptible to NBLS and LaGrue is resistant (Linscombe et al. 1993; Moldenhauer et al. 2001).
The U.S. breeding germplasm panel is comprised of 387 lines. This panel represents modern and advanced U.S. breeding germplasm from public breeding programs, as well as historical pedigrees within public U.S. rice breeding programs. It includes historical varieties and advanced breeding lines from Louisiana (131), Arkansas (105), Texas (84), Mississippi (46), California (10), and Missouri (1). The panel represents germplasm from all common U.S. grain [Long (n = 316), Medium (n = 48), Short (n = 4)] and herbicide classes traits [Conventional (n = 247), Clearfield (n = 132), and Provisia (n = 7)].
The validation populations, Multi-Parent A (MPA) and Multi-Parent B (MPB), were developed from elite U.S. rice varieties and advanced to the F3:F4 generation through single seed descent. The MPA population was developed from the cross CL111 x Roy J, and the MPB population was developed from the cross CL153 x LaKast. Each population consisted of 300 F3:F4 RILs. A subset of 70 lines from each population was identified as fixed at the CRSP-2.1 locus, with 35 lines fixed for the susceptible allele and 35 lines with the resistant allele. This material was rated for NBLS resistance under field conditions in 2019 and used for validating the CRSP-2.1 locus. Additionally, an expanded subset of RILs fixed at the CRSP-2.1 locus consisting of 95 and 76 lines from the MPA and MPB populations, respectively, were evaluated for NBLS resistance under field conditions in 2020.
Phenotyping methods
Field evaluations for NBLS resistance were conducted at the H. Rouse Caffey Rice Research Station (HRCRRS) near Crowley, LA. All materials were planted with a Hege seed drill, except the MPA and MPB materials in 2020, which were planted with an Almaco plot planter. Standard agronomic practices were conducted according to the Louisiana Rice Production Handbook (Louisiana Rice Production Handbook 2014). The MY2 RIL population was evaluated for NBLS resistance and agronomic traits in 2016, 2017, and 2018 and each rep consisted of a 1.8 m row. The germplasm panel was planted and evaluated for NBLS resistance and agronomic traits in 2017 and 2019 and each rep consisted of a 1.8 m row. The validation populations MPA and MPB were evaluated for NBLS using two row plots (0.4 m × 1.8 m) in 2019 and in 7 row plots (1.7 m × 4.4 m) in 2020. NBLS was evaluated in the primary, main crop in 2019 and 2020, as well as the ratoon crop in 2020.
Days to 50% heading (DTH) was calculated from the number of days between emergence and 50% panicle heading. Disease ratings were taken when natural disease pressure was most prevalent. The disease was scored from 0 to 9 based on number and size of lesions on the leaf blades; 0 = no lesions, 1 = pin-head lesions, 2 = dark brown specks that are 0.5 – 1.0 mm in diameter, 3 = linear reddish-brown spots that are 1–2 mm in length, 4 = ≤ 15 lesions per leaf that are 3–4 mm in length, 5 = ≤ 25 lesions per leaf that are 5 –6mm in length, 6 = 25 to 50 lesions per leaf that are 7–10 mm in length, 7 = < 75 lesions per leaf that are 10 –20 mm in length, 8 = 75 to 100 lesions per leaf with less than 25% of leaf area killed by overlapping lesions, and 9 = > 100 lesions per leaf with more than 25% of leaf area killed by overlapping lesions (Groth et al. 1993).
KASP genotyping and assay development
Single nucleotide polymorphism (SNP) genotyping was conducted at the HRCRRS using Kompetitive Allele Specific PCR (KASP) genotyping chemistry and the LGC SNPline genotyping platform (LGC Group 2016). Genome-wide SNP markers were selected from a set of 2000 KASP assays developed by LGC genomics (LGC Group 2016). Additional SNPs in the QTL region were subsequently identified from the International Rice Research Instituted (IRRI) Rice SNP-Seek Database version IRGSPv1 (Alexandrov et al. 2015; Mansueto et al. 2017) and the 7 K Illumina C7AIR SNP array (Morales et al. 2020). Genotyping of the MY2 population was conducted on single plants (F10) that were re-derived from single panicles in the 2016 field trial. Genotyping of MPA and MPB RIL populations was conducted on F3 plants. Leaf tissue was sampled from the flag leaf of each individual panicle for DNA extraction. DNA extraction of the MY2 population and the U.S. breeding germplasm panel was conducted using a modified CTAB method (Khan et al. 2013), and KASP assays were designed by LGC genomics (LGC Group 2016).
Quantitative trait loci mapping and statistical analysis
Initial QTL mapping of the MY2 population was conducted in 2016 using 79 markers distributed across the whole genome. Single-marker analysis of variance was performed using JMP Pro 14 (SAS Institute Inc. 2018). Upon identifying the CRSP-2.1 QTL, an additional 36 markers were added to the map. Twenty-four SNPs were added to fill gaps across the genome, and 12 SNPs were added to increase resolution of the QTL region. A linkage map was constructed using 115 markers, anchored by base pair (bp) position (IRGSP v1), and QTL mapping was conducted using IciMapping software version 4.1 (Meng et al. 2015, Sasaki, T., 2005). QTL mapping was conducted with the inclusive composite interval mapping function for additive effects (ICIM-ADD). Genetic distances were determined using the Kosambi mapping function, and the LOD threshold was calculated based on 1000 permutations with a type 1 error of 0.05. Heritability was calculated on an entry mean basis using the following equation:
where σg2 represents the genotypic variance, σe2 represents the error variance, σge2 represents the genotype x environment variance, r represents the number of replicates, and e represents the number of environments. A Student T test using least-squares means was performed in JMP Pro 14 (SAS Institute Inc. 2018) to determine significant differences between haplotype classes. The variance component analysis for MY2 and NTP NBLS score was conducted with a mixed-linear model in ASReml (Butler et al. 2017), where genotype, year, genotype year interaction, and replication nested in year were treated as random effects.
Haplotype characterization of U.S. germplasm
To characterize the haplotype diversity of the QTL region across the U.S. germplasm panel, a set of 25 SNPs was selected from the Illumina C7AIR array based on their minor allele frequency within U.S. germplasm. Thirteen of these SNPs were polymorphic between the MY2 RIL population parents. These 13 SNPs were screened as KASP assays on the U.S. germplasm panel. Haplotypes were defined within the panel based on the SNP profile across the 13 SNPs. Lines with the same SNP profile across the target region were assigned the same haplotype ID. Haplotypes were designated with “Hap” ID. The average NBLS resistance rating of the lines in a given haplotype class was used to assign a phenotypic value to each haplotype. To help ensure a reasonable representation of each haplotype class, only haplotype classes with 10 or more lines were used for phenotypic associations. Analysis of variance was performed to determine if statistical differences in phenotypes were present across haplotype classes, and a Student T test was conducted to determine statistical differences between specific haplotype classes. For each line, NBLS ratings were based on the average rating across two years (2017 and 2019).
Results
Phenotypic evaluation of the MY2 population
The MY2 RIL population was phenotyped for NBLS resistance under field conditions with natural disease pressure over three years (2016–2018) at the HRCRRS near Crowley, LA. The population consisted of 286 RILs derived from a cross between the variety LaGrue x Cypress (Oard et al. 2010). Phenotypic evaluation was based on a 0–9 visual rating scale, with 0 being the most resistant and 9 being the most susceptible (Groth et al. 1993). Based on the three-year average NBLS rating of each RIL, the population ranged from resistant (0) to moderately susceptible (4.56) (Fig. 1a). The overall mean of the population was 2.06 with a standard deviation of 1.38. The distribution of the population was bimodal, with a normal distribution at a mode of 3.5 and a half normal distribution at a mode of zero. The Cypress parent exhibited a moderately susceptible rating of 3.27, while the LaGrue parent was resistant with an average score of 0.42. Narrow brown leaf spot ratings were highly correlated across all years, with the correlation coefficient (r) ranging from 0.75 to 0.85 between any two years. The entry mean basis heritability across all three years was 0.90. The percent variance explained across all three years was highest for genotype at 60%, followed by genotype x year interaction at 9.4%, year at 5.9%, and rep within year at 0.3%, the error component was 24.4%.

a Narrow brown leaf spot rating distribution based on a three-year average in MY2 RIL population. The population ranged from resistant (0) to moderately susceptible (4.56). The resistant parent, LaGrue, averaged a 0.42 rating, and the moderately susceptible parent, Cypress, averaged a 3.27 rating. b Days to 50% Heading distribution based on a three-year average in MY2 RIL population. The population ranged from 75.69 to 95.11 days. The overall means of the population was 85.11. Cypress was the earlier heading parent at 87.2 days, and LaGrue headed later at 89.8 days
Days to 50% heading exhibited a normal distribution, ranging from 76 to 95 based on the three-year average. The overall mean of the population was 85 with a standard deviation of 3.72. Cypress parent headed at 87 days, and LaGrue headed at 90 days (Fig. 1b). Days to 50% heading values were correlated across all years, ranging from 0.53 to 0.60 between any two years. A negative correlation (r = − 0.39) was observed between heading and susceptibility to NBLS, i.e., earlier heading was associated with increased susceptibility.
QTL Mapping and genetic map construction
The population was initially genotyped with 79 SNPs across the genome to determine if any large-effect loci were present for NBLS resistance. Single marker analysis using the 2016 phenotype data identified two SNPs (SNP271 and SNP78) on Chromosome 2 that were highly significant (p < 0.001). SNP271 (24,042,628 IRGSP V.1) and SNP78 (25,795,772 IRGSP V.1) explained 45% and 67% of the phenotypic variation, respectively. An additional 36 SNPs were subsequently added to fill gaps across the genome and provide increased marker density across the QTL region.
A linkage map, consisting of 115 SNPs, was constructed using IciMapping mapping software (Meng et al. 2015). The total size of the linkage map was 1596.45 cM, and the average gap between markers was 16.4 cM (2.85 Mb). Composite interval mapping identified a total of 10 QTLs for NBLS and DTH (Table 1). A single QTL, CRSP-2.1, was detected for NBLS resistance on Chromosome 2, with the LaGrue parent contributing the resistant allele. CRSP-2.1 mapped between SNP790 (24,477,977) and SNP792 (24,693,023) and explained 81.4% of the phenotypic variance (PVE) (LOD = 116.9) across all three years (Table 1). CRSP-2.1 was detected within each year individually, explaining between 67.1 and 72.7% of the phenotypic variance within each year (Supplemental Table 1). The average disease rating of RILs with the resistant allele was 0.57 and ranged from 0 to 2.89. Whereas the average rating of RILs with the susceptible allele was 3.16 and ranged from 1.36 to 4.56 with a normal distribution. The normal distribution observed among the RILs with the susceptible allele suggests that there is underlying quantitative resistance within the RIL population in the absence of the large-effect resistance allele.
Nine QTL were detected across six chromosomes for DTH, ranging from 2.47 to 13.52% of the phenotypic variance explained across all three years. Two QTL (MY2dth-8.1 and MY2dth-10.1) were detected for each individual year and for combined analysis of all three years. The DTH QTL, MY2dth-2.2 (LOD = 12.5, PVE = 8.4%), was detected at the same region on Chromosome 2 as CRSP-2.1, with the Cypress allele conferring early heading (Table 1). MY2dth-2.2 was significant within 2016 and 2017 but not in 2018 when analysis was conducted on a within year basis (Supplemental Table 1). The parents of the MY2 populations only differed by three days to heading, however, the range of DTH across the population was 76 days to 96 days, suggesting that DTH trait is highly quantitative in this population. The quantitative nature of DTH in the MY2 population was also demonstrated by the detection of multiple QTLs, each with a relatively small effect.
Relationship between early heading and nbls resistance
The negative correlation between early DTH and NBLS resistance (r = − 0.39) and the presence of overlapping QTLs (CRSP-2.1 and MY2dth-2.2) for both the traits was further investigated. To explore this, the RIL population was divided into two groups based on the allelic state at the CRSP-2.1 region; 159 RILs with the Cypress allele were grouped and the 115 RILs with the LaGrue allele combined as a separate group. Twelve RILs were not considered in this analysis due to recombination within the CRSP-2.1 region. A correlation between DTH and NBLS was still observed within each group; however, the correlation value was reduced. The RILs with the Cypress allele had an average DTH of 84 and an average NBLS score of 3.16. The correlation between the traits was r = − 0.20. The RILs with the LaGrue allele had an average DTH of 87 and an average NBLS score of 0.59. The correlation between the traits was r = − 0.22 (Supplemental Fig. 1).
We subsequently divided the RIL population into two groups based on DTH. One group contained the 100 earliest RILs, and the other group contained the 100 latest RILs. A single marker analysis was conducted within each group to determine if the CRSP-2.1 QTL was significant within each group. SNP861 explained 82% of the phenotypic variation across the entire RIL population, 81% within the late heading RIL group, and 77% within the early heading RIL group (Supplemental Fig. 2). These observations demonstrate that the correlation between DTH and NBLS resistance is due to the co-localization of a DTH QTL (MY2dth-2.2) at the same region as the CRSP-2.1 QTL and that the observed resistance is genetic and not explained by disease avoidance.

a Distribution of Narrow Brown Leaf Spot ratings across the U.S. rice germplasm panel based on the average of two years. The average of the germplasm panel was 1.7 and ranged from resistant (0) to moderately susceptible (5). b Distribution of Days to 50% Heading across the U.S. rice germplasm panel based on the average of two years. The overall mean of the germplasm panel was 91.5 with a range of 72.2 to 115 days
High-resolution mapping of CRSP-2.1
Twelve RILs were identified as recombinants within the CRSP-2.1 QTL region (Chr. 2: 24,128,810 – 24,768,787 bp). To further define the QTL region, these recombinant RILs were phenotyped with additional replicates in 2017 and in 2018, and the RILs were grouped into recombinant classes based on their recombination breakpoints (Table 2). Recombinant inbred lines 101,267, 101,004, and 101,147 were in recombinant class 1, with recombination between SNP858 and SNP859. Recombinant inbred lines 101,267 and 101,004 had susceptible phenotypes, and the LaGrue allele at SNP858 and Cypress alleles from SNP859 to SNP877 (Table 2). Recombinant inbred line 101,147 had a resistant phenotype, and the Cypress allele at SNP858 and LaGrue alleles from SNP859 to SNP877. These observations suggest the gene is distal to SNP858. Recombinant class 2 included eight RILs that were recombinant between SNP862 and SNP868, with LaGrue alleles from SNP858 to SNP862 and Cypress alleles from SNP868 to SNP877. All RILs in this class were resistant, indicating that the gene is upstream of SNP868. Recombinant class 3 contained RIL 101,124, which was susceptible and contained Cypress alleles across the region, except at SNP875 and SNP877. These observations indicate that the causal gene underlying the CPRS-2.1 QTL is within a 532 kb region between SNP858 (24,128,810) and SNP868 (24,661,686) (Table 2). This region contains 77 genes, with the primary gene classes including 21 retrotransposons, 21 expressed proteins, and a cluster of six receptor kinase proteins and leucine-rich repeat proteins that are common classes of plant resistance genes (Supplemental Table 2).
Characterization of CRSP-2.1 QTL across U.S. Germplasm
To investigate the effect of CRSP-2.1 across public U.S. rice germplasm, a U.S. rice breeding germplasm panel, consisting of 387 lines, was evaluated for NBLS resistance and DTH data over two years. Narrow brown leaf spot ratings ranged from 0 (resistant) to 5 (susceptible), with a mean of 1.7 and a standard deviation of 1.1 (Fig. 2a). The correlation coefficient (r) for NBLS across years was 0.67. The percent variance explained across both years was highest for genotype at 36.4%, followed by year at 28.1%, genotype x year interaction at 8.3%, and rep within year at 0.6%, the error component was 28.7%. The mean of the panel for DTH was 92 days, with a standard deviation of 5.3, and a range of 72 to 115 (Fig. 2b). The correlation coefficient (r) for DTH across years was 0.75.
A set of 13 SNPs was identified across the 532 kb CRSP-2.1 QTL region that were polymorphic between the parents of the MY2 mapping population. These SNPs were screened as KASP assays on the public U.S. breeding germplasm panel and used to construct extended haplotypes across the CRSP-2.1 region. Fourteen unique haplotypes were identified in the panel (Table 3), with six haplotypes (Hap 1–6) accounting for 94% of the lines. The remaining eight haplotypes contained between one and six lines (Supplemental Table 3). A total of 336 lines had NBLS phenotype data available and were used to associate the NBLS phenotype for each haplotype class. Two-hundred and forty-two lines were phenotyped across both years (2017 and 2019), and 96 lines were phenotyped in 2017 alone. Phenotypic comparisons were made between haplotype classes that were present at a frequency of 0.03 or greater.
Haplotype 1 (Hap 1) was the most prevalent haplotype in the panel (44%, n = 170) and included the moderately susceptible RIL parent, Cypress (Table 3). Haplotype 1 was the only susceptible haplotype class and was significantly different than all other haplotypes (p < 0.0001) which showed NBLS resistance (Fig. 3). Haplotype 1 was more common within Clearfield herbicide-resistant germplasm (63%) than in non-Clearfield germplasm (41%). The average NBLS rating for lines with Hap 1 was 2.5 with a normal distribution (SD = 0.89) ranging from 0.2 (resistant) to 5 (moderately susceptible). Similar to the observation within the RIL population, this is consistent with underlying quantitative resistance in the genetic background of lines carrying the susceptible allele at the CRSP-2.1 locus. All other haplotypes, which were represented in 13 or more of the phenotyped lines, exhibited NBLS resistance, with average NBLS ratings ranging from 0.61 to 1.13 (Table 3). Based on these observations, we concluded that the only CRSP-2.1 susceptible allele is carried by Hap 1 and that all other haplotypes in the public U.S. rice germplasm panel confer resistance to NBLS. One genotype within the resistant haplotype 2 was observed as susceptible in the field ratings, we suspect that this is most likely explained by phenotyping error in the field or a mix-up during planting, leaf sampling, or DNA extraction.

Comparison of Narrow Brown Leaf Spot phenotypes across CRSP-2.1 haplotype groups that appear at a frequency of at least 0.03 in the U.S. rice germplasm panel. The number of lines in each haplotype group is indicated in parenthesis. The susceptible haplotype, Hap 1, present in the susceptible RIL parent Cypress was present in 44% of the germplasm panel. All other haplotype groups conferred resistant ratings with average ratings ranging from 0.61 to 1.13
Identification and validation of a single trait snp marker
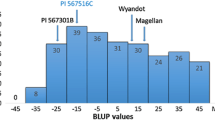
The SNP (SNP861) located at 24,386,096 bp perfectly differentiated the susceptible Hap 1 from all other haplotypes in the public U.S. rice germplasm panel and explained 52.7% of the phenotypic variation for NBLS resistance within the panel across both years (Table 3, Fig. 4). SNP861 produced consistent and reliable results across five independent genotyping experiments using the 387 U.S. lines. Out of a total of 1,920 data points generated, there were 17 missing data points (0.8%) and 4 cases (0.2%) where homozygotes were misidentified as heterozygous allele calls. SNP861 produced clear and distinct clusters when evaluated on segregating materials that contained all three genotypic classes, demonstrating that this SNP can easily be scored and is amenable to automated scoring on a computer (Supplemental Fig. 5). These validation steps demonstrate that SNP861 is informative and accurate across the target germplasm, and the reported KASP assay produces high-throughput, high-quality data necessary for large-scale breeding applications.

Phenotypic comparison based on the allele calls for SNP 861 across the U.S. rice germplasm panel. SNP 861 explained 52.7% of the phenotypic variation for Narrow Brown Leaf Spot resistance based on the two-year average and perfectly distinguished the susceptible haplotype (Hap 1) from all other resistant haplotypes
Validation of CRSP-2.1 locus across multiple populations
To validate the effect of the CRSP-2.1 locus across populations, two additional bi-parental populations segregating at the CRSP-2.1 locus were rated for NBLS resistance and genotyped at the CRPS-2.1 QTL region. The MPA population was developed from the cross CL111 x Roy J and segregated for the susceptible CRSP-2.1 Hap 1 allele and the resistant Hap 3 allele (Supplemental Table 3). The MPB population was developed from the cross CL153 x LaKast and segregated for the susceptible Hap 1 allele the resistant Hap 2 allele (Supplemental Table 3).
Within both populations, 35 lines that were homozygous for the susceptible CRSP-2.1 allele and 35 lines that were homozygous for the resistant CRSP-2.1 allele were identified and phenotyped for NBLS resistance in 2019. The CRSP-2.1 allele was highly significant in both populations (p < 0.0001). Within the MPA population, the CRSP-2.1 locus explained 74% of the phenotypic variation and RILs with the resistant allele had an average NBLS rating of 0.09, compared to 2.69 for RILs carrying the susceptible allele (Supplemental Fig 4a). Within the MPB population, the CRSP-2.1 locus explained 65% of the phenotypic variation, and RILs with the resistant allele had an average NBLS rating of 0.6, compared to 3.46 for RILs with the susceptible allele (Supplemental Fig 4b).
The MPA and MPB populations were also phenotyped for NBLS in 2020 in both the primary and ratoon crops. A subset of RILs consisting of 95 and 76 lines from the MPA and MPB populations, respectively, were homozygous at the CRSP-2.1 locus and used for evaluations. The CRSP-2.1 locus was highly significant in both populations and at both evaluation time points (p < 0.0001). In the MPA population, the CRSP-2.1 locus accounted for 29% and 38% of the phenotypic variation for NBLS resistance during the primary and ratoon crops, respectively. Homozygous lines harboring the resistant allele had average NBLS scores of 0.13 in the primary crop and 1.09 in the ratoon crop. Lines with the susceptible allele had average NBLS scores of 0.92 and 2.51 in the primary and ratoon crops, respectively (Supplemental Fig4c). In the MPB population, the CRSP-2.1 locus accounted for 60% of the phenotypic variation in the primary and 25% in the ratoon crop. Homozygous lines with the resistant allele had average NBLS scores of 0.73 and 1.90 in the primary and ratoon crops, respectively. Lines with the susceptible allele had average NBLS scores of 2.59 in the primary crop and 3.2 in the ratoon crop. (Supplemental Fig 4d).
Source of the Susceptible CRSP-2.1 Allele
Haplotype analysis of the CRSP-2.1 region identified a single susceptible haplotype and multiple resistant haplotypes across the public U.S. rice germplasm panel. The germplasm panel consisted of 170 lines with the susceptible Hap 1 allele. These lines can be classified into five groups based on pedigree relationships (Supplemental Table 3), Supplemental Fig. 3a and b). Group A consists of 15 lines, all of which trace their pedigrees back to the California variety L-201. Group B consisted of 133 lines that all trace their pedigrees back to Cypress, with the pedigree of Cypress tracing back to L-201 via the line L-202 (Tseng et al. 1984). Group C consists of 11 lines whose pedigrees trace back to the Louisiana variety Cheniere, whose pedigree also traces back to L-201 via the line L-202. Group D consists of six lines, including the Louisiana variety Lacassine and five lines developed from crosses containing Lacassine as a parent (Linscombe et al. 1992) (Supplemental Fig. 3b). Haplotype data in this study classify Lacassine as having the susceptible Hap 1, which is inconsistent with its pedigree, Newbonnet x Lemont, in which both parents contain a resistant CRSP-2.1 allele. Group E includes seven lines from Texas and Mississippi that represent four different pedigrees. These pedigrees all include exotic, introduced parents that could be additional sources of the susceptible Hap 1 allele. Overall, 158 lines (93%) that contain the susceptible Hap 1 allele can be traced directly to L-201 by pedigree, providing very strong evidence that L-201 is the original source of the susceptible allele in southern U.S. rice germplasm.
Discussion
To our knowledge, this is the first study examining the genetic architecture of narrow brown leaf spot resistance in rice and is the first report of a major gene underlying native variation. In this study, a novel NBLS resistance QTL, CRSP-2.1, was mapped to a 532 kb region and explained 81.4% of the phenotypic variation in NBLS resistance in the MY2 bi-parental RIL population and 52.7% of the variation across U.S. breeding germplasm. This region contains 77 genes, with the primary gene classes including 21 retrotransposons, 21 expressed proteins, two leucine-rich repeat (LRR) family proteins, and four receptor kinase proteins (Supplemental Table 2). The LRR and receptor kinase genes are clustered together in an 81 kb region (24,293,135 bp-24,374,496 bp) and are the strongest candidate genes underlying CRSP-2.1. Leucine-rich repeat genes belong to the large class of resistance genes and confer resistance to several races of rice blast (Magnaporthe oryzae) (Zhang et al. 2015; Yan et al. 2017) and bacterial blight in rice (Xanthamonas oryzae) (Lin et al. 1996). In addition to rice pathogens, LRR genes confer resistance to several pathogens in other cereal crops, such as powdery mildew (Blumeria graminis) in barley (Shirasu et al. 1999; Zhou et al. 2001), leaf rust (Puccinia sorghi) in maize (Collins et al. 1999) and leaf rust (Puccinia triticina) in wheat (Spielmeyer et al. 2000).
Interestingly, it appears that all the rice germplasm in the southern United States contained a resistant allele at CRSP-2.1 prior to the 1990s. The susceptible allele was introduced from California germplasm and experienced rapid proliferation driven by the widespread use of the variety Cypress as a breeding parent. Cypress was widely grown across the southern U.S. in the 1990s and has been used extensively as a parent in breeding programs in the United States. Of the 170 lines containing the susceptible allele in the germplasm panel, 133 lines include Cypress in their pedigree. Fifty of these 133 lines trace directly back to Cypress, and 82/133 lines were developed from CL161. CL161 is a Clearfield herbicide-resistant line that was developed from EMS mutagenesis of Cypress (Linscombe and Sha 2002). The Clearfield production system is widely used across the southern United States and globally, accounting for as much as 51% of the acres in Louisiana in 2019 (Harrell 2019). Thus, the pedigrees of all U.S. Clearfield rice germplasm traces back to Cypress. The Clearfield gene is also located on Chromosome 2 (18.2 Mb) and is loosely linked to the CRSP-2.1 locus (24.5 Mb). The pedigree relationship linking Clearfield germplasm to the susceptible CRSP-2.1 allele through Cypress, and the physical linkage of the Clearfield locus and CRSP-2.1 explains the increased frequency of the susceptible Hap 1 allele within Clearfield germplasm. Sixty-three percent of Clearfield germplasm contains the susceptible CRSP-2.1 allele compared to only 41% of non-Clearfield germplasm. Group D of the haplotype analysis includes lines that trace their parentage back to Lacassine. The pedigree of Lacassine (Newbonnet x Lemont) includes two parents that both carry Hap 2 with resistant CRSP-2.1 alleles, suggesting that either outcrossing or a seed mix-up occurred during the development of Lacassine which resulted in the inheritance of the susceptible Hap 1 (Supplemental Table 3). Lacassine was released in 1991 and was under development at the same time as Cypress (released 1992), during a period when L-202 was commonly used as a breeding parent (Linscombe, Personal Communication).
A major outbreak of NBLS occurred in 2006 and was, in part, attributed to the planting of susceptible varieties (Groth 2011). Over 90% of the total rice acreage in 2006 consisted of varieties containing Hap 1 with the susceptible CRSP-2.1 allele. The most prevalent varieties planted in 2006 were Cheniere (25.10%), Cocodrie (24.63%), CL131 (23.98%), CL161 (11.14%), and Cypress (5.34%) (Harrell 2019). All these varieties carry the Hap 1 and have L-202 in their pedigrees. The frequency of the susceptible CRSP-2.1 allele continues to be high among varieties planted in the southern U.S.; 80% of acres planted to inbred rice in Louisiana in 2019 carried the susceptible CRSP-2.1 allele (Harrell 2019). This highlights an opportunity to increase productivity and simultaneously reduce fungicide applications by incorporating CRSP-2.1 resistance into new varieties.
A primary objective of this work was to develop a SNP marker to facilitate selection of CRSP-2.1 resistance for use in our applied breeding program. Prior to deploying a trait marker in an applied breeding program there are three major validation stages. First, the locus must be demonstrated to confer the desired phenotype in the target germplasm and environments. In this study, this was accomplished using the three bi-parental populations evaluated over four years. Second, the identified trait marker must be validated across the diversity of the target breeding germplasm. In this study, this was accomplished using a panel of 387 lines that were assembled for the specific purpose of representing the diversity of modern public U.S. rice germplasm. This panel represents all modern varieties, advanced breeding lines, and nearly all parental lines that are likely to be used in variety development programs in the southern US. Thus, we are confident that any trait marker that is informative within this panel will also be informative within our breeding program and populations. The last key consideration is the technical performance of the trait marker. A trait marker ideally consists of a single marker that unequivocally differentiates the desirable and the undesirable haplotypes. The marker assay should be amenable to high-throughput and low-cost genotyping to facilitate the processing of large numbers of samples in a time-sensitive way to meet the needs of many applied breeding applications. In this study, we characterized the diversity of CRSP-2.1 haplotypes present in U.S. germplasm and determined there was one susceptible haplotype and multiple resistant haplotypes. We then identified a single SNP (SNP861) that perfectly distinguished the susceptible Hap 1 from all resistant haplotypes at the CRSP-2.1 QTL. SNP861 provided a high rate of data return and reproducible results when evaluated as a KASP assay. Additionally, the KASP assay produced clear and distinct clusters that made it easy to differentiate the three genotype classes in a segregating population, making it amenable to automated scoring. Based on the successful validation of each of these steps, this SNP can be reliably and effectively deployed in U.S. rice breeding programs and has been successfully utilized in our applied breeding program over the last two years.
In summary, this work demonstrates that NBLS resistance is controlled by a single major gene. CRSP-2.1 is the first NBLS resistance locus reported in rice and is demonstrated to confer resistance across a panel of 387 public U.S. rice lines as well as in three independent bi-parental populations. Future research efforts will focus on identifying the gene(s) underlying resistance at the CRSP-2.1 QTL and understanding how it functions to mediate plant response to NBLS.
Data availability
Datasets supporting the results are included as supplemental files.
Abbreviations
- Hap:
-
Haplotype
- SNP:
-
Single nucleotide polymorphism
- Bp:
-
Base pair
- QTL:
-
Quantitative Trait Loci
- RIL:
-
Recombinant Inbred Line
- CRSP:
-
Cercospora janseana
- DTH:
-
Days to 50% Heading
- NBLS:
-
Narrow brown leaf spot
References
Albu S, Schneider RW, Price PP, Doyle VP (2016) Cercospora cf. flagellaris and Cercospora cf. sigesbeckiae are associated with Cercospora leaf blight and purple seed stain on soybean in North America. Phytopathology 106(11):1376–1385. https://doi.org/10.1094/PHYTO-12-15-0332-R
Alexandrov N, Tai S, Wang W, Mansueto L, Palis K et al (2015) SNP-Seek database of SNPs derived from 3000 rice genomes. Nucleic Acids Res 43(D1):D1023–D1027. https://doi.org/10.1093/nar/gku1039
Borges LL, Ferreira TF, Lana MG, Caliman ID, Bluhm BH et al (2018) Multi-host species of Cercospora are associated with Cercospora leaf blight and purple seed stain of soybean. Trop Plant Pathol 43(2):170–177. https://doi.org/10.1007/s40858-017-0205-x
Bounds RS, Hausbeck MK (2008) Evaluation of disease thresholds and predictors for managing late blight in celery. Plant Dis 92(3):438–444. https://doi.org/10.1094/pdis-92-3-0438
Butler D G, Cullis BR, Gilmour A R, Gogel BG, Thompson R. 2017. ASReml-R Reference Manual Version 4. VSN International Ltd, Hemel Hempstead, HP1 1ES, UK.
Collins N, Drake J, Ayliffe M, Sun Q, Ellis J et al (1999) Molecular characterization of the maize Rp1-D rust resistance haplotype and its mutants. Plant Cell 11(7):1365–1376. https://doi.org/10.1105/tpc.11.7.1365
Crous PW, Groenewald JZ, Groenewald M, Caldwell P, Braun U et al (2006) Species of Cercospora associated with grey leaf spot of maize. Stud Mycol 55:189–197
Farr DF, Rossman Fungal Databases AY, U.S. National Fungus Collections, ARS, USDA. Retrieved December 6, 2020, from https://nt.ars-grin.gov/fungaldatabases/
Groth DE (2011) Why Cercospora, bacterial panicle blight and blast are more severe some years. Rice Res Stn News 8(3):1–7
Groth CA , Hollier. 2010. Narrow brown leaf spot. louisiana plant pathol. Identif. Manag. Ser. (Pub. 3105).
Groth DE, Rush MC, Giesler GG, Hollier CA. 1993. Foliar fungicides for use in the management of rice diseases. Bull. no. 840.
Hardke J, Bateman N, Wamishe Y. 2019. Arkansas Rice Update 08–31–2019.
Harrell D. 2019. Louisiana rice acreage by variety. LSU AgCenter - Louisiana Rice Acreage by Var. https://www.lsuagcenter.com/topics/crops/rice/statistics/rice-varieties (Accessed 21 November 2019).
Khan J, del Rio LE, Nelson R, Rivera-Varas V, Secor GA et al (2008) Survival, dispersal, and primary infection site for cercospora beticola in sugar beet. Plant Dis 92(5):741–745. https://doi.org/10.1094/pdis-92-5-0741
Khan NA, Bedre R, Parco A, Bernaola L, Hale A et al (2013) Identification of cold-responsive genes in energycane for their use in genetic diversity analysis and future functional marker development. Plant Sci 211:122–131. https://doi.org/10.1016/j.plantsci.2013.07.001
LGC Group. 2016. SNPline genotyping automation. https://www.lgcgroup.com/products/genotyping-instruments/snpline/#.XFCv91xKhaQ (Accessed 29 March 2019).
Lin XH, Zhang DP, Xie YF, Gao HP, Zhang Q (1996) Identifying and mapping a new gene for bacterial blight resistance in rice based on RFLP markers. Phytopathology 86(11):1156–1159. https://doi.org/10.1094/Phyto-86-1156
Linscombe SD, Jodari F, Mckenzie KS, Bollich PK, Groth DE et al (1992) Registration of ‘Lacassine’ Rice. Crop Sci 32(2):494. https://doi.org/10.2135/cropsci1992.0011183x003200020044x
Linscombe SD, Jodari F, McKenzie KS, Bollich PK, White LM et al (1993) Registration of ‘Cypress’ Rice. Crop Sci 33(2):355. https://doi.org/10.2135/cropsci1993.0011183X003300020039x
Linscombe S, Sha X. 2002. CL161 Rice variety. rice Stn. released var. 1917–2015. https://www.lsuagcenter.com/profiles/lblack/articles/page1484946379503 (Accessed 20 November 2019).
Louisiana rice production handbook. 2014. LSU AgCenter Louisiana Rice Production Handbook (J. Saichuk, editor).
Mani KK, Hollier CA, Groth DE (2016) Effect of planting date, fungicide timing and cultivar susceptibility on severity of narrow brown leaf spot and yield of rice. Crop Prot 90:186–190. https://doi.org/10.1016/j.cropro.2016.07.029
Mani KK, Hollier CA, Groth DE (2017) Effect of cultivar susceptibility and planting date on narrow brown leaf spot progression in rice. Crop Prot 102:88–93. https://doi.org/10.1016/j.cropro.2017.08.004
Mansueto L, Fuentes RR, Borja FN, Detras J, Abrio-Santos JM et al (2017) Rice SNP-seek database update: New SNPs, indels, and queries. Nucleic Acids Res 45(D1):D1075–D1081. https://doi.org/10.1093/nar/gkw1135
Meng L, Li H, Zhang L, Wang J (2015) QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J 3(3):269–283. https://doi.org/10.1016/j.cj.2015.01.001
Mian MAR, Missaoui AM, Walker DR, Phillips DV, Boerma HR (2008) Frogeye leaf spot of soybean: a review and proposed race designations for isolates of Cercospora sojina Hara. Crop Sci 48(1):14–24. https://doi.org/10.2135/cropsci2007.08.0432
Moldenhauer K, Gravois KA, Lee FN, Norman RJ, Bernhardt JL, et al. 2001. Registration of “LaGrue” Rice. Crop Sci. 41(Cv): 1991–2013.
Morales KY, Singh N, Perez FA, Ignacio JC, Thapa R et al (2020) An improved 7K SNP array, the C7AIR, provides a wealth of validated SNP markers for rice breeding and genetics studies. PLoS ONE 15(5):1–14. https://doi.org/10.1371/journal.pone.0232479
Oard JH, Moldenhauer K, Fjellstrom B, Nelson JC, Scheffler B et al (2010) Registration of the MY2 ‘Cypress’/‘LaGrue’ Rice Recombinant Inbred Line Mapping Population. J Plant Regist 4(3):261. https://doi.org/10.3198/jpr2009.11.0668crmp
Ryker T, Chilton S (1942) Inheritance and linkage of factors for resistance to two physiologic races of Cercospora oryzae in rice. J Am Soc Agron 34(9):836–840
Ryker T, Jodon N (1940) Inheritance of resistance to Cercospora oryzae in rice. Phytopathology 30(12):1041–1047
Sasaki T (2005) The map-based sequence of the rice genome. Nature 436:793–800. https://doi.org/10.1038/nature03895
SAS Institute Inc (2018) JMP® Pro 14. Cary, NC
Shirasu K, Lahaye T, Tan MW, Zhou F, Azevedo C, et al. 1999. A novel class of eukaryotic zinc-binding proteins is required for disease resistance signaling in barley and development in C. elegans. Cell 99(4): 355–366. doi: https://doi.org/10.1016/S0092-8674(00)81522-6.
Soares APG, Guillin EA, Borges LL, Da Silva ACT, De Almeida ÁMR et al (2015) More Cercospora species infect soybeans across the Americas than meets the eye. PLoS ONE 10(8):1–20. https://doi.org/10.1371/journal.pone.0133495
Spielmeyer W, Huang L, Bariana H, Laroche A, Gill BS et al (2000) NBS-LRR sequence family is associated with leaf and stripe rust resistance on the end of homoeologous chromosome group 1S of wheat. Theor Appl Genet 101(7):1139–1144
Tseng ST, Carnahan HL, Johnson CW, Oster JJ, Hill JE et al (1984) Registration of “L-202” Rice. Crop Sci 24(6):1213–1214. https://doi.org/10.2135/cropsci1984.0011183X002400060055x
Westerveld SM, McKeown AW, McDonald MR (2008) Relationship between nitrogen application and alternaria leaf blight [Alternaria dauci] and cercospora leaf blight [Cercospora carotae] of carrots. Can J Plant Pathol Can Phytopathol 28(2):341
Woodward JE, Brenneman TB, Kemerait RC Jr, Culbreath AK, S.N. B. (2010) Management of peanut diseases with reduced input fungicide programs in fields with varying levels of disease risk. Crop Prot 20:222–229
Yan L, Bai-Yuan Y, Yun-Liang P, Zhi-Juan J, Yu-Xiang Z et al (2017) Molecular screening of blast resistance genes in rice germplasms resistant to magnaporthe oryzae. Rice Sci 24(1):41–47. https://doi.org/10.1016/j.rsci.2016.07.004
Zhang X, Yang S, Wang J, Jia Y, Huang J et al (2015) A genome-wide survey reveals abundant rice blast R-genes in resistant cultivars. Plant J 84(1):20–28. https://doi.org/10.1111/tpj.12955.A
Zhou F, Kurth J, Wei F, Elliott C, Valè G et al (2001) Cell-autonomous expression of barley Mla1 confers race-specific resistance to the powdery mildew fungus via a Rar1-independent signaling pathway. Plant Cell 13(2):337–350. https://doi.org/10.1105/tpc.13.2.337
Acknowledgements
Jovanny Zabaleta, Department of Pediatrics, LSUHSC-New Orleans, processed the Illumina C7AIR array genotype data. Seed of the MY2 mapping population and other historical germplasm were originally obtained from the USDA-GSOR in Stuttgart, AR. Jennifer Dartez for technical contributions in the SNP genotyping. Valerie Dartez for helpful contributions in the manuscript preparation and proofreading. Diane Wang and Susan McCouch for valuable manuscript suggestions and comments. Karen Bearb, Corey Conner, Gavin Guidry, and Brady Williams for field and project support. Raul Guerra assistance with data analysis and R scripts.
Funding
The research project was supported in part by the Louisiana Rice Research Board, the USDA National Institute of Food and Agriculture, and the US National Science Foundation.
Author information
Authors and Affiliations
Contributions
CKA and ANF designed the experiments, coordinated research activities, analyzed the data, and wrote the manuscript. CKA conducted the field phenotyping, managed seed and materials, and conducted literature review. TC developed the MPA/B populations and analyzed the marker data involving the two populations. BA conducted all genotyping, managed the germplasm panel, and edited the manuscript. DEG. rated the RIL population for NBLS and supervised NBLS phenotyping methods. JKR rated the MPA/B populations and edited the manuscript. SDL coordinated field experiments and contributed to the pedigree analysis.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Consent for publication
Yes, all authors have provided consent for publication.
Additional information
Communicated by Yunbi Xu.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

{kind=link}
Cite this article
Addison, C.K., Angira, B., Cerioli, T. et al. Identification and mapping of a novel resistance gene to the rice pathogen, Cercospora janseana. Theor Appl Genet 134, 2221–2234 (2021). https://doi.org/10.1007/s00122-021-03821-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-021-03821-2