
Abstract
Key message
We introgressed wheatgrass germplasm from the octoploid amphiploid Triticum aestivum× Lophopyrum elongatum into wheat by manipulating the wheat Ph1 gene and discovered and characterized 130 introgression lines harboring single or, in various combinations, complete and recombined L. elongatum chromosomes.
Abstract
Diploid wheatgrass Lophopyrum elongatum (genomes EE) possesses valuable traits for wheat genetics and breeding. We evaluated several strategies for introgression of this germplasm into wheat. To detect it, we developed and validated multiplexed sets of Sequenom MassARRAY single nucleotide polymorphism (SNP) markers, which differentiated disomic and monosomic L. elongatum chromosomes from wheat chromosomes. We identified 130 introgression lines (ILs), which harbored 108 complete and 89 recombined L. elongatum chromosomes. Of the latter, 59 chromosomes were recombined by one or more crossovers and 30 were involved in centromeric (Robertsonian) translocations or were telocentric. To identify wheat chromosomes substituted for or recombined with L. elongatum chromosomes, we genotyped the ILs with the wheat 90-K Infinium SNP array. We found that most of the wheat 90-K probes correctly detected their targets in the L. elongatum genome and showed that some wheat SNPs are ancient and had originated prior to the divergence of the wheat and L. elongatum lineages. Of the 130 ILs, 52% were homozygous for Ph1 deletion and thus are staged to be recombined further. We failed to detect in the L. elongatum genome the 4/5 reciprocal translocation that has been reported in Thinopyrum bessarabicum and several other Triticeae genomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The grass tribe Triticeae comprises over 300 species (Love 1984). Most of them can be hybridized with wheat and represent a valuable reservoir of genes for the improvement of disease and pest resistance, tolerance of adverse environmental conditions, and other traits in wheat.
Commercially relevant species of wheat are polyploid. Durum wheat (Triticum turgidum ssp. durum) is tetraploid (subgenomes AABB), and bread wheat (T. aestivum) is hexaploid (subgenomes AABBDD). Triticum urartu and Aegilops tauschii contributed the wheat A and D subgenomes, respectively (Kihara 1944; McFadden and Sears 1946; Dvorak et al. 1993). The source of the B subgenome is unknown. Its closest relatives are Ae. speltoides (Dvorak and Zhang 1990) and Amblyopyrum muticum (Glémin et al. 2019).
Meiotic recombination in polyploid wheat takes place only between homologous chromosomes; recombination between homoeologous chromosomes is precluded by the expression of the Ph1 gene on chromosome 5B (Okamoto 1957; Riley and Chapman 1958; Sears and Okamoto 1958). The Ph1 gene also precludes recombination between wheat chromosomes and those of wheat relatives and therefore greatly restricts gene introgression from related species into wheat. Since Ph1 is completely dominant, gene introgression in polyploid wheat requires eliminating both copies of the gene, which has usually been accomplished by homozygosity for a Ph1 deletion. The ph1b deletion in bread wheat (Sears 1977) and ph1c deletion in durum wheat (Giorgi 1978) have been most extensively used.
The traditional approach to gene introgression in wheat has been to develop first a disomic addition (DA) or disomic substitution (DS) line for a targeted alien chromosome and then induce recombination between the alien chromosome and its wheat homoeologues by manipulating the Ph1 gene (Sears 1973; Dvorak and Gorham 1992). This approach is laborious and takes many years. Its execution requires specialized training. Moreover, the approach is difficult to use if complementary genes on different chromosomes control a trait.
An alternative approach is genome-wide introgression. This approach eliminates the need for the development of DA or DS lines prior to inducing chromosome recombination (Dvorak 1977). It also reduces the likelihood of failures due to epistasis because it allows introgression of several alien chromosomes or chromosome segments in a single introgression line (IL). This approach was used to introgress germplasm into wheat from Ae. speltoides (2n = 2x = 14) (Dvorak 1977), Ae. tauschii (2n = 2x = 14) (Cox et al. 1995; Pestsova et al. 2006), Amblyopyrum muticum (2n = 2x = 14) (King et al. 2017), Lophopyrum elongatum (2n = 2x = 14) (Lou et al. 2017), Thinopyrum bessarabicum (2n = 2x = 14) (Grewal et al. 2018), and Triticum timopheevii (2n = 4x = 28) (Devi et al. 2019).
Using an existing amphiploid as a source of alien germplasm for genome-wide introgression is complicated because such an amphiploid is usually homozygous for the Ph1 gene, which has to be removed. A likely consequence of this manipulation is availability of only a portion of the alien genome for introgression in a plant homozygous for a Ph1 deletion. Alternatively, the expression of Ph1 can be suppressed in the amphiploid by crossing it with the tetraploid Su1-Ph1 suppressor line (Li et al. 2017).
The octoploid amphiploid AgCS (2n = 8x = 56, AABBDDEE) produced from the cross T. aestivum cv Chinese Spring (CS) × L. elongatum (Rommel and Jenkins 1959) has a special value for wheat genetics and is a desirable target for introgression. AgCS has been used as the source of L. elongatum chromosomes for the development of complete sets of DA and DS lines in wheat (Dvorak 1980; Dvorak and Chen 1984; Tuleen and Hart 1988). These single-chromosome lines have proven useful in mapping salt-stress tolerance (Dvorak and Ross 1986; Dvorak et al. 1988; Omielan et al. 1991), perennial growth habit (Lammer et al. 2004), waterlogging tolerance (Taeb et al. 1993), and fusarium head-blight-resistance (Shen et al. 2004; Shen and Ohm 2006; Miller et al. 2011; Fu et al. 2012).
Genus Lophopyrum is closely related to genus Thinopyrum, and the two are often considered congeneric (Barkworth and Dewey 1985). Both genera are perennial; the Thinopyrum species are rhizomatous, and Lophopyrum species are caespitose (Love 1984). The species of Lophopyrum vary in ploidy from diploid (L. elongatum and L. haifense, 2n = 14) to decaploid (L. ponticum, 2n = 70). Lophopyrum elongatum is probably an inbreeding species, but most other Lophopyrum species, both diploid and polyploid, are outcrossers. Lophopyrum elongatum is distributed along the coastal areas of the Mediterranean, where it is found in salt marshes and is highly tolerant of salinity (McGuire and Dvorak 1981).
The detection of introgressed chromosomes and chromosome segments is challenging in wheat due to polyploidy and the large number of chromosomes. Accomplishing that in a reasonable timeframe and at a reasonable cost requires genotyping putative ILs with a multiplexed platform. Massively parallel SNP genotyping platforms have been developed for Ae. tauschii and wheat (Luo et al. 2009; Cavanagh et al. 2013; Luo et al. 2013; Wang et al. 2014; Allen et al. 2017), and some were successfully used in genotyping alien chromosomes in the wheat genetic background (Grewal et al. 2018; Zhang et al. 2018; Zhou et al. 2018; Devi et al. 2019; Li et al. 2019). The downside of these platforms is that they are inflexible and are not cost effective in the manipulation of single alien chromosomes or chromosome segments subsequent to their discovery.
An alternative approach is to build a SNP genotyping array that is specifically designed for a project and based on genome-specific SNPs (Akhunov et al. 2010). An example of this approach is the deployment of L. elongatum genome-specific SNPs in the development of high-resolution melting (HRM) markers specifically designed for the characterization of L. elongatum germplasm in wheat introgression lines (Lou et al. 2017). HRM markers are flexible and cost effective, but cannot be easily multiplexed, which makes their deployment on a large-scale time-consuming.
In the project described here, we developed an array of L. elongatum genome-specific SNP markers utilizing the Sequenom MassARRAY SNP genotyping platform with the specific objective of discovering introgressed chromosomes and chromosome segments in a population of wheat recombinant inbred lines (RILs) derived from AgCS by a genome-wide introgression approach. The Sequenom MassARRAY platform permits multiplexing as many as 40 markers per single genotyping reaction (Bradic et al. 2011). We multiplexed the markers based on their chromosome location so that the resulting seven assays can be used together for genome-wide genotyping or individually for single-chromosome genotyping. The L. elongatum Sequenom MassARRAY SNP markers thus balance throughput with flexibility.
The Sequenom technology utilizes a single extension primer to generate an allele-specific product with a distinct mass. The assay consists of an initial locus-specific PCR, followed by single-base extension using a mass-modified dideoxynucleotide terminator of an oligonucleotide primer, which anneals immediately upstream of the polymorphic site. MALDI-TOF mass spectrometry is used to determine which SNP allele is present based on the mass of the extension product (Gabriel et al. 2009). The Sequenom MassARRAY platform has been used in SNP genotyping in diploid rice (Masouleh et al. 2009), but it is also suitable for SNP genotyping in polyploid wheat (Berard et al. 2009).
Since there was no sequence available for the L. elongatum genome at the start of this project, we produced L. elongatum Illumina paired-end reads. We then aligned the reads on the coding sequences of 39,622 Ae. tauschii high-confidence genes annotated in the Aet v4.0 genome sequence (Luo et al. 2017). We compared the aligned sequences with the CS chromosome arm scaffolds (Mayer et al. 2014) to discover SNPs between the L. elongatum genome, on the one hand, and the three subgenomes of the CS genome, on the other hand.
To identify the wheat chromosomes replaced by or recombined with L. elongatum chromosomes in the introgression population, we genotyped each RIL harboring an introgressed L. elongatum chromosome or chromosome segment (henceforth, IL) with Illumina’s wheat 90-K Infinium SNP array (Wang et al. 2014). Infinium genotyping utilizes probes annealing to all homologous targets present in a DNA sample. The probes end one base before the SNP sites. A single-base extension leads to incorporation of labeled nucleotide, which is subsequently dually stained, depending on the base present at the SNP site. Probes are captured and scanned and relative intensity of each color is used to interpret the genotype. In genotyping of polyploids, such as wheat, Illumina’s polyploid module is able to discriminate two-color readouts that are in other than the 2A:0B, 1A:1B, and 0A:2B allelic ratios of a diploid (Akhunov et al. 2009). In our project, polyploid genotyping complexity was further exacerbated by the fact that the RIL population was produced by a three-way cross (AgCS, ph1b, ph1c), and an unequivocal interpretation of each genotype call was sometimes difficult. To identify the replaced or recombined wheat chromosome, we consequently resorted to an empirical approach, in which we compared the SNP genotype of an IL with those of corresponding L. elongatum DS lines derived from AgCS.
Here, we induced recombination between the seven L. elongatum chromosomes present in AgCS and wheat chromosomes and evaluated several strategies of genome-wide introgression. Based on the sequential Sequenom and wheat 90-K Infinium SNP genotyping, we discovered ILs and built a database of their graphical genotypes. In the database, we indicated the L. elongatum and wheat chromosomes involved in introgression events and provided information about chromosome number and the presence/absence of the Ph1 gene for each IL. Finally, we addressed the veracity of assumptions about genotyping alien germplasm with the wheat 90-K SNP markers.
Materials and methods
Plants
We obtained AgCS, which is an octoploid amphiploid CS × L. elongatum, from L. Evans (University of Manitoba, Winnipeg, Canada) in 1970. We presumed it to be the amphiploid (designated as 8A206) reported by Rommel and Jenkins (Rommel and Jenkins 1959). Later, we also obtained from L. Evans an accession of L. elongatum (2n = 14) (received as Agropyron elongatum), which we designated as accession D. C.B. Jenkins (University of Manitoba, Winnipeg, Canada) obtained the accession from G.L. Stebbins (University of California, Davis) who received it from Y. Cauderon (INRA, France). This accession came from near Ariana, Tunisia (Stebbins and Pun 1953). We used here 19 of the 21 possible DS lines derived from AgCS (Dvorak 1980; Dvorak and Chen 1984; Tuleen and Hart 1988) as controls in genotyping. DS 4E(4B) and DS 5E(5A) were not available. We used two Ph1 deletion lines: ph1b in the CS genetic background (Sears 1977) and ph1c in the background of durum wheat cv Cappelli (Giorgi 1978). Finally, we used durum wheat ‘Langdon’ (LDN), which we received from J. Faris (USDA-ARS, North Dakota State University, Fargo) and bread wheat ‘Rollag,’ which we received from J. Anderson (University of Minnesota, St. Paul).
Introgression population
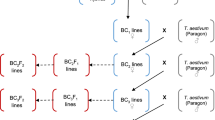
Since wheat cross-pollinates more extensively in the field than in the greenhouse, we grew all plants in the greenhouse, but without bagging spikes during flowering. First, we crossed AgCS with the tetraploid ph1c deletion line (Fig. 1). The hybrid seeds failed to develop endosperm, and we rescued immature embryos by culturing them on nutrient medium consisting of Murashige-Skoog Basal Salt Mixture (M5524-1L Sigma-Aldrich) with 3% sucrose and 7% agar.

Schematic representation of the development of the introgression population. The genome formulas are in parentheses. Capital letters indicate complete genomes and lower-case letters indicate partial genomes. Ph1 indicates a wild-type allele, and ph1 indicates a deletion of the Ph1 gene. The Ph1 or ph1 symbol in front of parentheses indicates homozygosity for the presence or absence of Ph1. Explicit genotypes follow parentheses. An × in a circle is a standard symbol for selfing. The numbers of plants at specific steps are indicated
We then crossed the hexaploid (AABBDE) F1 plants heterozygous for ph1c with the hexaploid ph1b deletion line. We selected F1 progeny homozygous for the absence of Ph1 based on the absence of a locus corresponding to EST BE442676. We showed that the locus was located within the ph1b and ph1c deletions. We assayed the presence/absence of BE442676 by PCR with the forward primer BE442676_GGGATTCGTTACTGGAAGCA and reverse primer BE442676_TGTAACAACAGAAGGTGGTCTA. We employed a 5B-specific SNP in BE442676 reported earlier (https://wheat.pw.usda.gov/SNP/new/index.shtml) (Akhunov et al. 2010) in the primer design. The PCR conditions were: 94 °C for 5 min, followed by 35 cycles of 94 °C for 0.5 min, 60 °C for 0.5 min, 72 °C for 3 min, and a final extension at 72 °C for 5 min. We resolved the PCR products by electrophoresis on a 1.5% agarose gel.
We backcrossed 13 F1 plants homozygous for the absence of Ph1 to the ph1b line, to maintain homozygosity for the Ph1 deletion (Fig. 1). We then either selfed the resulting BC1F1 plants, producing class I RILs, or crossed them to CS, to reintroduce Ph1, and then recurrently selfed them, producing class II RILs (Fig. 1). Alternatively, we crossed the 13 ph1 F1 plants immediately with CS and either recurrently selfed progeny, producing class III RILs, or backcrossed them once more to CS and selfed them, producing class IV RILs. Lastly, we used three Ph1/ph1 heterozygous F1 plants to increase the likelihood of introgressing intact L. elongatum genome chromosomes (Fig. 1). For two of these three plants, we either selfed them, producing class V RILs, or backcrossed F2 plants to CS and recurrently selfed them, producing class VI RILs. The third Ph1/ph1 plant differed from the previous two plants by being derived from using the Ph1/ph1c F1 as a male rather than as a female. Selfing this plant produced class VII RILs.
Genome-specific SNP discovery
We isolated DNA from L. elongatum acc. D and sequenced it with Illumina HiSeq 2000. We obtained about 50 Gb of paired-end reads with an average length of 150 bp and about 10 × genome coverage. We performed read-quality control and filtered out low-quality reads. To discover SNPs, we used the Ae. tauschii Aet v4.0 genome sequence (http://aegilops.wheat.ucdavis.edu/ATGSP/data.php) (Luo et al. 2017) as a reference. We mapped clean L. elongatum reads to the Ae. tauschii genome sequence using BWA (Li 2013) with the following parameters: -k 19 –A 1 –B 4 –O 6 –E 1 –U 17 –T 30. We removed PCR duplicates and filtered alignments using SAMtools (Li 2011). We called single nucleotide variants (SNVs) with SAMtools pileup with 20 as minimum mapping quality for alignments and 5 as minimum gapped reads for indel candidates, while setting other parameters at default. We further filtered SNV results using VCFtools (Danecek et al. 2011). We kept SNPs meeting the following criteria: coverage depth between 5 and 20 reads, SNP quality value > 20, and no other variation within a 100-bp range in sequences flanking a SNP. We extracted a set of 201-bp sequences from Ae. tauschii pseudomolecules as queries and aligned them to the CS chromosome arm survey sequences (Mayer et al. 2014). We filtered queries based on the following criteria: a hit should fall within a coding region (CDS), it should have at least 90% identity, and no more than one hit should be present per wheat subgenome. We then utilized the filtered queries for Sequenom MassARRAY marker design.
Sequenom MassARRAY marker design
We designed two PCR primers and one extension primer for each SNP using the Sequenom MassARRAY Designer software (Gabriel et al. 2009). To ensure genome specificity of the SNP markers, we searched homology by BLASTN against CS scaffolds and filtered the results according to the following criteria. Hits should have 100% identity, each primer should have a single hit in each wheat subgenome, and the lengths of PCR amplicons should be 200 bp or less. We designed primers including extension nucleotides, multiplexed them, and validated the multiplexed pools on the Sequenom MassARRAY platform by genotyping a panel of control lines including CS, Ae. tauschii, AgCS, L. elongatum acc. D, and the 19 L. elongatum DS lines. We considered a marker validated if the assay detected the wheat base in CS and Ae. tauschii, the E-genome base in L. elongatum, and both bases in AgCS and the relevant DS lines.
Some markers correctly detected the wheat base in all relevant stocks and the L. elongatum base in L. elongatum acc. D, but not in AgCS and the relevant DS lines. To determine whether the cause of these failures was an unfavorable haplotype ratio in genotyping polyploid templates or polymorphism between L. elongatum acc. D and the L. elongatum genome present in AgCS, we combined the CS DNA with the DNA of L. elongatum acc. D in a 3:1 mass ratio, respectively, to mimic the DNA of AgCS. We also combined the DNA of durum wheat cv Langdon with the DNA of L. elongatum in a 2:1 mass ratio to mimic the DNA of DS lines. We genotyped these reconstituted 6x and 8x DNAs along with DNA of actual AgCS and DS lines with Sequenom markers that had previously failed genotyping in AgCS and DS lines.
Genotyping
To identify ILs among the RILs, we isolated DNAs (Dvorak et al. 2006) from 554 RILs (Fig. 1), 19 DS lines, CS, ph1b, ph1c, AgCS, and L. elongatum acc. D and genotyped them with the Sequenom MassARRAY SNP markers. We concluded that a RIL was free of E-genome DNA at a SNP site if the SNP marker detected only the CS base. If a marker detected both bases, we concluded that the RIL possessed E-genome DNA at the site and designated such a RIL as an IL. Within each L. elongatum chromosome, we ordered the Sequenom SNP markers based on their registry (bp) in the Ae. tauschii pseudomolecule and constructed a graphical genotype for each IL.
To identify the wheat chromosome replaced by L. elongatum chromosome or recombined with it, we genotyped the ILs, 19 DS lines, AgCS, CS, L. elongatum acc. D, and the ph1c deletion line with Illumina’s wheat 90-K Infinium iSelect SNP genotyping array (Illumina Inc., San Diego, CA, USA) (Wang et al. 2014). Genotyping was performed at the DNA Technologies and Expression Analysis Core Laboratory, UC Davis Genome Center. We used the polyploid clustering module in the GenomeStudio v2.0 program (Illumina Inc., San Diego, CA, USA) for graph clustering and genotype calling.
To filter the genotype calls, we first removed markers that produced a no call (NC) result in a DS line or produced the same genotype in the trio or duo [DS 4E(4B) and 5E(5A) were not available] of DS lines sharing a common L. elongatum chromosome. We projected SNP markers onto the Ae. tauschii genome sequence by BLASTN homology searches using marker sequences as queries and the Ae. tauschii Aet v4.0 genome sequence as the target. Only those 90-K Infinium SNP markers that produced a hit on the Ae. tauschii pseudomolecule homoeologous with that on which the marker was mapped in wheat (Wang et al. 2014) were retained. We arranged these markers in the ascending order based on their coordinates along the Ae. tauschii Aet v4.0 pseudomolecules.
If a complete L. elongatum chromosome replaced a specific wheat homoeologue in an IL, we reasoned that the 90-K SNP genotype of the IL would be similar to that of the corresponding DS line. We therefore compared the genotype calls for the introgressed chromosome in an IL with genotype calls for the corresponding DS lines, counted the numbers of agreements and disagreements in each IL-DS line comparison while disregarding NCs, and used 2 × 2 contingency table and Fisher exact test to compute the p value. The three (two for homoeologous chromosome groups 4 and 5) p values were adjusted for multiple comparisons with the Bonferroni correction. We then expressed the degree of identity as a fraction of markers with the same genotype/total number of genotyping markers (we excluded NC markers). For a recombined chromosome, we computed these p values separately for the L. elongatum segment and for the wheat segment of the chromosome and made these pairwise comparisons both between and within chromosomes. An IL-DS line comparison with significantly greater degree of identity than the other comparisons indicated the wheat chromosome that was replaced or recombined in a specific introgression event.
To determine whether a line was homozygous ph1 or had at least one allele of the Ph1 gene, we genotyped each IL with PCR marker BE442676 (see above). A PCR amplification was considered as evidence for the presence of Ph1.
Chromosome number determination
To estimate the chromosome number in an IL, we germinated seeds in Petri dishes, excised about 1-cm-long root tips, and pre-treated them in distilled water at 2 °C for 24 h in a refrigerated water-bath. We then fixed them in a 3 alcohol/1 glacial acetic acid (v/v) fixative for 24 h, hydrolyzed them in 1 N HCl at 60 °C for 10 min, stained them in Schiff’s reagent for 30 min, and macerated them in a 0.2% pectinase (SIGMA, CAS#9032-75-1)/cellulase (SIGMA, CAS#9012-54-8) solution for 20 min. We determined chromosome numbers and the presence of telocentric chromosomes in standard root-tip squashes.
Results
Sequenom MassARRAY SNP marker development
We discovered 22,399 SNPs between L. elongatum and Aet v4.0 coding sequences. After filtration, we selected 914 SNPs for marker development. Since our goal was to multiplex about 10 to 15 SNP markers differentiating the E genome in AgCS from the three CS subgenomes per chromosome, we selected 221 evenly distributed markers along the Ae. tauschii chromosomes for validation (below). We validated 106 (48%) of these markers and multiplexed 97 of them into seven multiplexes corresponding to the seven Ae. tauschii chromosomes (Online Resource 1).
We used the following naming convention for the markers. The name starts with AgCS, indicating that the SNP was validated in AgCS. The single digit following AgCS indicates the Ae. tauschii chromosome in which the marker is located. The chromosome number is followed by the marker registry (bp) on the Ae. tauschii pseudomolecule. The registries start at the tips of the short arms of the Ae. tauschii chromosomes.
Of the 97 multiplexed SNP markers, 93 behaved as expected in RIL genotyping by showing: the wheat base in CS, ph1b, and ph1c; the E-genome base in L. elongatum; and both bases in the relevant DS lines (Online Resource 2). For example, marker AgCS7_21618791 produced the following genotypes: a G base in CS, ph1b, and ph1c, a T base in L. elongatum, and G and T bases in DS 7E(7A), DS 7E(7B), and DS 7E(7D) (Fig. 2a).

Sequenom marker validation and genotyping. We genotyped DNA isolated from Chinese Spring (CS), ph1b, ph1c, L. elongatum acc. D, and relevant DS lines with the Sequenom assays for markers AgCS7_21618791 (a) and AgCS4_61583948 (b). We used water as a non-template control (NTC). The yield of the allele with low and high mass was measured on the horizontal axis and vertical axis, respectively. Genotypes were located in the plot based on their ratio of the low-mass allele yield and high-mass allele yield. The dotted plot-split lines delimit the yields. If two alleles are 1:1, as in a heterozygous diploid, the ratio is expected to be near the diagonal line. If the high-mass and low-mass alleles are 2:1 or 1:2, as in a hexaploid (our situation), their ratios are expected to be in a sector either above or below the diagonal. a CS and deletion lines ph1b and ph1c (blue triangles) clustered in the bottom sector, indicating the presence of the wheat base. L. elongatum acc. D (brown inverted triangle) was located in the top sector, indicating the presence of the E-genome base. DS lines 7E(7A), 7E(7B), and 7E(7D) (green circles) clustered in the lower middle sector, indicating the presence of both bases in a 2:1 ratio. b Both E-genome and wheat bases were detected in the DS 4E(4A) line, but only the E-genome base was detected in the DS 4E(4D) line
Genotyping with the remaining four SNP markers differed from this pattern (labeled blue in Online Resource 2) by detecting two genotypes among the DS lines, not a single one, as expected. Assays with markers AgCS4_61583948 (Fig. 2b) and AgCS4_492665454 showed both the E-genome and CS bases in DS 4E(4A) but only the E-genome base in DS 4E(4D); DS 4E(4B) was not studied. This may be due to a Sequenom primer not correctly annealing or there may be polymorphism among the wheat subgenomes. We obtained similar results with markers AgCS1_20572405 and AgCS1_239592530 (also labeled blue in Online Resource 2).
The numbers of multiplexed markers ranged from 10 in homoeologous group 3 to 19 in homoeologous group 7 (Table 1). Markers were evenly distributed along the chromosomes as judged from their locations in the Ae. tauschii genome sequence. Distance between neighboring markers ranged from 33.93 to 62.72 Mb with a genome-wide average of 42.95 Mb. The most distal markers were close to chromosome tips in most Aet v4.0 pseudomolecules (Table 1). The longest distance between a marker and a chromosome tip was in the short arm of chromosome 6 (72 Mb) and the long arm of chromosome 5 (48 Mb).
Of the 116 markers that failed the validation process, 18 failed due to a PCR failure (Fig. 3a) or ambiguous clustering (Fig. 3b). The remaining 98 (43.9%) markers correctly genotyped the wheat base in CS, ph1b, and ph1c and the L. elongatum base in L. elongatum, but failed to detect the L. elongatum base in AgCS and the relevant DS lines. We evaluated the possibility that these failures were caused by the inability of these Sequenom SNP assays to detect the L. elongatum base when the wheat-to-E-genome base ratio was 2:1 (DS lines) or 3:1 (AgCS). All 98 markers detected the correct wheat base in CS, ph1b, ph1c, and the correct L. elongatum base in L. elongatum and in the 2:1 and 3:1 reconstituted DNAs (methods) that mimicked DNAs of AgCS and DS lines, but not in the actual AgCS and DS lines (Fig. 3c). Based on these results, we concluded that these Sequenom markers likely failed due to polymorphism between the genome of the L. elongatum acc. D and the E genome present in AgCS.

Failures of Sequenom marker validation. PCR failure (a), ambiguous clustering with an allele mass ratio near a sector boundary (b), failure to detect L. elongatum base in AgCS and DS lines (c). We constructed the plots as described in Fig. 2. In b, c, reconstituted 6x and reconstituted 8x refer to mixed LDN and L. elongatum DNAs and CS and L. elongatum DNAs, respectively. In c, the assay detected only the wheat base in AgCS and DS lines as indicated by the clustering of these genotypes with CS and the ph1b and ph1c lines rather than with reconstituted 6x and reconstituted 8x DNAs
Genotyping of CS, ph1b, ph1c, L. elongatum, and DS lines with the 97 multiplexed markers was remarkably robust. Of 651 data points collected, 650 (99.85%) showed the expected genotype (Online Resource 2). The assay was equally robust in genotyping heterozygotes plants. We crossed three ILs (44268, 44546, and 45297, Online Resource 2) with the bread wheat ‘Rollag’ and genotyped 28 F1 plants with Sequenom markers for chromosomes 2, 4, 5, and 7. Of 76 markers, 75 (98.7%) detected the L. elognatum allele in the heterozygous state.
Development and characterization of introgression lines
We initiated the development of RILs with 1355 plants in seven breeding strategies (Fig. 1). RILs in classes I, II, III, and IV were derived from 13 F1 plants homozygous for ph1, and those in classes V, VI, and VII were derived from three heterozygous plants (Fig. 1). Many lines went extinct during subsequent crosses or selfing due to sterility or other causes. We ultimately selected 554 RILs (Fig. 1) along with controls for genotyping with the 97 Sequenom MassARRAY SNP markers (Table 2).
Of the 554 RILs, 130 (23.5%) were ILs with one or more intact or recombined E-genome chromosomes (Online Resource 2, Fig. 4a); in the rest, we failed to detect alien germplasm (Table 2). The largest number, 61 ILs, were found among RILs in class I (27.7% of 220 class I RILs) (Table 2). Only 10.2% of the class II RILs had an introgressed chromosome (p < 0.0001, two-tailed 2 × 2 contingency table and Fisher exact test).

Characterization of ILs. a Nineteen Sequenom markers for homoeologous group 7 are aligned in the first row with the most distal marker in the short arm to the left. Markers flanking the centromere are in yellow. The genotypes of CS, L. elongatum acc. D, three relevant DS lines, and five ILs at the 19 SNP markers are shown. Pink cells indicate the detection of the CS base only, blue cells indicate the detection of the L. elongatum base only, and green cells indicate the detection of both CS and L. elongatum bases. IL 44546 harbors an introgressed 7ES, likely as Robertsonian fussions 7ES::7AL or telocentric 7ES, and ILs 44457 and 45497 are substitutions of telocentric chromosome 7EL for 7D or Robertsonian fusions 7DS::7EL. IL 45285 is characterized with a recombined chromosome harboring the tip of the short arm of 7E. Line 44528 exemplifies an IL with an entire chromosome 7E, which in this IL replaced wheat chromosome 7D. b Chromosome numbers in families derived from 114 ILs. The histogram shows the numbers of families in which the lowest chromosome number was as indicated on the horizontal axis (light blue) and the number of families in which the highest chromosome number was as indicated on the horizontal axis (dark blue). Also shown are the numbers of families with a modal chromosome number indicated on the horizontal axis (brown)
Ninety-three ILs, 16.8% of the 554 RILs genotyped, contained a single introgressed chromosome. The remaining 37 ILs (6.1% of the 554 RILs) contained two or more introgressed chromosomes. We cataloged 197 E-genome chromosomes among the 130 ILs; 108 complete and 89 recombined (Table 3). There were 30 centromeric breaks among the 89 recombined chromosomes (Table 2). Of chromosomes with crossovers, 73% had a wheat centromere and 27% had an E-genome centromere (p = 0.0004, Chi-square test for the expected 1:1 wheat-to-E-genome centromere ratio in recombination products). Most crossovers were in chromosomes 2E, 4E, and 5E, and most centromeric breaks were in chromosome 7E (Table 3).
Of 21 RILs in classes V to VII (Table 2), which we developed from Ph1/ph1 plants, 16 were ILs. We recorded four putative crossover events among these 16 lines, of which three double crossovers involving a single marker, detected in ILs 45495 and 44539, were likely genotyping errors. This low incidence of crossovers in these three classes was consistent with heterozygosity for Ph1 in the parental plants. The 16 ILs harbored 14 (56%) of the 30 centromeric breaks recorded among the 130 ILs. Thirteen (81%) of these 16 ILs had multiple introgressed chromosomes (Online Resource 2).
Only 15 of the 93 ILs with a single E-genome chromosome had a complete chromosome. Among the 37 ILs with multiple introgressed chromosomes, the most common was a set, 2E, 3E, 5E, and 6E; yet, at least some of these ILs originated independently; 3 such ILs were in classes I and II and 5 were in classes V and VI (Online Resources 2).
We examined pedigrees (Online Resource 2) of the 130 ILs to determine how many ILs may have contained chromosomes with independent recombination events or independent centromeric breaks. We considered two events independent if they showed different breakpoints. We also considered two events independent if they had the same breakpoints or haplotype but diverged after the second selfing generation. We estimated that the 59 chromosomes harboring breakpoints that most likely originated by crossovers originated from at least 34 independent events and that the 30 chromosomes harboring centromeric breaks originated from at least 26 independent events.
We determined chromosome numbers in root tips for 316 progeny of 114 of the 130 ILs (Online Resource 2). The mean chromosome number across ILs was 41.8 chromosomes, and the modal number was 42 chromosomes (Fig. 4b). Progeny segregated for chromosome number in 53 of 114 (46.5%) ILs. The lowest number was 2n = 40 recorded in progeny of three ILs, and the highest number was 2n = 46 recorded in progeny of two ILs, which had three and four different E-genome chromosomes. For each family, we determined progeny with the lowest, modal, and the highest chromosome number. While the density distribution of modal and highest chromosome numbers among the 114 families was symmetrical around 42 chromosomes, that of the lowest chromosome numbers was skewed toward hypoploid numbers (41 and 40 chromosomes) (Fig. 4b). Since most of the families had a modal number of 42 chromosomes, a vast majority of the 108 complete E-genome chromosomes must be substituted for wheat homoeologues in ILs. This includes ILs with multiple complete chromosomes (Online Resource 2).
To identify the wheat chromosomes substituted by or recombined with L. elongatum chromosomes, we genotyped the ILs, 19 L. elongatum DS lines, CS, AgCS, ph1c, and L. elongatum with the wheat 90-K Infinium SNP markers. Genotyping was successful in 118 of the 130 ILs. After removing failed markers, 80.1% of the remaining markers produced a genotype using DNA of L. elongatum acc. D as a template (Online Resource 3). We analyzed in detail genotyping of IL 44375 harboring the distal end of chromosome arm 2EL to assess the specificity of genotyping the L. elongatum genome present in AgCS with the wheat 90-K SNP markers. Of 1358 wheat 90-K SNP markers successfully projected onto Aet v4.0 pseudomolecule 2D (Online Resource 3), we selected 207 producing the following genotyping results (Online Resource 4). First, CS, the ph1c deletion line, and the DS lines excluding 2E(2A), 2E(2B), and 2E(2D) were genotyped as AA (pattern A) or BB (pattern B) and AgCS was genotyped as AB. If the A pattern occurred, we assumed that the L. elongatum genome in AgCS harbored the B allele. If the B pattern occurred, we assumed that the L. elongatum genome in AgCS harbored the A allele. Second, if the A pattern occurred, at least one of the DS lines involving 2E had one or both alleles genotyped as the B allele and if the B pattern occurred, at least one of the DS lines involving 2E had one or both alleles genotyped as the A allele. Of the 207 markers, 66 were in the 2E segment and 141 were in the wheat segment in the recombined chromosome in IL 44375. A total of 55 (83%) of the 66 markers showed the E-genome allele, but only 1 (0.7%) of the 141 markers showed the E-genome allele. This analysis showed that about 80% of the 90-K probes annealed to the orthologous haplotypes in the E genome and genotyped them corredctly.
To further pursue this surprisingly high specificity of wheat 90-K Infinium SNP markers in L. elongatum genotyping, we selected 743 SNP markers with the following genotypes: CS = AA or BB, L. elongatum acc. D = CS, and AgCS = AB and determined the ancestral/derived status for each base. If all wheat SNPs originated after the divergence of the wheat and L. elongatum lineages, there should always be the ancestral base at these SNPs in L. elongatum and derived base in the CS subgenomes. However, if some wheat SNPs are ancient and originated prior to the divergence of the two lineages, some SNP sites could show a derived base in L. elongatum and the ancestral base in the CS subgenomes. To test this hypothesis, we attempted to align the sequences of these 743 SNP markers on the barley (Mascher et al. 2017), Brachypodium distachyon v3.1, and rice v7.0 (Phytozome 12) sequences. We were successful with aligning and inferring the ancestral/derived status for 505 of them. At 188 (40%) of these 505 SNPs, L. elongatum appeared to have the derived base (Online Resource 5) indicating that these SNPs originated prior to the divergence of the wheat and L. elongatum lineages and were transmitted as a polymorphism into at least one wheat subgenome.
These preliminary analyses showed that most 90-K Infinium SNP assays are able to detect and genotype L. elongatum DNA and can be used to identify the substituted or recombined wheat chromosomes in ILs. We analyzed 7295 SNP markers which we could project onto Ae. tauschii v4.0 pseudomolecules (Online Resource 3). We compared the 90-K Infinium SNP genotypes for chromosomes previously identified as introgressed with Sequenom SNP markers (green in Online Resource 2 and Fig. 4a) with the genotypes of the corresponding DS lines (Online Resource 3). For example, based on genotyping with the Sequenom SNP markers, IL 44534 harbored an introgressed chromosome 2E (Online Resource 2). Progeny of the line had 2n = 41 or 42 chromosomes (Online Resource 2). Therefore, chromosome 2E most likely replaced a wheat chromosome in this IL. Of 1358 wheat 90-K Infinium markers successfully projected onto Ae. tauschii pseudomolecule 2D, 0.38, 0.31, and 0.91 (p < 0.0001, 2 × 2 contingency table and Fisher exact test with the Bonferroni correction) markers shared the same genotype in the comparison of the IL with DS lines 2E(2A), 2E(2B), and 2E(2D), respectively (Online Resource 3). Since the SNP genotype of the IL was most similar to that of DS 2E(2D), we concluded that L. elongatum chromosome 2E was substituted for wheat chromosome 2D in this IL. Another example is IL 44377 (2n = 41 or 42). In this IL, 2E Sequenom SNP markers indicated that a part of a wheat chromosome was replaced by the distal end of L. elongatum chromosome arm 2EL (Online Resource 2). The introgressed 2E chromosome segment included 749 90-K Infinium SNP markers. The introgressed segment shared the same genotype with DS lines 2E(2A), 2E(2B), and 2E(2D) in 0.89, 0.32, and 0.47 of the 749 SNP markers, respectively (p < 0.0001, 2 × 2 contingency table and Fisher exact test with the Bonferroni correction) (Online Resource 3). For the wheat segment of the recombined chromosome, 0.62 (p < 0.0001 compared to 0.89 of the E-genome segment), 0.46, and 0.51 of the 609 markers had the same genotype as DS lines 2E(2A), 2E(2B), and 2E(2D), respectively. We therefore concluded that 2E was recombined with wheat chromosome 2A in this IL.
Using this strategy, we identified the wheat chromosome replaced or recombined for 144 introgression events (Table 4). In some of the introgression events for which we failed to identify an involved wheat chromosome (Online Resource 2), the alien chromosome may had been added to the wheat genome or an alien chromosome segment may have been too short to be detectable. We summarized the results in Online Resource 2 for each of the 118 ILs we genotyped with the wheat 90-K Infinium array. The genotyping data for each introgressed chromosome as well as for corresponding duo or trio of DS lines, CS, and other controls are in Online Resource 3.
Wheat D-subgenome chromosomes were involved most frequently in complete chromosome (71.6%) and telocentric chromosome (57.1%) substitutions (Table 4). They were also frequently involved in recombination with L. elongatum chromosomes (31.4%) and centromeric fusions with them (60.0%). The opposite was true for the wheat B-subgenome chromosomes. They were substituted by or recombined with the L. elongatum chromosomes the least frequently.
Ph1 status
To determine how many ILs had Ph1, we genotyped DNA of 125 ILs with EST BE442676, which we showed is located within the ph1b and ph1c deletions. Genotyping of the remaining 5 ILs failed. Sixty (48%) of the 125 ILs produced positive PCR results indicating the presence of at least one copy of Ph1, and the remaining 65 (52%) ILs gave negative PCR results indicating ph1 homozygosity (absence of Ph1) (Online Resource 2). Only in two pairs of ILs (44510 and 44512, 44901, and 44903) derived from a common F2 parent did the lines differ in the outcome of the BE442676 PCR amplification showing that a negative PCR result was due to a genetic cause in most cases, not due to a PCR failure. There were 49 ph1 plants present among the classes I and II ILs. Forty-four (89.8%) of them were in class I, and only 5 were in class II (p = 0.004, 2 × 2 contingency table, two-tailed Fisher exact test). There were also more ph1 ILs in class I than in class III (9 of 31 IL in class III, p = 0.0001, 2 × 2 contingency table, two-tailed Fisher exact test). The presence of 11 plants with Ph1 in class I is an anomaly since the entire class I was to be derived from plants homozygous for ph1 backcrossed to ph1b. The 11 plants were derived from two F1 plants. Most likely these two plants were misclassified Ph1/ph1 heterozygotes. Only one of these 11 ILs harbored a recombined E-genome chromosome, which is consistent with the presence of Ph1 in these F1 plants.
Structure of the L. elongatum chromosomes
We used the genotyping data of the 130 ILs to delimit syntenic blocks and compare their order in the E-genome with their order in the Aet v4.0 genome sequence. Crossovers and centromeric breaks subdivided the seven E-genome chromosomes into 33 syntenic blocks (Online Resource 2), which we aligned on the Aet v4.0 genome sequence. Except for a putative inversion in the order of two blocks in the short arm of chromosome 3, the order of the syntenic blocks in the two genomes was same.
Discussion
Genotyping of alien germplasm with wheat SNPs
To characterize an introgression event, it is necessary to detect the presence of an alien chromosome or alien chromosome segment in the wheat genome and to identify the wheat chromosome that is substituted by or recombined with the alien chromosome. Some introgression projects have employed fluorescence genomic in situ hybridization (GISH) and molecular marker genotyping to accomplish these two tasks (Zhang et al. 2017, 2018; Grewal et al. 2018). We genotyped our RIL population with Sequenom markers, which we designed to detect specifically SNPs between the L. elongatum genome present in AgCS and the CS genome, to accomplish the first task. An asset of our approach was its time-effectiveness, but a drawback was a potential failure to detect introgression if the breakpoint was beyond the most distal Sequenom marker in a chromosome arm. The distance from the most distal marker to the chromosome arm end was short in most chromosome arms, but in some, such as in the long arm of chromosome 5 and the short arm of chromosome 6, it was substantial. Therefore, the RILs that we concluded were devoid of E-genome germplasm could contain small E-genome segments and should be screened further if a distally located gene is targeted in a project.
To accomplish the second task, we genotyped the ILs with the wheat 90-K Infinium SNP markers. We showed that a high percentage (about 80%) of the wheat 90-K Infinium SNP probes could genotype targets located on L. elongatum chromosomes homoeologous with wheat chromosomes on which the markers are located in wheat and Ae. tauschii and are presumably orthologous. Moreover, we showed that the origin of about 40% of wheat SNP markers at which the three CS subgenomes had the same genotype, but AgCS showed both alleles predated the divergence of the wheat and L. elongatum lineages. This was a special class of 505 SNPs, and it will be illuminating to determine the time of the origin of all types of SNPs in the 90-K assay. In the construction of a genetic map with the wheat Axiom 660-K SNP array in Agropyron cristatum, 2.9% of the markers successfully genotyped SNPs (Zhou et al. 2018). This successful genotyping in another Triticeae gene pool with wheat SNP markers is consistent with our finding and suggests that some polymorphisms within the wheat gene pool have deep roots within the radiation of the tribe.
Using the wheat 90-K Infinium SNP array, we were able to identify the wheat chromosome in 144 (73%) of the 197 introgression events. In some of the 53 events in which we did not identify the wheat chromosome, the L. elongatum chromosome may have been added to the wheat genome, as indicated by hyperaneuploid chromosome numbers in some of those ILs.
Development of the ILs
Of 554 RILs we developed and screened, 130 (23.5%) harbored one or more E-genome chromosomes or chromosome segments. In producing the 554 RILs, we either backcrossed homozygous ph1 plants to ph1b and recurrently selfed the progeny (class I RILs) or crossed them to CS to reintroduce the Ph1 allele prior to selfing or backcrossing to CS (class II RILs). The former strategy was a more successful introgression approach as 22.7% of class I RILs were found to be ILs compared to 7.6% of the class II RILs. One reason for this difference is that the backcross to ph1b maintained homozygosity for the deletion of Ph1, which provided an additional opportunity for recombination. Another reason, not mutually exclusive, is that meiotic pairing of the E-genome chromosomes in the absence of Ph1 could facilitate transmission of the alien chromosomes during selfing. That accounts for the fact that we also introgressed more complete chromosomes among class I RILs.
Sixteen ILs were identified by genotyping 21 RILs in classes V to VII, which were produced by selfing Ph1/ph1 heterozygous plants or crossing them to CS and selfing. Ten of these 16 ILs harbored a total of 14 chromosomes with centromeric breaks detected among the 130 ILs. We suggest that this enrichment of ILs for centromeric breaks in these three classes (p < 0.0001, 2 × 2 contingency table and Fisher exact test) was caused by the lack of pairing of the E-genome and D-subgenome chromosomes in the presence of Ph1, which destined them for misdivision.
Wheat D-subgenome chromosomes were more frequently substituted for or recombined with L. elongatum chromosomes than were the A- or B-subgenome chromosomes. This is expected since the F1 plants homozygous for Ph1 deletions contained a partial haploid D subgenome and a partial haploid L. elongatum genome. Haploidy and incompleteness of the two chromosome sets targeted them for substitution and recombination.
The population of ILs was hexaploid with a modal chromosome number of 2n = 42. The highest chromosome numbers were 45 in one IL and 46 in three ILs. Since most ILs had 42 chromosomes, lines with complete E-genome chromosomes are very likely substitution lines. This was true also for ILs with multiple E-genome chromosomes. For example, ILs 44534 and 44903 had four different E-genome chromosomes, but both had 2n = 42 chromosomes.
Nearly half of the ILs segregated for chromosome numbers. The density distribution of the highest chromosome numbers within the families was symmetrical around 42 chromosomes, but that of the lowest chromosome numbers within the families were greatly skewed toward hypoploid numbers. Such distributions are expected if the cause was failure of chromosomes to pair, since unpaired chromosomes have the tendency to be lost rather than being randomly distributed to the poles during meiosis (Sears 1954). Homozygosity for ph1 in 52% of the ILs was undoubtedly a major contributor to instability of ILs.
Structure of the L. elongatum chromosomes
During the validation of the SNP markers, we found all 97 markers to be on homoeologous chromosomes in the Ae. tauschii and L. elongatum genomes, suggesting that the gross chromosome structure is similar in the two genomes. Chromosome pairing in the Ae. tauschii × L. elongatum hybrids (Dvorak 1971) and quantitative comparisons of wheat and L. elongatum homoeologous chromosomes (Dvorak et al. 1984) led to the same conclusion. The L. elongatum genome is closely related to the genome of Th. bessarabicum (Jauhar 1988; Wang and Hsiao 1989). The two genomes differ by two translocations sharing a common chromosome (Jauhar 1988; Wang and Hsiao 1989). The translocations are between chromosomes 2J and 5J and between 4J and 5J (Grewal et al. 2018).
The reciprocal translocation between the long arms of chromosomes 4J and 5J (henceforth 4/5 translocation) is of particular interest as translocations involving the same two chromosome arms are present in several other Triticeae lineages (Liu et al. 1992) including that of T. monococcum (genomes AmAm) and the wheat A subgenome (Devos et al. 1995). Our Sequenom markers AgCS4_492665454, AgCS4_492752059, and AgCS4_495234484 are located distally to the 4/5 breakpoint in the sequence of wild emmer (Dvorak et al. 2018). The three markers should therefore be located on 5E if the E genome possesses the 4/5 translocation, but they are not, confirming the absence of the translocation in L. elongatum. If the A-genome 4/5 translocation and the J-genome 4/5 translocation were monophyletic, we would have to assume that the A-genome lineage and the Thinopyrum lineage share a common ancestor whereas the D-genome lineage and the Lophopyrum lineage, which both lack the 4/5 translocation, share a common ancestor. There is no evidence for either. If the breakpoints of the 4J/5J translocation indeed correspond to those of the 4A/5A translocation (Grewal et al. 2018), then the two translocations would have to be polyphyletic and originate by recurrent breaking of chromosomes 4 and 5 in Triticeae (Li et al. 2016).
Concluding remarks
We evaluated the utility of the Sequenom MassARRAY platform for the development of multiplexed SNP markers for introgression detection in wheat. We showed that the Sequenom MassARRAY platform provides a flexible and cost-effective environment for custom design of a genotyping array. The Sequenom MassARRAY platform was remarkably robust in genotyping homozygous RILs but also of heterozygous plants. Therefore, the markers will continue to be valuable in subsequent genetic and breeding applications of the ILs. We evaluated several strategies of introgression of L. elongatum germplasm into wheat and showed that backcrossing ph1/ph1 plants with alien chromosomes into a ph1 line followed by subsequent selfing produced a population of RILs containing 22.7% ILs. The added benefit of this strategy was that most of the resulting ILs were homozygous for ph1 and can be further recombined by crossing them with ph1b or an analogous stock and screening progeny for recombinants. We identified 37 ILs harboring multiple introgressed chromosomes. These lines represent a unique resource for studies of interactions among wheatgrass chromosomes and complement the existing single-chromosome DA and DS lines that have been developed from AgCS.
Availability of data and materials
All data generated in this study are included in this published article and its online resource files. Seeds of the introgression lines can be requested from JD.
References
Akhunov ED, Nicolet C, Dvorak J (2009) Single nucleotide polymorphism genotyping in polyploid wheat with the Illumina GoldenGate assay. Theor Appl Genet 119:507–517
Akhunov ED, Akhunova AR, Anderson OD, Anderson JA, Blake N, Clegg MT, Coleman-Derr D, Conley EJ, Crossman CC, Deal KR, Dubcovsky J, Gill BS, Gu YQ, Hadam J, Heo H, Huo N, Lazo GR, Luo MC, Ma YQ, Matthews DE, McGuire PE, Morrell PL, Qualset CO, Renfro J, Tabanao D, Talbert LE, Tian C, Toleno DM, Warburton ML, You FM, Zhang W, Dvorak J (2010) Nucleotide diversity maps reveal variation in diversity among wheat genomes and chromosomes. BMC Genom 11:702
Allen AM, Winfield MO, Burridge AJ, Downie RC, Benbow HR, Barker GLA, Wilkinson PA, Coghill J, Waterfall C, Davassi A, Scopes G, Pirani A, Webster T, Brew F, Bloor C, Griffiths S, Bentley AR, Alda M, Jack P, Phillips AL, Edwards KJ (2017) Characterization of a Wheat Breeders’ Array suitable for high-throughput SNP genotyping of global accessions of hexaploid bread wheat (Triticum aestivum). Plant Biotech J 15:390–401
Barkworth ME, Dewey DR (1985) Genomically based genera in the perennial Triticeae of North America: identification and membership. Am J Bot 72:767–776
Berard A, Le Paslier MC, Dardevet M, Exbrayat-Vinson F, Bonnin I, Cenci A, Haudry A, Brunel D, Ravel C (2009) High-throughput single nucleotide polymorphism genotyping in wheat (Triticum spp.). Plant Biotechnol J 7:364–374
Bradic M, Costa J, Chelo IM (2011) Genotyping with Sequenom. In: Orgogozo V, Rockman MV (eds) Molecular methods for evolutionary genetics. Humana Press, Totowa, pp 193–210
Cavanagh CR, Chao SM, Wang SC, Huang BE, Stephen S, Kiani S, Forrest K, Saintenac C, Brown-Guedira GL, Akhunova A, See D, Bai GH, Pumphrey M, Tomar L, Wong DB, Kong S, Reynolds M, da Silva ML, Bockelman H, Talbert L, Anderson JA, Dreisigacker S, Baenziger S, Carter A, Korzun V, Morrell PL, Dubcovsky J, Morell MK, Sorrells ME, Hayden MJ, Akhunov E (2013) Genome-wide comparative diversity uncovers multiple targets of selection for improvement in hexaploid wheat landraces and cultivars. Proc Natl Acad Sci USA 110:8057–8062
Cox TS, Sears RG, Bequette RK, Martin TJ (1995) Germplasm enhancement in winter wheat × Triticum tauschii backcross populations. Crop Sci 35:913–919
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R, Grp GPA (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158
Devi U, Grewal S, Yang CY, Hubbart-Edwards S, Scholefield D, Ashling S, Burridge A, King IP, King J (2019) Development and characterisation of interspecific hybrid lines with genome-wide introgressions from Triticum timopheevii in a hexaploid wheat background. BMC Plant Biol 19:193. https://doi.org/10.1186/s12870-019-1785-z
Devos KM, Dubcovsky J, Dvorak J, Chinoy CN, Gale MD (1995) Structural evolution of wheat chromosomes 4A, 5A, and 7B and its impact on recombination. Theor Appl Genet 91:282–288
Dvorak J (1971) Hybrids between a diploid Agropyron elongatum and Aegilops squarrosa. Can J Genet Cytol 13:90–94
Dvorak J (1977) Transfer of leaf rust resistance from Aegilops speltoides to Triticum aestivum. Can J Genet Cytol 19:133–141
Dvorak J (1980) Homoeology between Agropyron elongatum chromosomes and Triticum aestivum chromosomes. Can J Genet Cytol 22:237–259
Dvorak J, Chen KC (1984) Phylogenetic relationships between chromosomes of wheat and chromosome 2E of Elytrigia elongata. Can J Genet Cytol 26:128–132
Dvorak J, Gorham J (1992) Methodology of gene transfer by homoeologous recombination into Triticum turgidum: transfer of K+/Na+ discrimination from T. aestivum. Genome 35:639–646
Dvorak J, Ross K (1986) Expression of tolerance of Na+, K+, Mg2+, Cl−, and SO42− ions and seawater in the amphiploid of Triticum aestivum × Elytrigia elongata. Crop Sci 26:658–660
Dvorak J, Zhang HB (1990) Variation in repeated nucleotide sequences sheds light on the phylogeny of the wheat B and G genomes. Proc Natl Acad Sci USA 87:9640–9644
Dvorak J, McGuire PE, Mendlinger S (1984) Inferred chromosome morphology of the ancestral genome of Triticum. Plant Syst Evol 144:209–220
Dvorak J, Edge M, Ross K (1988) On the evolution of the adaptation of Lophopyrum elongatum to growth in saline environments. Proc Natl Acad Sci USA 85:3805–3809
Dvorak J, di Terlizzi P, Zhang HB, Resta P (1993) The evolution of polyploid wheats: identification of the A genome donor species. Genome 36:21–31
Dvorak J, Akhunov ED, Akhunov AR, Deal KR, Luo MC (2006) Molecular characterization of a diagnostic DNA marker for domesticated tetraploid wheat provides evidence for gene flow from wild tetraploid wheat to hexaploid wheat. Mol Biol Evol 23:1386–1396
Dvorak J, Wang L, Zhu TT, Jorgensen CM, Luo MC, Deal KR, Gu YQ, Gill BS, Distelfeld A, Devos KM, Qi P, McGuire PE (2018) Reassessmelnt of the evolution of wheat chromosomes 4A, 5A, and 7B. Theor Appl Genet 131:2451–2462
Fu SL, Lv ZL, Qi B, Guo X, Li J, Liu B, Han FP (2012) Molecular cytogenetic characterization of wheat-Thinopyrum elongatum addition, substitution and translocation ines with a novel source of resistance to wheat fusarium head blight. J Genet Genom 39:103–110
Gabriel S, Ziaugra L, Tabbaa D (2009) SNP genotyping using the Sequenom Massarray iPLEX platform. Curr Protoc Hum Genet 60:2.12.11–12.12.18
Giorgi B (1978) A homoeologous pairing mutant isolated in Triticum durum cv. Cappelli. Mut Breed Newsl 11:4–5
Glémin S, Scornavacca C, Dainat J, Burgarella C, Viader V, Ardisson M, Gautier S, Santoni S, David J, Ranwez V (2019) Pervasive hybridizations in the history of wheat relatives. Sci Adv 5:eaav9188
Grewal S, Yang CY, Edwards SH, Scholefield D, Ashling S, Burridge AJ, King IP, King J (2018) Characterisation of Thinopyrum bessarabicum chromosomes through genome-wide introgressions into wheat. Theor Appl Genet 131:389–406
Jauhar PP (1988) A reassessment of genome relationships between Thinopyrum bessarabicum and Thinopyrum elongatum of the Triticeae. Genome 30:903–914
Kihara H (1944) Discovery of the DD-analyser, one of the ancestors of Triticum vulgare (Japanese). Agric Hort (Tokyo) 19:13–14
King JL, Grewal S, Yang CY, Hubbart S, Scholefield D, Ashling S, Edwards KJ, Allen AM, Burridge A, Bloor C, Davassi A, da Silva GJ, Chalmers K, King IP (2017) A step change in the transfer of interspecific variation into wheat from Amblyopyrum muticum. Plant Biotechnol J 15:217–226
Lammer D, Cai XW, Arterburn M, Chatelain J, Murray T, Jones S (2004) A single chromosome addition from Thinopyrum elongatum confers a polycarpic, perennial habit to annual wheat. J Exp Bot 55:1715–1720
Li H (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27:2987–2993
Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv Prepr arXiv:13033997
Li WL, Challa GS, Zhu HL, Wei WJ (2016) Recurrence of chromosome rearrangements and reuse of DNA breakpoints in the evolution of the Triticeae genomes. G3-Genes Genomes Genet 6:3837–3847
Li H, Deal KR, Luo MC, Ji WQ, Distelfeld A, Dvorak J (2017) Introgression of the Aegilops speltoides Su1-Ph1 suppressor into wheat. Front Plant Sci 8:2163
Li H, Wang L, Luo MC, Nie F, Zhou Y, McGuire PE, Distelfeld A, Dai XT, Song CP, Dvorak J (2019) Recombination between homoeologous chromosomes induced in durum wheat by the Aegilops speltoides Su1-Ph1 suppressor. Theor Appl Genet 132:3265–3276
Liu CJ, Atkinson MD, Chinoy CN, Devos KM, Gale MD (1992) Nonhomologous translocations between group-4, group-5 and group-7 chromosomes within wheat and rye. Theor Appl Genet 83:305–312
Lou HJ, Dong LL, Zhang KP, Wang DW, Zhao ML, Li YW, Rong CW, Qin HJ, Zhang AM, Dong ZY, Wang DW (2017) High-throughput mining of E-genome-specific SNPs for characterizing Thinopyrum elongatum introgressions in common wheat. Mol Ecol Resour 17:1318–1329
Love A (1984) Conspectus of the triticeae. Feddes Repert 95:425–521
Luo MC, Deal KR, Akhunov ED, Akhunova AR, Anderson OD, Anderson JA, Blake N, Clegg MT, Coleman-Derr D, Conley EE, Crossman CC, Dubcovsky J, Gill BS, Gu YQ, Hadam J, Heo HY, Huo N, Lazo G, Ma YQ, Matthews DE, McGuire PE, Morrell PL, Qualset CO, Renfro J, Tabanao D, Talbert LE, Tian C, Toleno DM, Warburton ML, You FM, Zhang WJ, Dvorak J (2009) Genome comparisons reveal a dominant mechanism of chromosome number reduction in grasses and accelerated genome evolution in Triticeae. Proc Natl Acad Sci USA 106:15780–15785
Luo MC, Gu YQ, You FM, Deal KR, Ma YQ, Hu Y, Huo N, Wang Y, Wang JR, Chen SY, Jorgensen CM, Zhang Y, McGuire PE, Pasternak S, Stein JC, Ware DH, Kramer M, McCombie WR, Kianian SF, Martis MM, Mayer KFX, Sehgal SK, Li W, Gill BS, Bevan MW, Simkova H, Dolezel J, Song W, Lazo GR, Anderson OD, Dvorak J (2013) A 4-gigabase physical map unlocks the structure and evolution of the complex genome of Aegilops tauschii, the wheat D-genome progenitor. Proc Natl Acad Sci USA 110:7940–7945
Luo MC, Gu YQ, Puiu D, Wang H, Twardziok SO, Deal KR, Huo NX, Zhu TT, Wang L, Wang Y, McGuire PE, Liu SY, Long H, Ramasamy RK, Rodriguez JC, Van SL, Yuan LX, Wang ZZ, Xia ZQ, Xiao LC, Anderson OD, Ouyang SH, Liang Y, Zimin AV, Pertea G, Qi P, Ennetzen JLB, Dai XT, Dawson MW, Muller HG, Kugler K, Rivarola-Duarte L, Spannagl M, Mayer KFX, Lu FH, Bevan MW, Leroy P, Li PC, You FM, Sun QX, Liu ZY, Lyons E, Wicker T, Salzberg SL, Devos KM, Dvorak J (2017) Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 551:498–502
Mascher M, Gundlach H, Himmelbach A, Beier S, Twardziok SO, Wicker T, Radchuk V, Dockter C, Hedley PE, Russell J, Bayer M, Ramsay L, Liu H, Haberer G, Zhang XQ, Zhang QS, Barrero RA, Li L, Taudien S, Groth M, Felder M, Hastie A, Simkova H, Stankova H, Vrana J, Chan S, Munoz-Amatrian M, Ounit R, Wanamaker S, Bolser D, Colmsee C, Schmutzer T, Aliyeva-Schnorr L, Grasso S, Tanskanen J, Chailyan A, Sampath D, Heavens D, Clissold L, Cao SJ, Chapman B, Dai F, Han Y, Li H, Li X, Lin CY, McCooke JK, Tan C, Wang PH, Wang SB, Yin SY, Zhou GF, Poland JA, Bellgard MI, Borisjuk L, Houben A, Dolezel J, Ayling S, Lonardi S, Kersey P, Lagridge P, Muehlbauer GJ, Clark MD, Caccamo M, Schulman AH, Mayer KFX, Platzer M, Close TJ, Scholz U, Hansson M, Zhang GP, Braumann I, Spannagl M, Li CD, Waugh R, Stein N (2017) A chromosome conformation capture ordered sequence of the barley genome. Nature 544:427–433
Masouleh AK, Waters DLE, Reinke RF, Henry RJ (2009) A high-throughput assay for rapid and simultaneous analysis of perfect markers for important quality and agronomic traits in rice using multiplexed MALDI-TOF mass spectrometry. Plant Biotechnol J 7:355–363
Mayer KFX, Rogers J, Dolezel J, Pozniak C, Eversole K, Feuillet C, Gill B, Friebe B, Lukaszewski AJ, Sourdille P, Endo TR, Dolezel J, Kubalakova M, Cihalikova J, Dubska Z, Vrana J, Sperkova R, Simkova H, Rogers J, Febrer M, Clissold L, McLay K, Singh K, Chhuneja P, Singh NK, Khurana J, Akhunov E, Choulet F, Sourdille P, Feuillet C, Alberti A, Barbe V, Wincker P, Kanamori H, Kobayashi F, Itoh T, Matsumoto T, Sakai H, Tanaka T, Wu JZ, Ogihara Y, Handa H, Pozniak C, Maclachlan PR, Sharpe A, Klassen D, Edwards D, Batley J, Olsen OA, Sandve SR, Lien S, Steuernagel B, Wulff B, Caccamo M, Ayling S, Ramirez-Gonzalez RH, Clavijo BJ, Steuernagel B, Wright J, Pfeifer M, Spannagl M, Mayer KFX, Martis MM, Akhunov E, Choulet F, Mayer KFX, Mascher M, Chapman J, Poland JA, Scholz U, Barry K, Waugh R, Rokhsar DS, Muehlbauer GJ, Stein N, Gundlach H, Zytnicki M, Jamilloux V, Quesneville H, Wicker T, Mayer KFX, Faccioli P, Colaiacovo M, Pfeifer M, Stanca AM, Budak H, Cattivelli L, Glover N, Martis MM, Choulet F, Feuillet C, Mayer KFX, Pfeifer M, Pingault L, Mayer KFX, Paux E, Spannagl M, Sharma S, Mayer KFX, Pozniak C, Appels R, Bellgard M, Chapman B, Pfeifer M, Pfeifer M, Sandve SR, Nussbaumer T, Bader KC, Choulet F, Feuillet C, Mayer KFX, Akhunov E, Paux E, Rimbert H, Wang SC, Poland JA, Knox R, Kilian A, Pozniak C, Alaux M, Alfama F, Couderc L, Jamilloux V, Guilhot N, Viseux C, Loaec M, Quesneville H, Rogers J, Dolezel J, Eversole K, Feuillet C, Keller B, Mayer KFX, Olsen OA, Praud S, Iwgsc (2014) A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345:1251788
McFadden ES, Sears ER (1946) The origin of Triticum spelta and its free-threshing hexaploid relatives. J Hered 37(81–89):107–116
McGuire PE, Dvorak J (1981) High salt tolerance potential in wheatgrasses. Crop Sci 21:702–705
Miller SS, Watson EM, Lazebnik J, Gulden S, Balcerzak M, Fedak G, Ouellet T (2011) Characterization of an alien source of resistance to Fusarium head blight transferred to Chinese Spring wheat. Bot Botanique 89:301–311
Okamoto M (1957) Asynaptic effect of chromosome V. Wheat Inf Serv 5:6
Omielan J, Epstein E, Dvorak J (1991) Salt tolerance and ionic relations of wheat as affected by individual chromosomes of salt-tolerant Lophopyrum elongatum. Genome 34:961–974
Pestsova EG, Borner A, Roder MS (2006) Development and QTL assessment of Triticum aestivum-Aegilops tauschii introgression lines. Theor Appl Genet 112:634–647
Riley R, Chapman V (1958) Genetic control of the cytologically diploid behaviour of hexaploid wheat. Nature 182:713–715
Rommel R, Jenkins BC (1959) Amphiploids in Triticeae produced at the University of Manitoba from March 1958 to December 1959. Wheat Inf Serv 9–10:23
Sears ER (1954) The aneuploids of common wheat. Res Bull Univ Mossouri Agric Exper Station 572:1–59
Sears ER (1973) Agropyron-wheat transfers induced by homeologous pairing. In: Proceedings of the fourth international wheat genetics symposium, pp 191–200
Sears ER (1977) An induced mutant with homoeologous pairing in common wheat. Can J Genet Cytol 19:585–593
Sears ER, Okamoto M (1958) Intergenomic chromosome relationships in hexaploid wheat. In: Proceedings of the tenth international congress on genetics, pp 258–259
Shen X, Ohm H (2006) Fusarium head blight resistance derived from Lophopyrum elongatum chromosome 7E and its augmentation with Fhb1 in wheat. Plant Breed 125:424–429
Shen R, Kong LR, Ohm H (2004) Fusarium head blight resistance in hexaploid wheat (Triticum aestivum)-Lophopyrum genetic lines and tagging of the alien chromatin by PCR markers. Theor Appl Genet 108:808–813
Stebbins GLJ, Pun FT (1953) Artificial and natural hybrids in the gramineae, tribe hirdeae. VI. chromosome pairing in Secale cereale X Agropyron intermedium and the problem of genome homologies in the Triticinae. Genetics 38:600–608
Taeb M, Koebner RMD, Forster BP (1993) Genetic variation for waterlogging tolerance in the Triticeae and the chromosomal location of genes conferring waterlogging tolerance in Thinopyrum elongatum. Genome 36:825–830
Tuleen NA, Hart GE (1988) Isolation and characterization of wheat—Elytrigia elongata chromosome 3E and 5E addition and substitution lines. Genome 30:519–524
Wang RRC, Hsiao C (1989) Genome relationship between Thinopyrum bessarabicum and Thinopyrum elongatum revisited. Genome 32:802–809
Wang SC, Wong DB, Forrest K, Allen A, Chao SM, Huang BE, Maccaferri M, Salvi S, Milner SG, Cattivelli L, Mastrangelo AM, Whan A, Stephen S, Barker G, Wieseke R, Plieske J, Lillemo M, Mather D, Appels R, Dolferus R, Brown-Guedira G, Korol A, Akhunova AR, Feuillet C, Salse J, Morgante M, Pozniak C, Luo MC, Dvorak J, Morell M, Dubcovsky J, Ganal M, Tuberosa R, Lawley C, Mikoulitch I, Cavanagh C, Edwards KJ, Hayden M, Akhunov E (2014) Characterization of polyploid wheat genomic diversity using a high-density 90 000 single nucleotide polymorphism array. Plant Biotechnol J12:787–796
Zhang W, Cao YP, Zhang MY, Zhu XW, Ren SF, Long YM, Gyawali Y, Chao SM, Xu S, Cai XW (2017) Meiotic homoeologous recombination-based alien gene introgression in the genomics era of wheat. Crop Sci 57:1189–1198
Zhang W, Zhu XW, Zhang MY, Chao SM, Xu S, Cai XW (2018) Meiotic homoeologous recombination-based mapping of wheat chromosome 2B and its homoeologues in Aegilops speltoides and Thinopyrum elongatum. Theor Appl Genet 131:2381–2395
Zhou SH, Zhang JP, Che YH, Liu WH, Lu YQ, Yang XM, Li XQ, Jia JZ, Liu X, Li LH (2018) Construction of Agropyron Gaertn. genetic linkage maps using a wheat 660 K SNP array reveals a homoeologous relationship with the wheat genome. Plant Biotechnol J 16:818–827
Funding
This work was supported in part by the joint Bill and Melinda Gates Foundation and US National Science Foundation BREAD grant IOS-1212591, US Department of Agriculture Grant 58-2090-8-071, NIFA Hatch Program 1002302, and ARS/USWBSI Agreement 58-2090-8-071.
Author information
Authors and Affiliations
Contributions
JD, JX, PEM, M-CL, designed the experiments. JD, KRD, and PEM made initial crosses and JX developed the introgression population. LW, JX, FMY, TTZ, and M-CL discovered genome specific SNPs. JM and JX developed Sequenom markers and genotyped the introgression population. JX and JD performed the cytogenetic analyses. KRD developed Ph1 PCR assay and KRD and JX performed PCR and analyzed the results. RKR analyzed the evolutionary history of 743 wheat 90-K SNP markers. JD and JX wrote the first draft of the paper. All authors read and approved the final draft.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Communicated by Albrecht E. Melchinger.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
122_2020_3544_MOESM4_ESM.xlsx
Online Resource 4: Genotypes of 19 L. elongatum disomic substitution lines, CS, Chinese Spring × L. elongatum amphiploid AgCS, ph1c line, L. elongatum acc. D, and introgression line IL 44375 with wheat 90-K Infinium SNP markers (spreadsheet) (XLSX 43 kb)
Rights and permissions
About this article

Cite this article
Xu, J., Wang, L., Deal, K.R. et al. Genome-wide introgression from a bread wheat × Lophopyrum elongatum amphiploid into wheat. Theor Appl Genet 133, 1227–1241 (2020). https://doi.org/10.1007/s00122-020-03544-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-020-03544-w