
Abstract
Key message
A total of 33 additive stem rot QTLs were identified in peanut genome with nine of them consistently detected in multiple years or locations. And 12 pairs of epistatic QTLs were firstly reported for peanut stem rot disease.
Abstract
Stem rot in peanut (Arachis hypogaea) is caused by the Sclerotium rolfsii and can result in great economic loss during production. In this study, a recombinant inbred line population from the cross between NC 3033 (stem rot resistant) and Tifrunner (stem rot susceptible) that consists of 156 lines was genotyped by using 58 K peanut single nucleotide polymorphism (SNP) array and phenotyped for stem rot resistance at multiple locations and in multiple years. A linkage map consisting of 1451 SNPs and 73 simple sequence repeat (SSR) markers was constructed. A total of 33 additive quantitative trait loci (QTLs) for stem rot resistance were detected, and six of them with phenotypic variance explained of over 10% (qSR.A01-2, qSR.A01-5, qSR.A05/B05-1, qSR.A05/B05-2, qSR.A07/B07-1 and qSR.B05-1) can be consistently detected in multiple years or locations. Besides, 12 pairs of QTLs with epistatic (additive × additive) interaction were identified. An additive QTL qSR.A01-2 also with an epistatic effect interacted with a novel locus qSR.B07_1-1 to affect the percentage of asymptomatic plants in a row. A total of 193 candidate genes within 38 stem rot QTLs intervals were annotated with functions of biotic stress resistance such as chitinase, ethylene-responsive transcription factors and pathogenesis-related proteins. The identified stem rot resistance QTLs, candidate genes, along with the associated SNP markers in this study, will benefit peanut molecular breeding programs for improving stem rot resistance.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Stem rot disease, caused by the fungus Sclerotium rolfsii, is an important disease leading to yield loss to many important crops including cultivated peanut (Arachis hypogaea L.) (Punja 1988). Cultivated peanut is an allotetraploid (AABB, 2n = 4x = 40) derived from the chromosome doubling of an interspecific hybrid crossed between two diploids, A. duranensis (A genome) and A. ipaensis (B genome), which originated from South America (Bertioli et al. 2016). Peanut is mostly grown in tropical and subtropical regions in the world. Relatively high heat and humidity of those regions bring great challenges for peanut production. The typical yield loss caused by stem rot in peanut is approximately 10%, but it could reach up to 80% under favorable disease developing conditions (Kokalis-Burelle et al. 1997). In the USA, the economic loss of this disease affecting peanut production reached up to 59.7 million dollars in 2015 (Little 2015). Fungicide application and crop rotation are necessary for controlling stem rot disease, but at the cost of additional labor, management and environmental contamination. The use of stem rot resistant cultivars is an effective way to reduce yield loss caused by this disease (Anco 2017). Yet, most peanut cultivars do not possess high resistance to stem rot, and currently no source with immunity to stem rot has been identified.
QTL mapping is a powerful tool for dissecting genetic components controlling the expression of desirable traits or to develop molecular markers linked to the traits, which can be used for marker assisted selection (MAS) in breeding programs (Morrell et al. 2012). Many QTL mapping studies on resistance to stem rot disease, though caused by different fungal species, have been reported in several legume species, such as common bean (Mamidi et al. 2016; Vasconcellos et al. 2017) and soybean (Vuong et al. 2008; Zhao et al. 2015). In peanut, Bera et al. (2016) reported one QTL for stem rot resistance using a sparse linkage map based on 12 SSR markers in an F2 segregating population. Besides, Dodia et al. (2016) identified three SSR markers linked with stem rot resistance based on bulk segregation analysis (BSA). However, the stem rot resistance QTLs/markers identified in these studies were based on fewer than 60 SSR markers and highly heterozygous mapping populations, which led to large genomic intervals harboring the resistance QTLs making it hard to define candidate genes for further genetic analysis. Furthermore, the limited number of SSR markers was screened through different populations, which limited their further applications in breeding programs using materials of different genetic backgrounds. So far, most QTL mapping studies were focused on the additive effects of the genetic components, while other genetic effects such as epistasis and genetic × environment interaction were not fully addressed. Epistatic effect plays a significant role in determining phenotypic traits (Bocianowski 2013; Monnahan and Kelly 2015). Typically, host resistance to fungal invasion involves a series of genes in the defense pathways (Van Loon et al. 2006). Epistatic effects could be an important component in the disease resistance phenotype and thus should be taken into consideration in QTL mapping studies.
With the rapid advancement of high-throughput genotyping technologies, SNP array and next-generation sequencing (NGS)-enabled genotyping methods have greatly facilitated linkage analysis and QTL mapping in crop species (Rasheed et al. 2017). For instance, Zhou et al. (2014) constructed a high-density linkage map for cultivated peanut based on SNPs called from NGS. A 58 K SNP array in peanut called “Axiom_Arachis” was released (Clevenger et al. 2017; Pandey et al. 2017), which has been used to map disease resistance QTLs (Chu et al. 2019). The availability of genomes of both diploid peanut ancestors (Bertioli et al. 2016) and the cultivated peanut at PeanutBase (peanutbase.org; Bertioli et al. 2019) and Peanut Genome Resource (peanutgr.fafu.edu.cn; Zhuang et al. 2019) further facilitated researchers to efficiently map and discover genes of interest. For example, Jogi et al. (2016) identified a set of differentially expressed genes (DEGs) in four peanut cultivars with different stem rot resistance levels at 4 days post-inoculation using RNA sequencing. These genes are of great value for understanding the molecular responses of peanut plants to stem rot disease infection. However, since this RNA-seq experiment was conducted once in a growth chamber to investigate early plant–pathogen interactions, it may not have captured all the genes responsible for the resistance, especially those functional genes responding to disease infection in a field environment. In addition, some disease resistance genes are not differentially expressed during pathogen infection and thus would not be identified in the DEG pool. Even if the critical disease resistance genes were in the DEG pool, if there is no genetic sequence variation among germplasm accessions on or near the gene sequences, markers could not be designed according to these genes for marker assisted selection (MAS) in breeding. QTL mapping approach based on a high-density genetic map using multiple-year and multiple-location data can overcome these limitations and be helpful to identify key markers/genes for stem rot resistance.
The objectives of this study are to (1) phenotype the stem rot disease of a RIL population derived from a cross between a stem rot resistant peanut line NC 3033 and a susceptible cultivar Tifrunner (Beute et al. 1976; Holbrook et al. 2013; Holbrook and Culbreath 2007) at multiple locations and in multiple years; (2) identify additive and epistatic QTLs and evaluate QTL × environment interactions controlling stem rot resistance in a peanut; and (3) identify stem rot resistance candidate genes within QTL intervals. The stem rot resistance QTLs and genes identified in this study can be utilized by peanut researchers and breeders to further genetically characterize the stem rot resistance and to develop markers for MAS of stem rot resistance in peanut breeding programs.
Materials and methods
Mapping population, treatment and phenotyping
The mapping population consisting of 156 RIL lines was developed by crossing NC 3033 (stem rot resistant) and Tifrunner (stem rot susceptible). Briefly, F6-derived generation of this population was planted in Citra, Florida (2013), then F7 and F8 at Marianna, Florida (2014 and 2015), respectively, and F6, F7 and F8 at Tifton, Georgia (2013–2015), respectively, with two replications following a randomized complete block design with each block as one replication. Each year, plots were planted after 2 years of cotton and fields were prepared using standard tillage including moldboard tillage followed by disk harrowing. To minimize the impact of leaf spots, the fungicide program consisted of chlorothalonil-based fungicides applied at labeled rates every 2 weeks beginning about 30 days after planting and continuing for eight sprays. It is known that chlorothalonil-based fungicides did not affect S. rolfsii (Culbreath et al. 1995).
For the experiments conducted in Tifton, Georgia (GA), the field used for stem rot study was fumigated by injecting 100% chloropicrin (336 kg/ha) in the soil and covered with a plastic tarp for 7 days before planting. One highly virulent strain of S. rolfsii isolate (SR-18) was used for field inoculation. The mycelium was transferred to potato dextrose agar (PDA) plates and incubated for 2 days at 25 °C under light before the inoculation. PDA plugs were taken from the edge of the colony with a cork borer (1 cm in diameter). Two months after planting, approximately 8–10 healthy plants per plot were flagged and inoculated with the PDA plugs. Extra care was taken to place the PDA plug at the base of the plant with the mycelium in direct contact with the crown of the plant. Irrigation was applied before inoculation and for 2–3 consecutive days afterward. Disease rating was taken after plants were dug out and inverted (root of the plant was up and canopy was down) at harvest (130–150 days after planting, DAP); each marked plant was individually rated visually on a 0–10 scale for stem rot susceptibility where 0 = no disease symptoms; 1 = one or two small lesions (less than 0.7 cm in length) in crown area; 2 = one or more elongated, larger lesions (larger than 0.7 cm) in crown area; 3 = more lesions (typically more than five lesions) in the crown area; 4 = the entire crown area is girdled by lesions; 5 = the entire crown area is girdled and one branch is killed by the disease; 6 = up to 20% of the entire plant (stem, leaves, crown area, roots, pegs and pods) is colonized by the disease; 7 = 21–40% of the entire plant is colonized by the disease; 8 = 41–60% of the entire plant is colonized by the disease; 9 = 61–80% of the entire plant is colonized by the disease; and 10 = 81–100% of the entire plant is colonized by the disease.
For the experiments conducted in Florida (FL), stem rot fungal inoculum was prepared from two different sources: (1) S. rolfsii isolate SR-18 and (2) S. rolfsii isolate LE948 from Dr. Dufault (Khatri et al. 2017). To prepare the inoculum, the fungus was grown in petri dishes and then used to inoculate multiple autoclaved flasks with cracked corn and wheat for fungus increase at room temperature for 3 months, which were shaken daily for homogenizing. For inoculation, two different methods: point inoculation and broad spreading inoculation, were used. To provide a conducive environment for disease development, inoculation was conducted about 60 days after planting (DAP), when the canopies of the two rows of peanut plants were lapping. For point inoculation, in the middle of one row of each plot, a hole of 1 inch in depth in soil was made by pencil and 5 grams of each source of inoculum was buried in the hole. For each source of inoculum, two points of inoculations were conducted with 2 feet apart and marked with colored flags to label the site of inoculation and to differentiate different isolate source (blue flag for isolate LE948 and white flag for isolate SR-18). Therefore, in each row of the plot, four point-inoculations were conducted with two for each inoculum source per row. For point-inoculation phenotyping, the distance from the point of inoculation to the edge of disease progression was measured as disease index for each inoculation point. For broadcast inoculation method, 25 grams of inoculum of isolate LE948 was carefully spread on top of the canopy of the other row of each plot followed by shaking the canopy to allow the inoculum to fall to the soil. The disease severity was rated for both plant canopy (above ground) and root (underground) visually in 2013 experiment. In 2014 and 2015, the incidence of diseased plants per foot was recorded. Based on symptom development, the disease ratings were measured at 105 DAP in 2013; three times in 2014: 116 DAP, 140 DAP and at harvest after plants were dug and inverted (144 DAP); and two times in 2015: 126 DAP and at harvest after plant were dug and inverted (137 DAP).
The phenotypic data from FL experiments with broadcast inoculation were denoted as B followed by year in two digits and point inoculation with two isolates were denoted as “WF” (for isolate SR-18) and “BF” (for isolate LE948) followed by year in two digits. Ratings on plants above ground and underground were denoted as “BA” and “BU,” respectively. In addition, multiple times of ratings (three and two times in 2014 and 2015, respectively) were conducted and the datasets were differentiated by “_” followed by a number (Table S1). The data from experiments in GA with disease score and percentage of asymptomatic plants were denoted as “SC” and “PCT,” respectively, followed by years in two digits (Table S1). Therefore, in total, 24 sets of phenotypic data were collected, including 18 sets (BF13, BA13, BU13, B14_1, BF14_1, WF14_1 B14_2, BF14_2, WF14_2, B14_3, BF14_3, WF14_3, B15_1, BF15_1, WF15_1, B15_2, BF15_2 and WF15_2, Table S1) of data from the FL experiments and 6 sets (SC13, PCT13, SC14, PCT14, SC15 and PCT15) of data from GA.
Statistical analysis of phenotyping results
The distributions of the 24 sets of phenotypic data were tested by Shapiro–Wilk test (Shapiro and Wilk 1965). The distribution of dataset with P > 0.05 after Shapiro–Wilk test was considered normally distributed. The phenotype distribution was plotted using “ggplot2” package in R (Wickham 2016). Correlation coefficients among the 24 datasets and their statistical significance were tested by Spearman correlation (Zar 2005). The nonparametric Kruskal–Wallis one-way analysis of variance (ANOVA) was conducted for all datasets to determine each factor’s effects on the disease rating. For the datasets collected from GA, six Kruskal–Wallis one-way ANOVAs were conducted for factors of genotype, year and interaction of genotype and year for both disease score and percentage of asymptomatic plants (Table S2-1). For the datasets collected from FL, 19 Kruskal–Wallis one-way ANOVAs were conducted for datasets combined according to different year and inoculation methods for multiple possible factors (Table S2-2) and 14 Kruskal–Wallis one-way ANOVAs were conducted for datasets combined according to different inoculation method only (Table S2-3). The post hoc multiple comparisons were conducted using Dunn’s Test with Benjamini–Hochberg false discovery rate (FDR) adjustment (Benjamini and Hochberg 1995; Dunn 1964). The heritability of each dataset was estimated following the equation: \( h^{2} = \frac{{\sigma_{g}^{2} }}{{\sigma_{g}^{2} + \frac{{\sigma_{e}^{2} }}{r}}} \), in which \( \sigma_{g}^{2} \) and \( \sigma_{e}^{2} \) represented the genotypic variance and residual variance, respectively, while the r stood for the number of replicates. The variance of genetic effect and residual variance were estimated using a generalized linear model in R software (R Core Team 2015).
Genotyping and map construction
DNA of the parents and the 156 F6:7 RILs of the population was extracted using the Qiagen DNeasy Plant mini kit® and sent to Affymetrix for genotyping. SNP calls were curated using the Axiom Analysis Suite Software® (Thermo Fisher Scientific Inc. 2016) based on the clustering of data for the entire population and the parents. Also included were 111 fluorescence tagged SSRs (Guo et al. 2012) previously genotyped for this population. All RILs were checked for segregation distortion using a χ2 test and an expected 1:1 segregation ratio. Markers and RILs with more than 10% missing data were removed as well as the RILs with more than 20% heterozygote calls. A genetic map was constructed using JoinMap v4.1 (Van Ooijen 2006) with a minimum logarithm of the odds (LOD) of 3.0 and the Kosambi function.
QTL analysis
WinQTLCart v2.5 (https://brcwebportal.cos.ncsu.edu/qtlcart/WQTLCart.htm) software was used for mapping additive QTL using the composite interval mapping (CIM) function (Wang et al. 2012). The 24 datasets were input as individual traits for the mapping. The QTLs were scanned with a walking speed of 1 cM, control marker number of 5 and window size of 10 cM. Permutation analysis of 1000 times was used to determine the significant LOD score threshold (α = 0.05), and QTLs with a LOD score higher than this threshold were accepted as significant QTLs (Churchill and Doerge 1994).
QTLNetwork v2.0 (ibi.zju.edu.cn/software/qtlnetwork) was used to map QTLs with additive effect, epistatic effect (additive × additive) and QTL × environment interactions through a mixed model-based composite interval mapping (MCIM) method (Yang et al. 2008). The 24 single experiment datasets, 3-year combined data of disease score at GA, 3-year combined data of percentage of asymptomatic plants at GA, 2-year combined data (2014 and 2015) of broadcast at harvest collected at FL and 2-year combined data of point inoculation score at harvest at FL were input separately into the software for the mapping. The marker interval analysis was used to scan QTL, and a two-dimensional scan was used to find epistasis based on detected QTLs and QTL interval interaction analysis. Finally, Markov chain Monte Carlo (MCMC) algorithm was used to estimate the QTL main additive effect, epistatic effect and environment interaction effect (Wang et al. 1994). The 1000-time permutation analysis was used to determine the critical (α = 0.05) F-value for QTL detection. The significance level of 0.05 was adopted for candidate interval selection, putative QTL detection and QTL effects determination. For QTL detection, the testing window size, walk speed and filtration window size were set as 10 cM, 1 cM and 10 cM, respectively.
QTL IciMapping v4.2 (http://www.isbreeding.net/software/?type=detail&id=29) was further used to detect additive QTL and epistasis QTL using inclusive composite interval mapping (ICIM) method (Meng et al. 2015; Li et al. 2007; Li et al. 2008; Li et al. 2015). The single experiment datasets and multiple-year combined datasets used for QTLNetwork were inputted to QTL IciMapping for scanning using BIP and MET function, respectively. The mapping steps and P values for entering variables (PIN) for additive QTL scan were set to 1 cM and 0.001, respectively, while 5 cM mapping step and 0.0001 PIN were used for epistasis QTL mapping. The LOD thresholds for all QTL were determined by 1000-time permutation (α = 0.05).
The overlapped QTLs were identified from different sets of phenotypic data or different software if the LOD peaks of the QTLs were close on the linkage group and their confidence intervals were overlapped to each other. These overlapped original QTLs were then integrated using QTL meta-analysis by BioMercator v4.2 to get non-overlapping consensus QTL (moulon.inra.fr/index.php/fr/seminairedoc/cat_view/21-logiciels/101-abi-project-and-software/104-biomercator) (De Oliveira et al. 2014). The variance of position of the overlapping QTLs was taken into consideration to predict the most likely number of QTLs in the overlapping region (Goffinet and Gerber 2000; Sosnowski et al. 2012). The consensus QTLs after meta-analysis were plotted on linkage groups using “circlize” package in R (Gu et al. 2014).
Discover candidate genes within stem rot resistance QTL intervals
In order to reveal candidate gene for stem rot resistance within the QTL regions, probe sequence of SNP markers and primer sequence of SSR markers flanking the QTL were positioned on the cultivated peanut reference genomes (peanutbase.org, Bertioli et al. 2019) by BLAST analysis (Altschul et al. 1990) with an e-value cutoff of 1e−6. The positions of the top hits on the reference genomes by the QTL flanking markers, corresponding to linkage groups, were used to define the QTL interval to search for candidate genes (peanutbase.org). Gene models within the QTL regions and with annotated functions related to disease resistance were selected as stem rot resistance candidate genes. Specifically, genes with annotated functions such as chitinase, wall receptor kinases, ethylene-responsive transcription factors, peroxidases, MYB transcription factors, pathogenesis-related (PR) protein, nucleotide-binding site leucine-rich repeat (NBS-LRR) proteins and Glutathione S-transferase (GST) that are considered to be associated with biotic stress response or pathogen infection were searched (Adrian and Jeandet 2012; Marrs 1996; McHale et al. 2006; Rajyaguru et al. 2017; Vasconcellos et al. 2017).
To test whether disease-related genes were enriched in the QTL regions, a hypergeometric test was conducted using Hypergeometric package in R (Johnson et al. 2005), which takes into account the statistical significance of the number of k resistance-related genes in QTL regions (out of total of n genes in QTL regions) from the total of N genes in the genome containing K resistance-related genes. The probability of taking a resistance gene from QTL regions was \( P = \frac{{\left( {\begin{array}{*{20}c} K \\ k \\ \end{array} } \right)\left( {\begin{array}{*{20}c} {N - K} \\ {n - k} \\ \end{array} } \right)}}{{\left( {\begin{array}{*{20}c} N \\ n \\ \end{array} } \right)}} \).
Results
Phenotypic data analysis
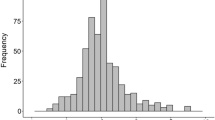
The distributions of the 24 sets of phenotypic data varied, and none of these datasets strictly followed a normal distribution (Fig. S1, Table 1). Noticeably, the datasets collected in early ratings were all left-skewed with the peak near zero disease score, while the distributions of ratings at harvest stage were closer to a normal distribution (Fig. S1). This indicated that most lines were not infected or had not shown symptoms yet at early disease rating time point.
Significant phenotype correlation (P < 0.05) was observed across most datasets (245 out of 276 experiment pairs) for the RIL population (Table S3). Specifically, all the disease symptom scores showed a high negative correlation (\( \left| r \right| \) > 0.6, P < 0.01) with the percentage of asymptomatic plants among the datasets from GA. Furthermore, the disease symptom scores in different years showed high positive correlation with each other (\( \left| r \right| \) > 0.5, P < 0.01), which suggested the high reliability of this phenotyping method. Moreover, all datasets from GA showed significant correlation (P < 0.05) with datasets in FL except some of the early rating time points in 2014 and 2015 (B14_1, BF14_1, WF14, 1 BF15_1, WF15_1). Their correlation coefficient with early ratings was also lower than ratings at harvest in FL. All the datasets from FL were positively correlated with each other. The ratings at harvest in the same year showed the highest correlation with each other (\( \left| r \right| \) > 0.6, P < 0.01).
Kruskal–Wallis one-way ANOVA showed that significant phenotypic differences (P < 0.01) between genotypes were observed in all datasets (Table S2). For broadcast inoculation datasets in FL, there was a significant phenotypic difference (P < 0.01) between years and rating time points. In 2013, the disease severity rated before digging on harvest date (BA13) was significantly lower (P < 0.01) than rating after digging (BU13) on the same date. These results indicated that the disease symptom was more visible after digging than before; thus, rating after digging should be recommended for the disease evaluation. For point inoculation, there was a significant phenotype difference observed between different rating time points, inoculums and years. Multiple pairwise comparisons showed that phenotype rated at harvest was significantly higher than earlier ratings (P < 0.01). Moreover, SR-18 inoculum consistently induced more severe disease symptoms every year compared to LE948. For disease rating and percentage of asymptomatic lines in GA datasets, significant phenotypic differences (P < 0.01) were observed between genotypes and between years. But the difference between years was mainly caused by the 2015 dataset because the dataset in 2015 is significantly lower and the datasets from 2013 and 2014 were similar. These results suggested that disease severity rating at harvest (preferable after digging out the plants) using SR-18 inoculum is more reliable for characterizing stem rot disease resistance.
Heritability calculated from disease scores in GA (SC13, SC14, SC15) and disease rating at harvest in FL (B14_3, BF14_3, WF14_3, B15_2, BF15_2, WF15_2) were relatively higher than other datasets (averaging 74%), which showed that phenotyping on disease severity, disease migration and occurrence number at harvest can capture the most genetic variance and it is effective for stem rot symptom phenotyping.
Stem rot resistance QTL detection
The linkage map consisted of 1524 markers (1451 SNP markers and 73 SSR markers) (described in detail in Chavarro et al. submitted). The 1524 markers were assigned to 29 linkage groups spanning 3381.96 cM (Table S4).
In order to capture all possible genes involved in stem rot disease resistance, all datasets were used for QTL mapping. For additive QTL mapping, a total of 26, 14 and 41 original additive QTLs were detected by WinQTLCart, QTLNetwork and QTL IciMapping, respectively (Table S5). The average LOD threshold determined by permutation was 3.57 (ranging from 2.98 to 4.79). QTLs were detected in 19 out of the 24 single experiment datasets except for B14_1, BF14_1, BF14_3, BF15_1 and WF15_2 by at least one software. The heritability of B14_1 and BF14_1 was relatively low (< 30%), but the heritability of BF14_3, BF15_1 and WF15_2 was all above 69% (Table S6). Besides, the high heritability of phenotype datasets did not translate into high PVE by QTLs detected using that dataset (correlation of 0.45), indicating that there may be many small effect loci contributed to these traits. Overlaps of confidence intervals of original QTLs detected from different experimental datasets or different software were integrated by QTL meta-analysis. As a result of QTL meta-analysis, a total of 33 consensus additive QTLs were identified on 15 linkage groups with an average LOD score, confidence interval size and PVE of 4.27, 5.46 cM and 10.13%, respectively (Fig. 1, Table S7). Notably, 16 out of the 33 consensus additive QTLs showed a PVE higher than 10%. In total, 13 out of the 33 consensus additive QTLs were repeatedly detected in different experiments or by different QTL analysis software above. Among the 13 repetitively detected QTLs, 11 of them could be detected from experiments in different years or locations. Importantly, six out of the 33 consensus additive QTLs (qSR.A01-2, qSR.A01-5, qSR.A05/B05-1, qSR.A05/B05-2, qSR.A07/B07-1 and qSR.B05-1) had a PVE higher than 10% and could be detected in different years or locations. Among the 33 consensus additive QTLs, the NC3033 alleles contributed to stem rot resistance on 18 QTLs and Tifrunner alleles on 14 QTLs were responsible for resistance (Table S7). Interestingly, there was one consensus additive QTL, qSR.A01-2, which had favorable alleles from either parents depending on the trait. Tifrunner allele contributed to improving the percentage of asymptomatic, while NC3033 allele was responsible for reducing disease symptom and disease dispersal. Three software with different QTL mapping algorithms were used for the QTL detection in this study, and only 10 out of the 33 consensus additive QTLs were detected by multiple software. There were 11 QTLs specifically detected by QTL IciMapping, 2 QTLs specifically detected by QTLNetwork, and 10 QTLs only detected by WinQTLCart.

Distribution of stem rot QTLs on linkage groups. Blue lines in the outer track represent the marker position on each linkage group; bars in the inner track indicated the LOD score for each QTL; the red lines under the track circle represent the presence of QTL on corresponding linkage group; the green lines linked different QTLs show the epistatic interaction between QTLs (color figure online)
The QTL with epistatic effect and QTL × environment interactions were explored using QTLNetwork and QTL IciMapping. As a result, 12 pairs of epistatic QTLs were detected but no significant QTL × environment interaction was discovered (Fig. 1, Table S8). The majority (23 of 24) of the epistatic QTLs were novel QTLs that can only be detected when considering the digenic interaction. Only qSR.A01-2, who was previously detected as an additive QTL using the 2013 disease score dataset, also showed epistatic interaction with qSR.B07_1-1 to affect the percentage of asymptomatic plants per row.
Stem rot resistance candidate genes
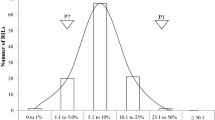
In order to identify candidate genes that may contribute to stem rot resistance, marker sequences of the linkage map were blasted to the cultivated peanut genome (peanutbase.org). Finally, 1514 markers were successfully aligned to the chromosomes with more than 98% identity. Flanking markers of all stem rot QTLs were aligned to the reference genome for candidate gene search. The average chromosome coverage of the 56 additive and epistatic QTLs on the reference genome is ~ 4.73 Mb, with 26 QTLs covering less than 1 Mb on the genome (Table S5). A total of 6134 genes were identified within all QTL regions. As a result of candidate gene search, a total of 193 disease resistance-related genes were identified on 38 QTLs (Fig. 2, Table S9). Noticeably, a large number of disease resistance protein genes including nucleotide-binding site leucine-rich repeat (NBS-LRR) proteins located within the regions of qSR.B01-1(15 genes), qSR.B02-1(24 genes) and qSR.B02-2 (33 genes) (Fig. 2). Hypergeometric test on the disease resistance-related genes for all QTLs showed that chitinase and fungus defense-related genes, ethylene-responsive transcription factor and wall receptor kinase were enriched (P < 0.05) in stem rot QTL regions (Table 2). Enrichment of these disease-related genes in stem rot resistance QTL regions suggested that these genes might play a role in stem rot resistance.

Distribution of candidate resistance genes within stem rot QTL
Discussion
Stem rot disease is one of the major diseases threatening peanut production, and breeding for resistant cultivars is the most economical and environmentally friendly way to reduce the yield loss. However, no stem rot resistance genes have been cloned and the genetics of stem rot resistance is largely unknown. The aim of this study was to map the key resistance genes to stem rot disease in peanut and identify useful markers for breeding programs by QTL mapping. Multiple years and locations phenotypic data were collected, and a SNP-based linkage map was used to identify stem rot resistance QTL and QTL interactions.
The stem rot pathogenesis includes pathogen penetration, infection and transmission. We used different disease scoring methods such as measuring disease incidence and spread distance along with multiple rating time points in our study, which enabled us to capture resistance loci involved in these stages. In FL experiments, disease dispersal index measurement on point inoculation at the harvest is more reliable because the data showed higher correlation with disease symptom ratings (Table S3). Phenotyping at harvest with the root/pots digging out is optimal rating time for evaluating stem rot disease because the disease symptoms are fully developed and visible to visual rating. The heritability calculated using these data was also significantly higher compared to data collected at early stages after infection (Table 1). Only 12 out of the total 56 QTLs can be detected using datasets at early stages (Table S5). Therefore, it is recommended to use disease symptom rating or disease dispersal index measurements taken at harvest for phenotyping stem rot disease on peanut. Two S. rolfsii isolates were used in our study, and significant phenotypic difference was observed between the two sources. The SR-18 inoculum induced a higher average disease score, indicating that this isolate is a more virulent pathogen. For the disease severity score recorded in GA experiments, the percentage of asymptomatic plants per row was consistently higher in the resistant parent NC 3033 and the disease symptom score of NC 3033 was generally lower except for the 2015 experiment (Table 1). However, for the disease dispersal index phenotype recorded in FL experiments, Tifrunner showed a lower score in most experiments compared to NC 3033. We also noticed that the population with Tifrunner genotype at 12 QTLs showed a more resistant phenotype on average. We speculate that Tifrunner had an overall resistance to fungus penetration, which contributed to the lower score in disease dispersal index. But once the S. rolfsii enters the plant, it will cause more severe disease symptoms.
The linkage map used in any QTL mapping study determines its power for detecting QTLs and its usefulness for the community. Many linkage maps have been constructed in cultivated peanut to map various important traits in the last decades; however, most of them were constructed by SSR/AFLP markers with a low marker density (Chen et al. 2016; Herselman et al. 2004; Huang et al. 2015; Sujay et al. 2012). Furthermore, many markers developed from specific populations are not transferable to other studies, and the QTL results are usually applicable to specific populations. SNP markers are abundant in plant genomes and suitable for high-throughput detection platforms (Schlötterer 2004). Several high-density linkage maps have been constructed by SNP markers generated by NGS (Hu et al. 2018; Zhao et al. 2016; Zhou et al. 2014). Compared to the sequencing experiments, genotyping using SNP arrays is relatively time and cost efficient. A 58 K SNP array was designed for cultivated peanut (Clevenger et al. 2017; Pandey et al. 2017), which greatly facilitates the high-throughput genotyping in the peanut research community. Using this peanut array, we were able to construct a high-density linkage map (1524 markers covering 3381 cM) for stem rot QTL mapping.
Complex regulatory networks are involved in plant stress response (Fujita et al. 2006; Shinozaki et al. 2003). We hypothesize that the interaction between different genes may also play a role in stem rot resistance genetics. Therefore, epistatic (additive × additive) and QTL × environment effect were also explored in this study. A total of 12 pairs of epistatic QTLs were detected (Fig. 1, Table S7). One of the QTL qSR.A01-2 was detected as an additive QTL related to disease severity and disease dispersal in 2013 experiment. In 2015 experiment, it interacted with a new locus qSR.B07_1-1 to affect the resistance to stem rot (percentage of asymptomatic plants). The average PVE of each pair of epistatic QTLs was ~ 3.8%, but together they contributed to significant amount of phenotypic variation. For example, two pairs of epistatic QTLs detected using combined datasets of percentage of infected plant from 2014 to 2015 contributed to 7.58% PVE. Eight pairs of epistatic QTLs detected using combined datasets of percentage of asymptomatic plant from 2013 to 2015 contributed to 26.54% PVE. Taking the resistant genotypes of the epistatic QTLs into consideration in MAS could further improve selection efficiency in breeding programs. However, no significant QTL × environment interaction was identified. This is probably because we analyzed the multi-year datasets from FL and GA separately due to the different phenotyping methods used in these locations.
Flanking marker sequences of previously identified stem rot resistance QTLs and sequences of stem rot resistance contigs were aligned to the reference genome to compare with our QTL result (Bera et al. 2016; Jogi et al. 2016; Dodia et al. 2019). The stem rot QTL qstga01.1 reported by Bera et al. (2016) was aligned to chromosome A01 (90386756-100852249 bp) which happened to overlap with our QTL qSR.A01-1 within a 4.56 Mb overlapping region. Two resistance candidate genes that related to peroxidase (arahy.2QEP5L) and MYB transcription factors (arahy.H5DQEZ) were found in this overlapping region. Fifteen resistance-related contigs were reported by Jogi et al. (2016), and seven of them could be aligned to 14 gene models in the reference genome. Among the 14 gene models, arahy.3X96H9, arahy.NJY7IU, arahy.3WH5A3 and arahy.UE6XF8 located in the region of qSR.B01-1, qSR.A06-2, qSR.B06_1-8 and qSR.B06_1-3, respectively. The gene arahy.UE6XF8 encoded E3 ubiquitin protein ligase, while arahy.NJY7IU encoded chitinase which involved in defense response to fungus. The arahy.3X96H9 gene also encoded a disease resistance response protein, and arahy.3WH5A3 encoded a chalcone synthase. Seven stem rot resistance QTLs were reported recently by Dodia et al. (2019) using genotyping-by-sequencing-based genetic mapping, and three of these QTLs were overlapped with three QTLs detected in our study. The q12DAI_S1B06.1 overlapped with qSR.B06_1-5 and four genes (arahy.IYK5A5, arahy.K3BVMJ, arahy.I2ZHTU and arahy.804PS6) both encoding ethylene-responsive transcription factor were discovered in this region. The q12DAI_S3B04.2 and q6DAI_S3B04.2 both overlapped with qSR.B04-1 and gene arahy.ZN5I3T encoded NBS-LRR disease resistance protein located in the overlapped region. The overlap of QTL regions with previous studies and resistance-related genes confirmed the efficacy of QTL identification in our study. Candidate genes listed in Table S9 can be selected for resistance function characterization experiments.
In summary, numerous QTLs for stem rot resistance were identified based on a comprehensive phenotyping and analysis of both additive and epistatic effects. These results provide further understanding of stem rot resistance in peanut. Stem rot resistance QTLs and candidate genes identified here based on the high-density SNP linkage map will benefit peanut breeding programs implementing MAS for stem rot resistance.
References
Adrian M, Jeandet P (2012) Effects of resveratrol on the ultrastructure of Botrytis cinerea conidia and biological significance in plant/pathogen interactions. Fitoterapia 83:1345–1350
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Anco D (2017) Peanut disease management. South carolina pest management handbook for field crops. South Carolina State Library, Columbia, pp 190–201
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 57:289–300
Bera SK, Kamdar JH, Kasundra SV, Ajay BC (2016) A novel QTL governing resistance to stem rot disease caused by Sclerotium rolfsii in peanut. Aust Plant Pathol 45:637–644
Bertioli DJ, Cannon SB, Froenicke L, Huang G, Farmer AD, Cannon EK, Liu X, Gao D, Clevenger J, Dash S (2016) The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat Genet 47:438
Bertioli DJ, Jenkins J, Clevenger J, Dudchenko O, Gao D, Seijo G, Leal-Bertioli SCM, Ren L, Farmer AD, Pandey MK, Samoluk SS, Abernathy B, Agarwal G, Ballén-Taborda C, Cameron C, Campbell J, Chavarro C, Chitikineni A, Chu Y, Dash S, El Baidouri M, Guo B, Huang W, Kim KD, Korani W, Lanciano S, Lui CG, Mirouze M, Moretzsohn MC, Pham M, Shin JH, Shirasawa K, Sinharoy S, Sreedasyam A, Weeks NT, Zhang X, Zheng Z, Sun Z, Froenicke L, Aiden EL, Michelmore R, Varshney RK, Holbrook CC, Cannon EKS, Scheffler BE, Grimwood J, Ozias-Akins P, Cannon SB, Jackson SA, Schmutz J (2019) The genome sequence of segmental allotetraploid peanut Arachis hypogaea. Nat Genet 51:877–884
Beute M, Wynne J, Emery DJCS (1976) Registration of NC 3033 Peanut Germplasm1 (Reg. No. GP 9) 16:887-887
Bocianowski J (2013) Epistasis interaction of QTL effects as a genetic parameter influencing estimation of the genetic additive effect. Genet Mol Biol 36:93–100
Chen W, Jiao Y, Cheng L, Huang L, Liao B, Tang M, Ren X, Zhou X, Chen Y, Jiang H (2016) Quantitative trait locus analysis for pod- and kernel-related traits in the cultivated peanut (Arachis hypogaea L.). BMC Genet 17:25
Chu Y, Chee P, Culbreath A, Isleib TG, Holbrook CC, Ozias-Akins P (2019) Major QTLs for resistance to early and late leaf spot diseases are identified on chromosomes 3 and 5 in peanut (Arachis hypogaea). Front Plant Sci 10:883
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Clevenger J, Chu Y, Chavarro C, Agarwal G, Bertioli DJ, Leal-Bertioli SCM, Pandey MK, Vaughn J, Abernathy B, Barkley NA, Hovav R, Burow M, Nayak SN, Chitikineni A, Isleib TG, Holbrook CC, Jackson SA, Varshney RK, Ozias-Akins P (2017) Genome-wide SNP genotyping resolves signatures of selection and tetrasomic recombination in peanut. Mol Plant 10:309–322
Core Team R (2015) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Culbreath AK, Brenneman TB, Bondari K, Reynolds KL, McLean HS (1995) Late leaf spot, southern stem rot, and peanut yield responses to rates of cyproconazole and chlorothalonil applied alone and in combination. Plant Dis 79:1121–1125
De Oliveira Y, Sosnowski O, Charcosset A, Joets J (2014) BioMercator 4: a complete framework to integrate QTL, meta-QTL, and genome annotation. In: European conference on computational biology, Strasbourg
Dodia S, Rathnakumar A, Mishra G, Radhakrishnan T, Binal J, Thirumalaisamy P, Narendra K, Sumitra C, Dobaria J, Abhay K (2016) Phenotyping and molecular marker analysis for stem-rot disease resistance using F2 mapping population in groundnut. Int J Trop Agric 34:1135–1139
Dodia SM, Joshi B, Gangurde SS, Thirumalaisamy PP, Mishra GP, Narandrakumar D, Soni P, Rathnakumar AL, Dobaria JR, Sangh C, Chitikineni A (2019) Genotyping-by-sequencing based genetic mapping reveals large number of epistatic interactions for stem rot resistance in groundnut. Theor Appl Genet 132(4):1001–1016
Dunn OJ (1964) Multiple comparisons using rank sums. Technometrics 6:241–252
Fujita M, Fujita Y, Noutoshi Y, Takahashi F, Narusaka Y, Yamaguchi-Shinozaki K, Shinozaki K (2006) Crosstalk between abiotic and biotic stress responses: a current view from the points of convergence in the stress signaling networks. Curr Opin Plant Biol 9:436–442
Goffinet B, Gerber S (2000) Quantitative trait loci: a meta-analysis. Genetics 155:463–473
Gu Z, Gu L, Eils R, Schlesner M, Brors B (2014) circlize implements and enhances circular visualization in R. Bioinformatics 30(19):2811–2812
Guo Y, Khanal S, Tang S, Bowers JE, Heesacker AF, Khalilian N, Nagy ED, Zhang D, Taylor CA, Stalker HT, Ozias-Akins P, Knapp SJ (2012) Comparative mapping in intraspecific populations uncovers a high degree of macrosynteny between A- and B-genome diploid species of peanut. BMC Genom 13:608
Herselman L, Thwaites R, Kimmins F, Courtois B, Van Der Merwe P, Seal SE (2004) Identification and mapping of AFLP markers linked to peanut (Arachis hypogaea L.) resistance to the aphid vector of groundnut rosette disease. Theor Appl Genet 109:1426–1433
Holbrook CC, Culbreath AK (2007) Registration of ‘Tifrunner’peanut. Plant Regist 1(124):10–3198
Holbrook C, Isleib T, Ozias-Akins P, Chu Y, Knapp S, Tillman B, Guo B, Gill R, Burow MJPS (2013) Development and phenotyping of recombinant inbred line (RIL) populations for peanut (Arachis hypogaea). Peanut Sci 40:89–94
Hu X, Zhang S, Miao H, Cui F, Shen Y, Yang W, Xu T, Chen N, Chi X, Zhang Z (2018) High-density genetic map construction and identification of QTLs controlling oleic and linoleic acid in peanut using SLAF-seq and SSRs. Sci Rep 8:5479
Huang L, He H, Chen W, Ren X, Chen Y, Zhou X, Xia Y, Wang X, Jiang X, Liao B (2015) Quantitative trait locus analysis of agronomic and quality-related traits in cultivated peanut (Arachis hypogaea L.). Theor Appl Genet 128:1103–1115
Jogi A, Kerry JW, Brenneman TB, Leebens-Mack JH, Gold SE (2016) Identification of genes differentially expressed during early interactions between the stem rot fungus (Sclerotium rolfsii) and peanut (Arachis hypogaea) cultivars with increasing disease resistance levels. Microbiol Res 184:1–12
Johnson NL, Kemp AW, Kotz S (2005) Univariate discrete distributions. Wiley, New York
Khatri K, Kunwar S, Barocco R, Dufault NJPS (2017) Monitoring fungicide sensitivity levels and mycelial compatibility groupings of Sclerotium rolfsii Isolates from Florida peanut fields. Peanut Sci 44:83–92
Kokalis-Burelle N, Porter D, Rodriguez-Kabana R, Smith D, Subrahmanyam P (1997) Compendium of peanut diseases. American Phytopathological Society, Saint Paul
Li H, Ye G, Wang J (2007) A modified algorithm for the improvement of composite interval mapping. Genetics 175(1):361–374
Li H, Ribaut JM, Li Z, Wang J (2008) Inclusive composite interval mapping (ICIM) for digenic epistasis of quantitative traits in biparental populations. Theor Appl Genet 116(2):243–260
Li S, Wang J, Zhang L (2015) Inclusive composite interval mapping of QTL by environment interactions in biparental populations. PLoS ONE 10(7):e0132414
Little E (2015) Georgia plant disease loss estimates. The University of Georgia Cooperative Extension Bulletin, University of Georgia, Athens
Mamidi S, Miklas PN, Trapp J, Felicetti E, Grimwood J, Schmutz J, Lee R, McClean PE (2016) Sequence-based introgression mapping identifies candidate white mold tolerance genes in common bean. Plant Genome 9:2
Marrs KA (1996) The functions and regulation of glutathione S-transferases in plants. Annu Rev Plant Biol 47:127–158
McHale L, Tan X, Koehl P, Michelmore RW (2006) Plant NBS-LRR proteins: adaptable guards. Genome Biol 7:212
Meng L, Li H, Zhang L, Wang J (2015) QTL IciMapping: integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J 3(3):269–283
Monnahan PJ, Kelly JK (2015) Epistasis is a major determinant of the additive genetic variance in Mimulus guttatus. PLoS Genet 11:e1005201
Morrell PL, Buckler ES, Ross-Ibarra J (2012) Crop genomics: advances and applications. Nat Rev Genet 13:85
Pandey MK, Agarwal G, Kale SM, Clevenger J, Nayak SN, Sriswathi M, Chitikineni A, Chavarro C, Chen X, Upadhyaya HD (2017) Development and evaluation of a high density genotyping ‘Axiom_Arachis’ array with 58K SNPs for accelerating genetics and breeding in groundnut. Sci Rep 7:40577
Punja ZK (1988) Sclerotium (Athelia) Rolfsii, a pathogen of many plant species. In: Sidhu GS (ed) Genetics of plant pathogenic fungi. Advances in plant pathology. Academic Press, Cambridge, pp 523–534
Rajyaguru RH, Thirumalaisamy P, Patel KG, Thumar JT (2017) Biochemical basis of genotypic and bio-agent induced stem rot resistance in groundnut. Legume Res Int J 40:2
Rasheed A, Hao Y, Xia X, Khan A, Xu Y, Varshney RK, He Z (2017) Crop breeding chips and genotyping platforms: progress, challenges, and perspectives. Mol Plant 10:1047–1064
Schlötterer C (2004) The evolution of molecular markers—just a matter of fashion? Nat Rev Genet 5:63
Shapiro SS, Wilk MB (1965) An analysis of variance test for normality. Biometrika 52:591–611
Shinozaki K, Yamaguchi-Shinozaki K, Seki M (2003) Regulatory network of gene expression in the drought and cold stress responses. Curr Opin Plant Biol 6:410–417
Sosnowski O, Charcosset A, Joets J (2012) BioMercator V3: an upgrade of genetic map compilation and quantitative trait loci meta-analysis algorithms. Bioinformatics 28:2082–2083
Sujay V, Gowda M, Pandey M, Bhat R, Khedikar Y, Nadaf H, Gautami B, Sarvamangala C, Lingaraju S, Radhakrishan T (2012) Quantitative trait locus analysis and construction of consensus genetic map for foliar disease resistance based on two recombinant inbred line populations in cultivated groundnut (Arachis hypogaea L.). Mol Breed 30:773–788
Van Loon LC, Rep M, Pieterse CM (2006) Significance of inducible defense-related proteins in infected plants. Annu Rev Phytopathol 44:135–162
Van Ooijen JW (2006) JoinMap 4®, software for the calculation of genetic linkage maps in experimental populations. Kyazma BV, Wageningen
Vasconcellos RC, Oraguzie OB, Soler A, Arkwazee H, Myers JR, Ferreira JJ, Song Q, McClean P, Miklas PN (2017) Meta-QTL for resistance to white mold in common bean. PLoS ONE 12:e0171685
Vuong TD, Diers BW, Hartman GL (2008) Identification of QTL for resistance to Sclerotinia stem rot in soybean plant introduction 194639. Crop Sci 48:2209–2214
Wang C, Rutledge J, Gianola D (1994) Bayesian analysis of mixed linear models via Gibbs sampling with an application to litter size in Iberian pigs. Genet Sel Evol 26:91
Wang S, Basten C, Zeng Z (2012) Windows QTL cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh
Wickham H (2016) ggplot2: elegant graphics for data analysis. Springer, Berlin
Yang J, Hu C, Hu H, Yu R, Xia Z, Ye X, Zhu J (2008) QTLNetwork: mapping and visualizing genetic architecture of complex traits in experimental populations. Bioinformatics 24:721–723
Zar JH (2005) Spearman rank correlation. Encycl Biostat 7:121
Zhao X, Han Y, Li Y, Liu D, Sun M, Zhao Y, Lv C, Li D, Yang Z, Huang L (2015) Loci and candidate gene identification for resistance to Sclerotinia sclerotiorum in soybean (Glycine max L. Merr.) via association and linkage maps. Plant J 82:245–255
Zhao Y, Zhang C, Chen H, Yuan M, Nipper R, Prakash C, Zhuang W, He G (2016) QTL mapping for bacterial wilt resistance in peanut (Arachis hypogaea L.). Mol Breed 36:13
Zhou X, Xia Y, Ren X, Chen Y, Huang L, Huang S, Liao B, Lei Y, Yan L, Jiang H (2014) Construction of a SNP-based genetic linkage map in cultivated peanut based on large scale marker development using next-generation double-digest restriction-site-associated DNA sequencing (ddRADseq). BMC Genom 15:351
Zhuang W, Chen H, Yang M, Wang J et al (2019) The genome of cultivated peanut provides insight into legume karyotypes, polyploid evolution and crop domestication. Nat Genet 51(5):865
Funding
This study was funded by the Florida Peanut Producers Association, the National Peanut Foundation and USDA National Institute of Food and Agriculture, Hatch Project 1011664.
Author information
Authors and Affiliations
Contributions
JW coordinated the research. YC, TGI, POA and CH developed the RIL population. ND and TB provided the inoculum and advised on the inoculation procedure. BT and JW supervised the phenotyping in Florida. YCT, HZ, ZP, XY, YL and JW conducted the experiments and collected the phenotypic data in Florida. TB and POA supervised the research in Georgia. RC and TB conducted the experiments and collected the phenotypic data in Georgia. CC and POA conducted the linkage map analysis. ZL analyzed the whole data sets and prepared the manuscript draft. All authors revised the manuscript and read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Data supporting the results are provided in the additional files and analyzed/organized in tables. The original data and the materials are available upon reasonable request to the corresponding author at wangjp@ufl.edu.
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Communicated by Albrecht E. Melchinger.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1
The distributions of phenotypic data from all datasets (TIFF 44237 kb)
Table S1
Description of experiments in this study (XLSX 10 kb)
Table S2
Kruskal-Wallis ANOVA test for different datasets (XLSX 11 kb)
Table S3
The correlation of phenotypic data from all experiments (XLSX 12 kb)
Table S4
Information of the linkage groups used for stem rot QTL mapping (XLSX 10 kb)
Table S5
The original additive QTLs detected for stem rot resistance by three types of software (XLSX 19 kb)
Table S6
Summary of original additive QTLs’ contribution to phenotype variations (XLSX 10 kb)
Table S7
The consensus additive QTLs detected for stem rot resistance after QTL meta-analysis (XLSX 15 kb)
Table S8
The epistatic QTLs for stem rot resistance detected by QTL IciMapping and QTLNetwork (XLSX 17 kb)
Table S9
The candidate genes identified within all stem rot QTL intervals (XLSX 27 kb)
Rights and permissions
About this article

Cite this article
Luo, Z., Cui, R., Chavarro, C. et al. Mapping quantitative trait loci (QTLs) and estimating the epistasis controlling stem rot resistance in cultivated peanut (Arachis hypogaea). Theor Appl Genet 133, 1201–1212 (2020). https://doi.org/10.1007/s00122-020-03542-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-020-03542-y