
Abstract
Key message
We discussed the most recent efforts in wheat functional genomics to discover new genes and their deployment in breeding with special emphasis on advances in Asian countries.
Abstract
Wheat research community is making significant progress to bridge genotype-to-phenotype gap and then applying this knowledge in genetic improvement. The advances in genomics and phenomics have intrigued wheat researchers in Asia to make best use of this knowledge in gene and trait discovery. These advancements include, but not limited to, map-based gene cloning, translational genomics, gene mapping, association genetics, gene editing and genomic selection. We reviewed more than 57 homeologous genes discovered underpinning important traits and multiple strategies used for their discovery. Further, the complementary advancements in wheat phenomics and analytical approaches to understand the genetics of wheat adaptability, resilience to climate extremes and resistance to pest and diseases were discussed. The challenge to build a gold standard reference genome sequence of bread wheat is now achieved and several de novo reference sequences from the cultivars representing different gene pools will be available soon. New pan-genome sequencing resources of wheat will strengthen the foundation required for accelerated gene discovery and provide more opportunities to practice the knowledge-based breeding.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Crop breeding is a continuous effort to ensure sustainable food production despite the challenges posed by growing global population, changing climate and emerging virulent strains of pathogens and pests. Historically, crop breeding relied on selection of visually important traits and has gone through several phases as the knowledge about the genetic control of such traits evolved and as the new technologies and innovations emerged. Knowledge-based breeding is the next epoch to deliver food in the most challenging time of human history due to the population growth and extreme weather events. Wheat (Triticum aestivum L.) is among the three most cultivated food crops which provide calories to one-fifth of the global population (FAO 2018). Identification of the genes involved in productivity traits and their accumulation followed by improved selection methods in the future cultivars will be central to crop breeding. Genomics and its associated technologies could play a pivotal role to achieve this goal (Bevan et al. 2017).
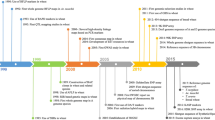
Wheat is both the easiest and most difficult crop to develop tools and resources required to practice knowledge-based breeding. For example, a huge array of wheat aneuploid stocks, which are not available to any other crop, were developed by Sears (1954). These aneuploids were developed in genetic background of wheat cv. Chinese Spring include possibly complete sets of nullisomics, trisomics, monosomics, tetrasomics and which were later accumulated by 41 out of the 42 possible telocentrics (Sears and Sears 1978). Such an extraordinary cytogenetic flexibility provided elegance and precision in genetic studies currently unattainable in few if any other higher organisms. This allowed a precise and often predictable genetic control over introduced diversity in the species, and major factors controlling major adaptability traits were physically mapped. The series of experiments then in wheat aneuploid lead to the discovery of genes for several important traits including waxiness, maturity (Driscoll and Jensen 1964), endosperm proteins (Shepherd 1968), vernalization (Halloran and Boydell 1967) and many more (Law 1966). These outcomes were so tempting that an effort was initiated in 1968 to catalogue wheat genes known as the Catalogue of Gene Symbols for Wheat by Prof. Robert McIntosh (McIntosh 1973). This seems unlikely when we see the progress of reference genome sequencing of wheat cultivar Chinese Spring. It took nearly 13 years to release a high-quality reference sequence in 2018 after establishing a consortium in 2005 (Appels et al. 2018). Such a difficulty in sequencing the genome of one of the most important food crops, when competitor food crops were sequenced quite early, was due to the very large genome size (~ 17 Gb; almost 6× bigger than human and 40× bigger than rice), polyploidy (6×) and highly repetitive genome contents (~ 80%).
An important use of genomics is to understand the full spectrum of genes involved in expression of a phenotype. The concept of marker-assisted selection using major alleles of vernalization, photoperiod response and grain size genes has contributed a lot in genetic improvement of crops (Würschum et al. 2015, 2017). There are several convincing evidence that new genomics resources and approaches could identify genetic basis for yield under target environment (Rasheed and Xia 2019). During the last few years, several high-throughput genotyping platforms in crops have been established as a result of technological innovations in genomics and allied disciplines (Li et al. 2018b). Use of these advances in genotyping technologies for crop breeding during the last two decades had hastened the finding of novel alleles (Rasheed et al. 2017). Here, we reviewed the gene discovery in wheat and the technological innovations that influenced the gene discovery studies with special emphasis to research conducted in Asian countries.
Population genomics resources with focus on Asian wheats
We have briefly described the major timelines and sequential activities in sequencing the bread wheat genome (Rasheed and Xia 2019). The Asian countries contributed in physical mapping and survey sequencing of chromosomes 2A (India) and 6B (Japan), 7D (China), and reference genome sequence of Aegilops tauschii (China) (Zhao et al. 2017). The reference genome sequence of bread wheat cv. Chinese Spring is now greatly facilitating the further studies to underpin the genes involved in major breeding traits and unravelling the quantitative genetic framework in wheat adaptability to target environments. The genome-wide resequencing of 120 wheat cultivars and landraces from China revealed that A and B subgenomes of modern Chinese cultivars were mainly derived from European landraces, while D subgenome is mostly derived from Chinese landraces (Chen et al. 2019a, b). The genomic regions were positively selected during modern wheat breeding in China included 48 high-confidence (HC) genes. The strongest signals were reported on chromosome 6B which contain genes TaNPF6.1-6B, a nitrate transporter gene; TaNAC24, associated with drought and heat stress adaptability; and TaRVE3, a circadian clock and flowering time regulator (Chen et al. 2019a, b). Apart from this genomic resource from China, several global efforts have successfully developed genomic resources that have implications for wheat breeding in Asia. A high-density SNP array analysis of 4506 cultivars and landraces originating from 105 countries provided a high-resolution phylogeographic insight on wheat breeding in Asia (Balfourier et al. 2019). The Asian wheats were culminated into two prominent groups including wheats from Southeast Asia (SEA), and Central Asia and Africa (CAA). The CAA cluster, mostly consisted of spring-type landraces, also had a prominent sub-cluster of the Indian Peninsula (INP) wheats. The INP cluster consisted of spring-type landraces from Pakistan, India and Nepal. The wheat landraces from the geographic region between the Black Sea and Caspian Sea were grouped under Caucasus (CAU) cluster. On the other hand, the landraces from Japan and China were clustered in the SEA group. A significant proportion (71%) of the modern cultivars was derived from the East European and Mediterranean pool, while ~ 94% of the landraces were derived from Asian pool (Balfourier et al. 2019). This indicated that breeding programs in Asia have higher reliance on European germplasm, especially those from Italy.
Pont et al. (2019) sequenced a panel of 500 worldwide wheat accessions using exome capture and provided insight into the selection and adaptation of wheat during the last 10,000 years that shaped the modern day wheat. The key findings include that (1) the genetic diversity in wheat is consistent with human migration pattern out of the Fertile Crescent, and Egypt to Maghreb (Northern Africa) a coastal route, (2) vernalization, historical groups and geographic origins (Europe, Asia, Oceania, Africa and America) were the three major driving forces partitioning genetic diversity, and (3) only the regions on chromosomes spanning 168 Mb containing selective sweeps were identical between the Asian and European germplasm, which indicated that genetic improvement targets were independent in these two germplasm pools. In another study, He et al. (2019) re-sequenced 890 diverse hexaploid and tetraploid wheat accessions to identify the wild-relative introgressions favoring global wheat adaptation. They concluded that introgressions from wild emmer were more frequent in wheat accessions from Europe than from Eastern Asia.
Previously, Chinese wheat landrace collection (Zhou et al. 2018b) and mini core collection (Hao et al. 2017) were genotyped with DArTseq markers and 9 K SNP array, respectively. Hao et al. (2017) traced back several genetic blocks to a century old famous Strampelli cross and identified significantly higher diversity in the gene coding regions in modern wheat cultivars. Zhou et al. (2018b) revealed that selective sweeps were distributed over 148 chromosomal regions in at least one geographic area. They postulated that wheat in China was adapted during its agricultural trajectory to increasingly mesic and warm climatic areas and spread from the northwestern Caspian Sea region to South China. Recently, the selection signals were identified and compared between cultivars and landraces from Pakistan and China using a 90 K SNP array (Liu et al. 2019b). Of the 477 genome-wide selective sweeps, 109 were shared in both Pakistan and China germplasm. It was further revealed that no germplasm admixture was observed among modern wheat germplasm from Pakistan and China despite being neighboring countries.
International Maize and Wheat Improvement Center (CIMMYT) has significant impact in developing improved wheat germplasm used in breeding or directly adapted for cultivation in several Asian countries. An extensive genomic analysis of 44,624 accessions from CIMMYT and phenotyping of 50 traits evaluated in South Asia, Africa and Americas identified important loci for grain quality, disease resistance and adaptability in stressed environments (Juliana et al. 2019). Genetic diversity studies for germplasm from Afghanistan (Manickavelu et al. 2014) and Japan (Kobayashi et al. 2016) using GBS markers, and from Kazakhstan using 90 K SNP array (Shavrukov et al. 2014) are also available. These studies not only had global breeding implications, but also provided a strong genomics foundation for wheat breeding in Asia. This would help to design strategies to efficiently manage economically important stresses like three rust diseases, wheat blast and resilience to climate change. All these studies provided insight into the population structure, genetic diversity, linkage disequilibrium decay and the identification chromosomal regions under artificial selection that would provide relevant guidelines to improve wheat and facilitate the germplasm use in breeding.
From QTL to gene discovery: strategies and inventory
Quantitative trait loci (QTL) mapping and genome-wide association studies (GWAS) from Asia dominated the research scenario in wheat genomics. These studies are the routine activities to discover the genetic basis of important phenotypes and result in underlying allelic variations, marker-traits associations and frequency of favorable alleles in the target germplasm and provide foundation for wheat functional genomics (Rasheed and Xia 2019). For example, almost all the economically important traits have been dissected using GWAS; however, the prominent limitations of GWAS are not ruled out. These limitations include problems of confounding population structure with low-frequency causal alleles leading to false-negative results and other unaccounted factors including low-accuracy genotype calls at some loci (Browning and Yu 2009) and small population size (Finnoet al. 2014). Therefore, further validation is necessary, which is not usually practiced in GWAS experiments. The validation could be cross-population, where candidate loci are either validated in bi-parental populations or independent germplasm collections; and the other could be biological validation, such as genetic transformation, gene silencing or gene knockout and gene editing (Liu and Yan 2019). In a GWAS experiment, TaRPP13L1 was associated with flour color in Chinese wheat cultivars, and the effect of the gene was validated in a doubled haploid (DH) mapping population and in two tetraploid wheat mutants carrying premature stop codons of the TaRPP13L1 gene (Chen et al. 2019b). Similarly, several marker-trait associations from GWAS experiments were validated in bi-parental populations for black point resistance (Liu et al. 2016, 2017), flour color (Zhai et al. 2016a, b), kernel number per spike (Shi et al. 2017) and thousand grain weight (Sun et al. 2017).
Another approach that is now widely used for gene discovery is the bulked segregant analysis (BSA) or bulked segregant RNA-seq (BSR-seq), which is a combination of RNA sequencing (RNA-seq) and BSA (Zou et al. 2016). BSA, BSR-seq or several of their variants are efficient methods to rapidly map QTL or genes. The process includes the pooling of DNA samples from individuals of a population showing contrasting phenotypes, which are subsequently sequenced to provide a simple and rapid way to identify consistent polymorphic regions in contrasting pools. This strategy not only enables discovery of SNPs linked to the target genes using NGS but also provides patterns of genes differentially expressing between pools. This approach has recently identified four candidate genes within a QTL interval, Qsm.hebau-4A, providing resistance against Orange wheat blossom midge (OWBM), which is a serious threat to wheat production in China, Canada, Russia, US and Japan (Hao et al. 2019). The discovery of candidate genes for OWBM resistance in Chinese wheat cv. Jimai 24 and development of six markers for diagnosis could be useful for breeding against midge resistance. The stripe rust resistance in Zhoumai 22 designated as YrZH22 has been confined to a narrow genetic interval spanning 4 Mb physical genomic region using BSR-seq approach (Wang et al. 2017a). There are several new genes discovered using BSR-seq such as powdery mildew resistance gene Pm4b (Wu et al. 2018a, b), Pm61 (Hu et al. 2019), leaf senescence gene els1 (Li et al. 2018a), stripe rust resistance gene Yr26 (Wu et al. 2018a), YrMM58, YrHY1 (Wang et al. 2018b), dwarfing gene Rht12 (Sun et al. 2019) and nitrogen-dependent lesion mimic gene Ndhrl1 (Li et al. 2016c). A diversity of genes underpinning important traits has been discovered using bulked sample approach, which indicate its success in initial mapping of gene with high certainty. It is expected that BSA or BSR-seq approaches will become increasingly important in crop genetics and breeding and have the potential to replace the analysis of entire population in several cases (Zou et al. 2016).
Although a huge array of genes has been cloned in wheat, very few were map-based cloned. Map-based cloning in bread wheat is still difficult due to the complexity of the wheat genome (Rasheed and Xia 2019). Alternatively, comparative genomics provides new opportunities for identification of genes because there is high collinearity in genetic organization among grass genomes. Using homology-based cloning approaches, a series of genes underpinning grain weight, grain yield and flowering time have been isolated in wheat. Actually, this arena of wheat genomics has dominated the research scenario in China. A good example is the cloning of more than 40 genes for grain morphology in rice using map-based cloning of quantitative trait loci (QTLs) and screening of T-DNA tagging libraries. The biological processes or function of these genes have been assigned which are related to the regulation of cell proliferation and cell elongation (Li et al. 2018b; Li and Yang 2017). To date, 33 grain size and weight-related genes have been isolated by homology-based cloning and used for developing diagnostic markers (Table 1). Several genes, including TaSus2-2A, TaTAR2.1-3A, TaCWI-4A, 6-SFT-A2, TaSus1-7A and TaTKW-7A, were closely (< 10 Mb) linked with yield-related loci identified in linkage and association mapping. Apart from the yield-related genes, some genes were related to flowering time, TaPRR73 (Zhang et al. 2016), TaZIM-A1 (Liu et al. 2019a); agronomic traits, TaPPH-7A (Wang et al. 2019a); root growth and plant height, TaARF4 (Wang et al. 2018a); and drought tolerance, TaSnRK2.9-5A (Ur Rehman et al. 2019).
Functional genomics and map-based cloning in wheat
The availability of new genomics resources like high-quality reference genome sequence, high-quality gene models, transcriptome databases, high-density SNP arrays and advancements in NGS is facilitating fine mapping and making map-based cloning more feasible in wheat. Yao et al. (2019) identified a grain yield QTL underpinning head length and spikelet number which was fine-mapped to a 0.2 cM interval, and the candidate gene was designated as Head Length 2 (HL2). Similarly, a gene for late heading was fine-mapped in an EMS mutant line and was designated as TaHdm605 (Zhang et al. 2018). A large-scale transcriptome analysis of 90 wheat lines identified the variations associated with spike architecture (Wang et al. 2017b). They identified three genes, TaTFL1-2D, TaHOX2-2B and TaAGLG1-5A, as regulators of spike architecture and biologically validated through wheat transgenic assays. In another study, the genes responding to abiotic stresses were selected from a yeast cDNA library constructed from a heat- and drought-tolerant wheat cv. Hanxuan 10 (Zhang et al. 2010). A gene TaPR-1-1 within the pathogenesis-related (PR) protein family was selected which conferred tolerance to abiotic stresses upon transgenesis in Arabidopsis and yeast. The screening of cDNA yeast library following abiotic stresses was found to be an efficient functional genomics tool to identify stress-tolerant genes.
The map-based cloning approach was used for Male Sterile 2 (Ms2), a gene inducing male sterility in wheat (Ni et al. 2017; Xia et al. 2017). The biological validation of the Ms2 using wheat transformation and induced mutations identified a terminal-repeat retrotransposon in miniature (TRIM) element in the promoter of Ms2. This TRIM element was responsible for the gene activation and consequent male sterility. The cloning of Ms2 provided substantial potential to exploit male sterility in wheat breeding for hybrid seed production. Similarly, a gene related to leaf angle, TaSPL8, was cloned using map-based cloning approach in wheat. TaSPL8 is responsible for the activation of genes associated with auxin and brassinosteroid pathways and cell elongation, and knockout mutants of this gene caused erected leaves due to the loss of the lamina joint, compact architecture and increased spike number (Liu et al. 2019c). Previously, a powdery mildew resistance gene, Pm21, from Haynaldia villosa was transferred to bread wheat in the form of 6VS.6AL translocation, and a Serine/threonine kinase gene was putatively cloned as the functional gene for Pm21 (Cao et al. 2011). The Pm21 gene is important for wheat production in China, and currently Pm21 containing cultivars are cultivated on ~ 4 million ha. Two complementary studies have recently used map-based cloning approach and identified Pm21 encode a typical CC–NBS–LRR protein which confers broad spectrum resistance to wheat powdery mildew disease (He et al. 2018a; Xing et al. 2018). These studies were supported by biological validations using multiple means.
Fusarium head blight (FHB) is a fungal disease of cereals which has become a major disease in most wheat producing areas. Despite intensive searches, no completely resistant germplasm has been identified except some landraces from China including Sumai 3, a Chinese wheat cultivar, which is known to carry Fhb1 gene. Sumai 3 is recognized as the best source of FHB resistance and has been widely used as a parent in many breeding programs. Rawat et al. (2016) reported that a pore-forming toxin-like (PFT) gene was the candidate for Fhb1 among 13 putative genes using positional cloning, mutation analysis, gene silencing and transgenic overexpression strategies, but PFT was subsequently found to be present in many FHB-susceptible cultivars casting doubt on the role of the gene (He et al. 2018b; Zhu et al. 2018). In two recent studies, a histidine-rich calcium-binding (TaHRC or His) gene adjacent to PFT from Ning 7840 and Wangshuibai, respectively, was identified as a better Fhb1 candidate. Su et al. (2019) proposed that the Fhb1 product is a susceptibility factor and FHB resistance results from loss-of-function. However, Li et al. (2019) concluded that Fhb1 is a gain-of-function gene and that the newly generated protein acts as a regulator of host immunity. Even though the discrepancies exist, all the three studies agreed upon that PFT gene was expressed at low level after anthesis and was expressed even less after FHB inoculation.
Functional marker development and application
The term of functional markers is used for the diagnostic PCR-based markers designed from causal polymorphisms within functional genes. Ultimately, the allelic variations of a specific functional genes are identified using functional markers (Liu et al. 2012). We have recently reviewed all the aspects of functional marker development and application (Rasheed and Xia 2019); therefore, this topic will be discussed briefly here. Functional markers are the markers of choice for practical breeding and can be used to pyramid genes or to introgress a specific gene. The use of functional markers in genomic selection could also improve selection accuracy. The availability of 72 functional markers in kompetitive allele-specific PCR (KASP) format enabled the high-throughput genotyping of large germplasm collections (Rasheed et al. 2016). Furthermore, this number has been recently increased to 157 KASP markers for various important phenotypic traits (Rasheed et al. unpublished data). Khalid et al. (2019) used KASP functional markers to genotype a diversity panel of synthetic-derived wheats characterized in optimal and water-limited conditions in Pakistan. They concluded that several functional markers of developmental genes like Vrn-A1, Rht-D1 and Ppd-B1 were associated with agronomic traits in both moisture conditions. The KASP assays of genes related to grain morphology were validated on CIMMYT elite lines and genes TaGS3-D1, TaTGW6 and TaSus1 showed significant effect on grain weight in historical CIMMYT germplasm (Sehgal et al. 2019). Zhao et al. (2019b) genotyped 1152 diverse global wheat accessions with KASP functional markers representing 47 genes controlling grain yield, quality, adaptation and stress tolerance. It was concluded that 22 alleles with positive effect of such genes were selected, while 14 alleles with negative effect were selected in global germplasm collection. This work comprehensively reported the human-mediated selection on favorable alleles of important genes in germplasm from Asia, Europe, North America and CIMMYT.
Another important aspect in application of KASP functional marker is the higher cost because KASP mastermix was only available from LGC™, a commercial proprietary. This led several groups to develop some open source SNP genotyping method. A recent example is the development of semi-thermal asymmetric reverse PCR (STARP) (Long et al. 2016) and Amplifluor (Jatayev et al. 2017) methods, which can be used with any commercial mastermix. This would have significant impact on the cost of the SNP genotyping. Currently, our group in Chinese Academy of Agricultural Sciences (CAAS) is converting and optimizing all the KASP assays to STARP assays to significantly reduce the cost and promote the use of high-throughput genotyping in wheat marker-assisted selection.
Gene discovery and marker application to wheat breeding: Japanese perspective
Common wheat should be harvested before the rainy season in Japan to avoid severe damages such as pre-harvest sprouting and Fusarium head blight disease. Recent climate changes bring the rainy time even in the Hokkaido area at the wheat harvest season. In addition, Japan is a very long country from north to south, and thus wheat cultivation should adapt to the various environmental conditions of each area. Due to these climate characteristics peculiar to Japan, early heading and pre-harvest sprouting tolerance are two of the most important targets in Japanese wheat breeding.
Use of the photoperiod-insensitive alleles has contributed to breed the early flowering cultivars. Ppd-D1a, which distributes widely in European and Japanese wheat cultivars, was originally derived from a Japanese landrace ‘Akakomugi’ (Worland 1996). Recently, a causal gene for photoperiod insensitivity of a Japanese wheat variety ‘Chogokuwase’ was identified as wheat PHYTOCLOCK1 (WPCL1) on 3AL (Mizuno et al. 2016). A 142-bp deletion in the MYB domain induces the extra-early flowering phenotype of ‘Chogokuwase’. WPCL1 is a homoeologue of the candidate for an early flowering mutant of einkorn wheat (Mizuno et al. 2012), and the Arabidopsis homologue LUX/PCL1 constitutes the “evening complex” repressing the PRR9 expression. Control of heading time via the Vrn-1-Ppd-1 allelic combinations could be important for further wheat breeding of early heading cultivars without frost injury in Japan.
Pre-harvest sprouting is induced by germination of wheat grains on the mother plants before harvest and greatly decreases the quality of the end products. Two important QTL for pre-harvest sprouting tolerance, QPhs.ocs-3A.1 on 3A and Phs1 on 4A, were recently cloned; one is MOTHER OF FT AND TFL1 (MFT) on chromosome 3A (Nakamura et al. 2011) and another TaMKK3-A on 4AL (Torada et al. 2016). QPhs.ocs-3A.1 and Phs1 have stable and large effects on seed dormancy (Mori et al. 2005; Torada et al. 2008). One of the Japanese wheat varieties ‘Zenkojikomugi’ provides the highest dormancy, and ‘Zenkojikomugi’-derived population has been used for breeding cultivars with strong grain dormancy (Chono et al. 2015). Frequency of the ‘Zenkojikomugi’-type SNP in MFT, which is located in the promoter region and increases the transcription level (Nakamura et al. 2011), is very high in southern part of Japan, but low in the northern part (Chono et al. 2015). ‘Zenkojikomugi’ contains two seed dormant alleles at the two QTLs on chromosomes 1B and 4A in addition to the 3A QTL (Cao et al. 2016). Phs1 was identified as a QTL for seed dormancy in a mapping population between Canadian dormant cultivar and nondormant Japanese cultivar (Torada et al. 2008), and a single nonsynonymous amino acid substitution (N260T) in the kinase domain of TaMKK3-A was related with the dormancy difference (Torada et al. 2016). If the N260T substitution can be introduced into each of the TaMKK3 homoeologues, developing of hyper-dormant cultivars could be expected in further wheat breeding (Nakamura 2018). Qsd1, a major seed dormancy QTL related to barley domestication, encodes an alanine aminotransferase family member (Sato et al. 2016). Common wheat contains three homoeologues of the barley Qsd1, and the B-genome copy of Qsd1 of a semi-dormant winter wheat cultivar ‘Kitahonami’ represents three amino acid substitutions compared with those of nondormant spring cultivars (Onishi et al. 2017).
Other genes and QTLs can be also expected to improve pre-harvest sprouting tolerance in wheat. TaABA8′OH1 encoding an ABA 8′-hydroxylase is highly expressed during seed development (Chono et al. 2013). A Japanese wheat cultivar ‘Tamaizumi’ contains an insertion mutation in TaABA8′OH1-D, and a gamma-ray irradiated mutant line TM1833 from ‘Tamaizumi’ lacks TaABA8′OH1-A. The double mutant TM1833 of TaABA8′OH1-A and TaABA8′OH1-D exhibits higher ABA contents in seed embryos and lower germination than ‘Tamaizumi’, indicating that reduction in the ABA 8′-hydroxylase activity in developing grains could effectively improve pre-harvest sprouting tolerance in wheat (Chono et al. 2013). Several QTLs for ABA sensitivity in germinated seeds has been found in common wheat. Significantly higher responsiveness is observed in Japanese wheat cultivars including some dormant seed-producing cultivars such as ‘Zenkojikomugi’ and ‘Fujimikomugi’ (Yokota et al. 2016). The QTLs for ABA responsiveness at the early seedling stage are assigned to chromosomes 1B, 2A, 3A, 5A, 6D and 7B and could contribute to the development of dehydration and pre-harvest sprouting tolerance (Kobayashi et al. 2010; Iehisa et al. 2014a). Near-isogenic lines for each ABA responsiveness-related QTL should be developed to validate the relationship between ABA responsiveness at the early seedling stage and pre-harvest sprouting tolerance in further studies.
Starch characteristics are important to end-product grain quality in common wheat. Two starch synthesis genes, encoding granule-bound starch synthase I (GBSSI) and starch synthase IIa (SSIIa), play important roles in determining the starch properties in developing wheat grains. Each of the two genes contains three homoeologues in the hexaploid genome, and effects of each copy on the starch properties are different (Shimbata et al. 2012; Inokuma et al. 2016). Combinations of the GBSSI and SSIIa alleles show diverse properties of the starch granules, and the double null mutant line has high levels of maltose and sucrose in the shrunken seeds (Nakamura et al. 2006; Inokuma et al. 2016). A soft winter wheat cultivar ‘Kitahonami’ shows a superior flour yield compared with other Japanese soft wheat cultivars, and QTLs on chromosomes 3B and 7A exhibit highly significant and consistent effects on the flour yield (Ishikawa et al. 2014). A newly developed hard wheat cultivar ‘Yumechikara’ has characteristics of extra-strong wheat (Tabiki et al. 2011). The dough properties and bread-making-quality-related characters have been further improved using different Glu-B3 alleles (Ito et al. 2015). Marker-assisted breeding makes it efficient to improve the end-product grain quality in common wheat.
New marker system to harness D-genome diversity
A diversity panel of wheat varieties from Japan is a useful resource for evaluating their genetic diversity and population structure. The genotyping-by-sequencing (GBS) approach clearly classified the Japanese wheat varieties into four clusters; (1) varieties from Hokkaido area, (2) modern cultivars from the northeast part of Japan, (3) modern cultivars from the southeast part of Japan and (4) classical varieties and landraces (Kobayashi et al. 2016). The SNP density on the D-genome chromosomes was remarkably lower than those on the A- or B-genome chromosomes. The low polymorphism on the D genome was found in RILs between a European wheat cultivar ‘Mironovskaya 808’ and ‘Chinese Spring’ using array-based marker and GBS approaches (Iehisa et al. 2014b; Kobayashi et al. 2016). To overcome the scarcity of the D-genome markers, a new marker system has been developed using genotyping by multiplexing amplicon sequencing and genome-tagged amplification (Ishikawa et al. 2018). The newly developed markers using genome-specific amplicon sequencing are uniformly distributed across the D-genome chromosomes.
The RNA-seq approach is cost-effective for genome-wide SNP discovery in wheat relatives. The members of Triticeae (synonym Hordeae) are considered as a single genetic system which shares homeologous chromosome structure and collinearity (Mayer et al. 2011; Wicker et al. 2011). The highly conserved synteny among homeologous chromosomes of the Triticeae species facilitates the genetic analysis of the wild relatives for which genome sequences are not available. For example, through the RNA-seq assembly from leaf transcripts of Aegilops tauschii, a large number of genome-wide polymorphisms were found among accessions and the SNPs and indels were successfully anchored to the chromosomes of Ae. tauschii and wheat D genome as well as those of barley (Iehisa et al. 2012; Nishijima et al. 2016). Similar RNA-seq approach was applicable to other diploid wheat relatives such as einkorn wheat, Ae. umbellulata and section Sitopsis species (Okada et al. 2018a; Miki et al. 2019; Michikawa et al. 2019). The genome-wide polymorphisms identified by this strategy could be converted to the PCR-based markers, and the converted markers are useful to construct linkage and high-density maps (Iehisa et al. 2014c; Nishijima et al. 2016; Michikawa et al. 2019). BSA combined with RNA-seq allowed to develop efficiently molecular markers linked to a chromosomal region associated with the target phenotype in tetraploid and hexaploid wheats (Trick et al. 2012; Ramirez-Gonzalez et al. 2015; Wu et al. 2018a, b; Nishijima et al. 2018). In the polyploid wheat species, the RNA-seq-based BSA requires precise assignment of reads from homoeologues to each subgenome, and thus having adequate reference sequences is important for the successful assignment.
Development of synthetic wheat lines and use of wild relatives in breeding in Japan
The evolutionary process of polyploid wheat (bread and durum wheats) is largely influenced by allopolyploid speciation. For the birth of a new allopolyploid species, normal growth and fertility of the interspecific hybrids are necessary. The interspecific hybridization between tetraploid wheat and Ae. tauschii results in normal triploid hybrids with the ABD genome, and unreduced gametes should be formed in pollens and eggs of the ABD hybrids. This evolutionary process can be repeated by artificial hybridization of the parental species, and the produced allohexaploid plants are called as synthetic hexaploid wheats (Matsuoka and Nasuda 2004).
Sometimes its triploid hybrids with tetraploid wheat show several types of abnormalities such as failure in germination, hybrid necrosis and hybrid sterility (Matsuoka et al. 2007). An array of synthetic hexaploid wheats developed between durum wheat cv. Langdon and Ae. tauschii accessions is classified into following four types based on abnormal growth phenotypes: type-II and type-III hybrid necrosis, hybrid chlorosis and severe growth abortion (Mizuno et al. 2010). The different types of growth abnormalities in developing synthetic wheats by triploid hybrids could act as postzygotic hybridization barriers leading to failure in development of synthetic wheat. Based on cytological and transcriptome analyses, an autoimmune response-like reaction appears to be associated with necrotic cell death in the two types of hybrid necrosis and hybrid chlorosis (Mizuno et al. 2010, 2011; Nakano et al. 2015). Causal genes for type-II and type-III necrosis in the D genome were assigned to the short arms of chromosomes 2D and 7D, respectively (Mizuno et al. 2010; Sakaguchi et al. 2016). The D-genome causative gene of hybrid chlorosis is located on 7DS (Hirao et al. 2015). However, chromosomal location of these hybrid necrosis and chlorosis genes in the AB genome still remains unknown. The autoimmune response-like reaction could be also assumed in hybrid necrosis observed in intraspecific crosses of common wheat and interspecific crosses of einkorn wheat (Takamatsu et al. 2015).
In the Japanese seedbank, namely National BioResource Project KOMUGI (https://shigen.nig.ac.jp/wheat/komugi/top/top.jsp), more than 12,000 accessions of wheat and its relatives are stored. This collection contains more than 4000 accessions of wild wheat relatives and a lot of cytogenetic experimental lines. Despite the presence of various hybrid incompatibilities, synthetic hexaploid wheat lines could be produced through interspecific crosses between ‘Langdon’ and Ae. tauschii accessions (Takumi et al. 2009; Kajimura et al. 2011). The wide variation in heading time in the parental Ae. tauschii accessions is maintained in the hexaploid background of the synthetic lines, although the ‘Langdon’ genome may have an enhancement on the effect of D-genome-derived variation in derived synthetics. A QTL for flowering time was commonly found on chromosome 7D in both Ae. tauschii and synthetic hexaploid wheat (Nguyen et al. 2013; Koyama et al. 2018). Grain shape is remarkably differentiated between the two subspecies of Ae. tauschii, and a few QTLs mainly control the grain shape differentiation (Nishijima et al. 2017). The D-genome QTL underpinning grain morphology could be at least partly functional in hexaploid wheat (Okamoto et al. 2013). Grains of the synthetic hexaploid wheat lines with the AABBDD genome generally exhibit a soft texture, whereas the U-genome addition to durum wheat generates hard-textured grains in synthetic hexaploid lines with the AABBUU genome (Okada et al. 2018b). The diversity in grain morphology traits of Ae. umbellulata will be useful for improvement of grain hardness diversity in hard-textured common wheat. Therefore, wild wheat accessions with useful genes and characters could be evaluated under the allohexaploid background. Phenotypes of the synthetic hexaploid wheat are distinct from modern cultivars of common wheat. To fill the gap, multiple synthetic derivatives (MSD) population was recently developed (Gorafi et al. 2018). The MSD population harboring chromosomal fragments from Ae. tauschii in the background of a common wheat cultivar, which make it an important resource for finding useful Ae. tauschii loci functioning under the wheat background (Elbashir et al. 2017; Gorafi et al. 2018). Genetic and epigenetic changes in the synthetic lines and MSDs could be elucidated in detail in further studies for efficient breeding use of the wild wheat relatives.
High-throughput phenotyping to support functional genomics
Adaptability of crops in fluctuating environment is an unclear concept, mainly influenced by genetics behind the physiological and morphological attributes, which continuously change during the growth cycle. The future course of action in accelerating genetic gain will depend on repeatable high-throughput phenotyping to get precise phenotypic information from broad range of environments (Araus and Cairns 2014). Intensive phenome data from multiple sites are important for deep understanding of genetic basis of important crop traits. Time-series phenotyping of large plant populations at multiple locations for important climate smart traits could maximize the selection accuracy (Rutkoski et al. 2016). It is difficult, even impractical, to evaluate genotypes for multiple times using present phenotyping approaches and facilities especially in Asia. There has been significant developments in using high-throughput phenotyping (HTPP) facilities outside of Asia, such as phenotyping facility at the Maricopa Agricultural Center (USA), Australian Phenotyping Facility in Canberra and integration networks between phenotyping communities for sharing facilities and data such as Australian, French, Italian, German and UK phenotyping networks under the umbrella of International Phenotyping Network. China has also recently established China Phenotyping Network which is the only phenotyping-assistance body in Asia. The majority of Asia is lagging behind in development and adopting modern phenotyping facilities.
During recent years, many computer vision-based tools and sensors have become vital for phenotyping of various crop traits with high accuracy and throughput (Fiorani and Schurr 2013). Taking concept of imaging-based quantification, several developments have been made for HTP phenotyping of crop under controlled and field conditions (Blais 2004; Zhao et al. 2019a). These advance phenotyping methods focus on a range of measurements such as from single organs level (leaves and roots) to whole plant using 2D/3D shoot imaging approaches in greenhouse facility and even whole plant or canopy surveillance of large field trials (Schmundt et al. 1998; Walter and Schurr 2005). These measurements are mainly in the form of RGB pixels, multi-/hyper-spectral bands, wavelength, time and direction. After converting these measurements into useful biological knowledge (crop traits) for closing phenome to genome knowledge gap using computer vision technology, scientists can get dynamic information about disease, photosynthesis, chlorophyll status, transpiration rate, physiochemical properties of crop through spectral vegetation indices for stress detection and genetic studies (Chaerle et al. 2006; Genty and Meyer 1995; Jahnke et al. 2009) (Fig. 1). Recently, noninvasive technologies have been used for high-throughput data generation for many crops in greenhouse facilities using environmental stimulations and field conditions (Tardieu et al. 2017). Modern greenhouse phenotyping installations include magnetic resource imaging, position emission technology and computer tomography or combination of all above under greenhouse conditions. Whereas, the category of field-based phenotyping platforms is diverse and encompass at many levels. It includes all kind of ground-based platform including fixed field phenotyping facilities such as cable suspended multisensory system (Kirchgessner et al. 2017), the Field Scanalyzer (Virlet et al. 2017) and specially modified carts-based pheno-mobile, pheno-carts as well as linearly moving pheno-poles, capable of carrying multiple sensors across the field (Deery et al. 2014; White and Conley 2013) (Fig. 1). The increase in image resolution and miniaturization of sensors cost together with usefulness of unmanned aerial vehicles (UAVs) has also allowed researchers to oversee performance of genotypes in breeding trials for crop management. UAV platforms that are flexible alternatives including poli-copter, helicopter and fixed wing configurations are attracting attention due to low cost along with their carrying ability for a range of payload (Gago et al. 2015). In addition, nanosatellites and microsatellites with high-resolution hyper-spectral and RGB cameras have been also becoming attractive alternative to low-altitude UAVs due to its large coverage capability for multiple sites at the same time (Yang et al. 2017). Use of these modern phenotyping approaches will bring a deep network to overcome the phenome to genome knowledge gap. The advances of plant phenotyping can benefit from other disciplines of remote sensing, robotics, computer vision and artificial intelligence.

High-throughput phenotyping network to complement deep genetic analysis. This is an example of big data generation on multiple phenotypes from a single organ to whole plant/canopy level with high repeatability and cost efficiency, using modern high-through facilities at both greenhouse-based laboratories and field conditions. Recent developments in phenotyping approaches such as observing the plants through 2D/3D images by using simple RGB camera, fluorescent, hyper/multispectral sensors in fixed or portable ground and aerial platforms have increased the efficiency of data collection. While computer vision-based approaches help in converting the raw information embedded in RGB and hyper-/multispectral images into useful biological knowledge and have increased phenome knowledge. Rapid and comprehensive phenome information of important traits such as seed morphology, seed physiology, spike number, plant height, biomass, chlorophyll contents, photosynthesis rate, senescence rate, normalized different vegetation index (NDVI), canopy temperature and disease scoring can increase the efficiency of genetic analysis for crop improvement
In Asia, only China is vastly developing central and institutional-based HTP phenotyping facilities such as NERCITA in Beijing (National Engineering Research Center for Information Technology in Agriculture) and Plant Phenomics Research Center (PPRC) in Nanjing Agricultural University. Several initiatives formed a strong international cooperation: the most important ones are with European countries, such as China–UK, China–France and China–Germany Phenomics Centers at Nanjing Agricultural University for development of joint phenotyping facilities. NERCITA has developed greenhouse and field-based remote sensing facilities through robotics to quantify important attributes of cereal and fruit crops in the context of precision agriculture (Wen et al. 2017; Yongjun et al. 2017; Yue et al. 2017; Zhang et al. 2015a, b; Zhou et al. 2018a). Whereas, PPRC is developing several phenotyping platforms ranging from growth chamber to field and is providing central phenotyping facilities for crop researcher and breeders. PPRC aims to develop and provide access to facilities that address multiscale plant phenotyping in different agro-climatic scenarios in China. In the context of practical use of modern HTPP, CAAS (Beijing) is establishing aerial and pheno-cart-based ground HTP phenome data for genomic prediction modeling and for precise selection in their breeding programs. Recently, a genomic selection paper has been published using UAV-based plant height data (Hassan et al. 2019). There are also some other reports from China on the use of HTP phenotyping platforms for assessment of important traits in different crops and their applications in quantitative genomic analysis and within field selection (Guo et al. 2015; Hassan et al. 2018a, b; Wang et al. 2019b). In Japan, there are also some developments in phenotyping technology through sensors and robotics, but application is very limited in crop breeding. Recently, a report has been published on assessment of sorghum plant height using UAV-based platform and its application in genomic modeling from the Institute of Sustainable Agro-ecosystem Services, The Tokyo University (Watanabe et al. 2017).
Asia needs more projects and initiatives which could be centralized by establishing Asian Phenotyping Network. In this context, China could help to initiate some cooperations with other Asian countries to introduce high-throughput technologies, to change the landscape of phenotyping infrastructure, the awareness about the importance of precision in phenome data for future research and integration of scientific community in Asia. This will create opportunities to coordinate data generation, analysis and integration across the platforms. Finally, this will allow us to bridge the phenotype-to-genotype gaps to characterize and improve a wide range of crops for food security in Asia.
Analytical approaches to utilize benefits of large-scale genomics
Advances in computer simulation and analytical genetics are crucial to improve selection accuracy in complex phenotypic traits and can assist wheat breeders in their important breeding decisions. Computer simulation is a product of integrated disciplines such as computer science, quantitative genetics, statistics, mathematics and molecular biology (Li et al. 2012). Computer simulation can be a tool which allows breeders to integrate theoretical, empirical and practical knowledge into dynamic model in silico (i.e., genetic model). The genetic model contains all necessary inputs (e.g., trait, environment, linkage map, marker information, gene or QTL location, QTL, pleiotropic and epistatic effects). This genetic model can be used to simulate long-term effects of breeding decision in early phase of breeding programs (Wang et al. 2005; Wang et al. 2003). Information such as choice of parents, crossing and breeding strategy, experimental population size, number of breeding cycles, number of replications and sampling method was required to meet specific breeding objective (Wang et al. 2005). In last two decades, computer simulation and modeling approaches have brought a new paradigm in wheat, rice and maize. For example, through computer simulation, Wang et al. (2003) compared two CIMMYT practical wheat breeding strategies called modified pedigree/bulk selection (MODPED) and selected bulk selection method (SELBLK) by using QU-GENE software. The genetic model accounted for pleiotropy, epistasis and G × E. The computer simulation revealed that SELBLK method resulted in 3.9% higher genetic gain than MODPED. Simulation study also showed SELBLK method is more cost-effective because of retaining less crosses from F1 to F8 and also requires one-third less land when compared with MODPED. In another study, Wang et al. (2009a) used computer simulation and modeling to compare single backcross breeding strategy with other selection and crossing schemes in different genetic composition parent lines. The favorable traits in simulation experiment were assumed as polygenic and were selected by phenotypic selection. The simulation outcomes indicated that the single backcross breeding strategy was superior over the two or more backcrosses and could transfer more than 60% favorable genes from donor parent to improve the adaptation of recurrent parent.
Moreover, computer simulations were also used to predict cross-performance in wheat for two quality traits, maximum dough resistance (Rmax) and extensibility (Wang et al. 2005). Simulation study used eight Silverstar sister lines which were morphologically very similar but different in allelic compositions. For simulation, the genetic model considered multiple alleles, linkage, pleiotropy and digenic epistasis. Simulation results showed Silverstar 3 and 7 were suitable parents for crossing for high Rmax objective while the other lines did not have alleles b and d at Glu-A3 and Glu-D1, respectively. In another experiment, Wang et al. (2009b) used simulation modeling to pyramid favorable alleles or genes into one target genotype for long coleoptile in wheat through a design breeding approach.
Li et al. (2013) compared the usage of doubled haploids (DH) with conventional wheat breeding strategy of CIMMYT wheat breeding program. Simulation results revealed that using DH lines derived from F1-DH and F3-DH in wheat did not improve the genetic gain per cycle, per year and per dollar for adaptation traits from shuttle breeding as compared with conventional breeding.
It is predicted from the last decade that MAS failed to improve quantitative traits that are polygenic (Bernardo 2008; Xu and Crouch 2008). However, MAS has been effective to improve monogenic traits with large QTL effects tagged with a known marker (Zhong et al. 2006). The limitation of MAS comes from its two components, (1) finding a significant marker-trait association and (2) estimation of QTL effects. To overcome this problem of polygenic traits, development of statistical methods which simultaneous includes all markers without significant tests and estimate marker effects of the trait under study made genomic selection (GS) popular. Thus, GS emerged as an alternative solution to improve traits with complex genetic architecture as well as to overcome previous selection methods such as phenotypic selection and MAS. GS consists of two parts; training population (TP) and breeding population (BP). TP is that which has been both phenotyped and genotyped and used to train prediction model to predict the genetic merit of BP (only genotyped) based on genomic estimated breeding values (GEBVs; also refers as breeding value). Predicated genotypes could be used as new parents for a new breeding cycle or enter multi-environment trials for variety development, but selection of genetically superior parents for making crosses is still a daunting task. More recently, Yao et al. (2018) demonstrated through computer simulations that GS can be used to predict cross-performance (i.e., cross with a high progeny mean and high genetic variance) based on parental selection criteria such as midparent GEBV and usefulness in bi-parental progenies. For simulation experiment, a total of 57 parents which included high-quality and high-yielding cultivars from China, USA and Australia were used. For crosses prediction, selection index (incorporating yield, maximum resistance and stability time) was developed as a new trait. The simulation results showed that the best crosses or candidate parents can be selected by the usefulness rating which resulted in higher genetic gain than those selected from midparent GEBVs for smaller selection proportion. Although significant progress is being made in wheat breeding and simulation, practical implementation in crop improvement programs is still in its infancy.
Conclusion and future outlook
The extensive use of NGS and other advancements are making inroads for gene discovery in Asia, but the progress is only limited to few countries like China and Japan. In the wheat pan-genome project (10+ wheat genome project), only one cultivar is from Asia (Norin 61 from Japan), while genetic diversity from other Asian countries is not represented. Therefore, there is a strong need to develop genomic resources based on genetic diversity from Asia to further translate this knowledge in applied breeding. Similarly, a comprehensive knowledge of diversity from Asia, including cultivars, landraces and wild relatives, is necessary to deepen our knowledge of the underlying evolution, domestication and adaptation of wheat in Asia. A more collaborative network among researchers from Asia is also needed on crop phenomics, like European Plant Phenotyping Network (EPPN), so that such facilities are accessible to other countries which are unable to afford high-tech facilities.
References
Appels R, Eversole K, Feuillet C, Keller B, Rogers J, Stein N, Pozniak CJ et al (2018) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361:eaar7191
Araus JL, Cairns JE (2014) Field high-throughput phenotyping: the new crop breeding frontier. Trends Plant Sci 19:52–61
Balfourier F, Bouchet S, Robert S, De Oliveira R, Rimbert H, Kitt J, Choulet F, Paux E (2019) Worldwide phylogeography and history of wheat genetic diversity. Sci Adv 5:eaav0536
Bernardo R (2008) Molecular markers and selection for complex traits in plants: learning from the last 20 years. Crop Sci 48:1649–1664
Bevan MW, Uauy C, Wulff BB, Zhou J, Krasileva K, Clark MD (2017) Genomic innovation for crop improvement. Nature 543:346–354
Blais F (2004) Review of 20 years of range sensor development. J Electron Imaging 13:231–243
Browning BL, Yu Z (2009) Simultaneous genotype calling and haplotype phasing improves genotype accuracy and reduces false-positive associations for genome-wide association studies. Am J Hum Genet 85:847–861
Cao A, Xing L, Wang X, Yang X, Wang W, Sun Y et al (2011) Serine/threonine kinase gene Stpk-V, a key member of powdery mildew resistance gene Pm21, confers powdery mildew resistance in wheat. Proc Natl Acad Sci 108:7727–7732
Cao L, Hayashi K, Tokui M, Mori M, Miura H, Onishi K (2016) Detection of QTLs for traits associated with pre-harvest sprouting resistance in bread wheat (Triticum aestivum L.). Breed Sci 66:260–270
Chaerle L, Leinonen I, Jones HG, Van Der Straeten D (2006) Monitoring and screening plant populations with combined thermal and chlorophyll fluorescence imaging. J Exp Bot 58:773–784
Chang JZ, Zhang JA, Mao XG, Li A, Jia JZ, Jing RL (2013) Polymorphism of TaSAP1-A1 and its association with agronomic traits in wheat. Planta 237:1495–1508
Chen H, Jiao C, Wang Y, Wang Y, Tian C, Yu H, Wang J, Wang X, Lu F, Fu X, Xue Y, Jiang W, Ling H, Lu H, Jiao Y (2019a) Comparative population genomics of bread wheat (Triticum aestivum) reveals its cultivation and breeding history in China. https://www.biorxiv.org/519587
Chen J, Zhang F, Zhao C, Lv G, Sun C, Pan Y, Guo X, Chen F (2019b) Genome-wide association study of six quality traits reveals the association of the TaRPP13L1 gene with flour colour in Chinese bread wheat. Plant Biotechnol J. https://doi.org/10.1111/pbi.13126
Chono M, Matsunaka H, Seki M, Fujita M, Kiribuchi-Otobe C, Oda S et al (2013) Isolation of a wheat (Triticum aestivum L.) mutant in ABA8′-hydroxylase gene: effect of reduced ABA catabolism on germination inhibition under field condition. Breed Sci 63:104–115
Chono M, Matsunaka H, Seki M, Fujita M, Kiribuchi-Otobe C, Oda S et al (2015) Molecular and genealogical analysis of grain dormancy in Japanese wheat varieties, with specific focus on MOTHER OF FT AND TFL1 on chromosome 3A. Breed Sci 65:103–109
Deery D, Jimenez-Berni J, Jones H, Sirault X, Furbank R (2014) Proximal remote sensing buggies and potential applications for field-based phenotyping. Agronomy 4:349–379
Dong CH, Ma ZY, Xia XC, Zhang LP, He ZH (2012) Allelic variation at the Tazds-A1 locus on wheat chromosome 2a and development of a functional marker in common wheat. J Integr Agric 11:1067–1074
Dong LL, Wang FM, Liu T, Dong ZY, Li AL, Jing RL et al (2014) Natural variation of TaGASR7-A1 affects grain length in common wheat under multiple cultivation conditions. Mol Breed 34:937–947
Dong Y, Zhang Y, Xiao Y, Yan J, Liu J, Wen W et al (2016) Cloning of TaSST genes associated with water soluble carbohydrate content in bread wheat stems and development of a functional marker. Theor Appl Genet 129:1061–1070
Driscoll CJ, Jensen N (1964) Chromosomes associated with waxlessness, awnedness and time of maturity of common wheat. Can J Genet Cytol 6:324–333
Elbashir AAE, Gorafi YSA, Tahir ISA, Kim J, Tsujimoto H (2017) Wheat multiple synthetic derivatives: a new source for heat stress tolerance adaptive traits. Breed Sci 67:248–256
FAO (2018). http://www.fao.org/faostat/en/#home
Finno CJ, Aleman M, Higgins RJ, Madigan JE, Bannasch DL (2014) Risk of false positive genetic associations in complex traits with underlying population structure: a case study. Vet J 202:543–549
Fiorani F, Schurr U (2013) Future scenarios for plant phenotyping. Annu Rev Plant Biol 64:267–291
Fu L, Xiao Y, Yan J, Liu J, Wen W, Zhang Y, Xia X, He Z (2019) Characterization of TaCOMT genes associated with stem lignin content in common wheat and development of a gene-specific marker. J Integr Agric 18:939–947
Gago J, Douthe C, Coopman R, Gallego P, Ribas-Carbo M, Flexas J et al (2015) UAVs challenge to assess water stress for sustainable agriculture. Agric Water Manag 153:9–19
Genty B, Meyer S (1995) Quantitative mapping of leaf photosynthesis using chlorophyll fluorescence imaging. Funct Plant Biol 22:277–284
Gorafi YSA, Kim JS, Elbashir AAE, Tsujimoto H (2018) A population of wheat multiple synthetic derivatives: an effective platform to explore, harness and utilize genetic diversity of Aegilops tauschii for wheat improvement. Theor Appl Genet 131:1615–1626
Guo Y, Sun J, Zhang G, Wang Y, Kong F, Zhao Y, Li S (2013) Haplotype, molecular marker and phenotype effects associated with mineral nutrient and grain size traits of TaGS1a in wheat. Field Crops Res 154:119–125
Guo W, Fukatsu T, Ninomiya S (2015) Automated characterization of flowering dynamics in rice using field-acquired time-series RGB images. Plant Methods 11:7
Halloran GM, Boydell CW (1967) Wheat chromosomes with genes for vernalization response. Can J Genet Cytol 9:632–639
Hanif M, Gao FM, Liu J, Wen W, Zhang Y, Rasheed A et al (2015) TaTGW6-A1, an ortholog of rice TGW6, is associated with grain weight and yield in bread wheat. Mol Breed 36:1–8
Hao C, Wang Y, Chao S, Li T, Liu H, Wang L, Zhang X (2017) The iSelect 9 K SNP analysis revealed polyploidization induced revolutionary changes and intense human selection causing strong haplotype blocks in wheat. Sci Rep 7:41247
Hao Z, Geng M, Hao Y, Zhang Y, Zhang L, Wen S, Wang R, Liu G (2019) Screening for differential expression of genes for resistance to Sitodiplosis mosellana in bread wheat via BSR-seq analysis. Theor Appl Genet. https://doi.org/10.1007/s00122-019-03419-9
Hassan MA, Yang M, Rasheed A, Jin X, Xia X, Xiao Y, He Z (2018a) Time-series multispectral indices from unmanned aerial vehicle imagery reveal senescence rate in bread wheat. Remote Sens 10:809
Hassan MA, Yang M, Rasheed A, Yang G, Reynolds M, Xia X, Xiao Y, He Z (2018b) A rapid monitoring of NDVI across the wheat growth cycle for grain yield prediction using a multi-spectral UAV platform. Plant Sci 282:95–103
Hassan MA, Yang M, Fu L, Rasheed A, Zheng B, Xia X, Xiao Y, He Z (2019) Accuracy assessment of plant height using an unmanned aerial vehicle for quantitative genomic analysis in bread wheat. Plant Methods 15:37
He XY, He ZH, Zhang LP, Sun DJ, Morris CF, Fuerst EP, Xia XC (2007) Allelic variation of polyphenol oxidase (PPO) genes located on chromosomes 2A and 2D and development of functional markers for the PPO genes in common wheat. Theor Appl Genet 115:47–58
He XY, Zhang YL, He ZH, Wu YP, Xiao YG, Ma CX, Xia XC (2008) Characterization of phytoene synthase 1 gene (Psy1) located on common wheat chromosome 7A and development of a functional marker. Theor Appl Genet 116:213–221
He H, Zhu S, Zhao R, Jiang Z, Ji Y, Ji J, Qiu D, Li H, Bie T (2018a) Pm21, encoding a typical CC-NBS-LRR protein, confers broad-spectrum resistance to wheat powdery mildew disease. Mol Plant 11:879–882
He Y, Zhang X, Zhang Y, Ahmad D, Wu L, Jiang P, Ma H (2018b) Molecular characterization and expression of PFT, an FHB resistance gene at the Fhb1 QTL in wheat. Phytopathology 108:730–736
He F, Pasam R, Shi F, Kant S, Keeble-Gagnere G, Kay P et al (2019) Exome sequencing highlights the role of wild-relative introgression in shaping the adaptive landscape of the wheat genome. Nat Genet 51:896–904
Himi E, Maekawa M, Miura H, Noda K (2011) Development of PCR markers for Tamyb10 related to R-1, red grain color gene in wheat. Theor Appl Genet 122:1561–1576
Hirao K, Nishijima R, Sakaguchi K, Takumi S (2015) Fine mapping of Hch1, the causal D-genome gene for hybrid chlorosis in interspecific crosses between tetraploid wheat and Aegilops tauschii. Genes Genet Syst 90:283–291
Hou J, Jiang Q, Hao C, Wang Y, Zhang H, Zhang X (2014) Global selection on sucrose synthase haplotypes during a century of wheat breeding. Plant Physiol 164:1918–1929
Hou J, Li T, Wang Y, Hao C, Liu H, Zhang X (2017) ADP-glucose pyrophosphorylase genes, associated with kernel weight, underwent selection during wheat domestication and breeding. Plant Biotechnol J 15:1533–1543
Hu MJ, Zhang HP, Cao JJ, Zhu XF, Wang SX, Jiang H et al (2016) Characterization of an IAA-glucose hydrolase gene TaTGW6 associated with grain weight in common wheat (Triticum aestivum L.). Mol Breed 36:1–11
Hu J, Li J, Wu P, Li Y, Qiu D, Qu Y et al (2019) Development of SNP, KASP, and SSR Markers by BSR-Seq technology for saturation of genetic linkage map and efficient detection of wheat powdery mildew resistance gene Pm61. Int J Mol Sci 20:E750
Iehisa JCM, Shimizu A, Sato K, Nasuda S, Takumi S (2012) Discovery of high-confidence single nucleotide polymorphisms from large-scale de novo analysis of leaf transcripts of Aegilops tauschii, a wild wheat progenitor. DNA Res 19:487–497
Iehisa JCM, Matsuura T, Mori IC, Takumi S (2014a) Identification of quantitative trait locus for abscisic acid responsiveness on chromosome 5A and association with dehydration tolerance in common wheat seedlings. J Plant Physiol 171:25–34
Iehisa JCM, Ohno R, Kimura T, Enoki H, Nishimura S, Okamoto Y et al (2014b) A high-density genetic map with array-based markers facilitates structural and quantitative traits locus analyses of the common wheat genome. DNA Res 21:555–567
Iehisa JCM, Shimizu A, Sato K, Nishijima R, Sakaguchi K, Matsuda R et al (2014c) Genome-wide marker development for the wheat D genome based on single nucleotide polymorphisms identified from transcripts in the wild wheat progenitor Aegilops tauschii. Theor Appl Genet 127:261–271
Inokuma T, Vrinten P, Shimabata T, Sunohara A, Ito H, Saito M et al (2016) Using the hexaploid nature of wheat to create variability in starch characteristics. J Agric Food Chem 64:941–947
Ishikawa G, Nakamura K, Ito H, Saito M, Sato M, Jinno H et al (2014) Association mapping and validation of QTLs for flour yield in the soft winter wheat variety Kitahonami. PLoS ONE 9:e111337
Ishikawa G, Saito M, Tanaka T, Katayose Y, Kanamori H, Kurita K, Nakamura T (2018) An efficient approach for the development of genome-specific markers in allohexaploid wheat (Triticum aestivum L.) and its application in the construction of high-density linkage maps of the D genome. DNA Res 25:317–326
Ito M, Maruyama-Funatsuki W, Ikeda TM, Nishio Z, Nagasawa K, Tabiki T (2015) Dough properties and bread-making quality-related characteristics of Yumechikara near-isogenic wheat lines carrying different Glu-B3 alleles. Breed Sci 65:241–248
Jahnke S, Menzel MI, Van Dusschoten D, Roeb GW, Bühler J, Minwuyelet S et al (2009) Combined MRI–PET dissects dynamic changes in plant structures and functions. Plant J 59:634–644
Jatayev S, Kurishbayev A, Zotova L, Khasanova G, Serikbay D, Zhubatkanov A et al (2017) Advantages of Amplifluor-like SNP markers over KASP in plant genotyping. BMC Plant Biol 17:254
Jiang Q, Hou J, Hao C, Wang L, Ge H, Dong Y, Zhang X (2011) The wheat (T. aestivum) sucrose synthase 2 gene (TaSus2) active in endosperm development is associated with yield traits. Funct Integr Genom 11:49–61
Jiang Y, Jiang Q, Hao C, Hou J, Wang L, Zhang H et al (2015) A yield-associated gene TaCWI, in wheat: its function, selection and evolution in global breeding revealed by haplotype analysis. Theor Appl Genet 128:131–143
Jiang H, Zhao L-X, Chen X-J, Cao J-J, Wu Z-Y, Liu K et al (2018) A novel 33-bp insertion in the promoter of TaMFT-3A is associated with pre-harvest sprouting resistance in common wheat. Mol Breed 38:69
Juliana P, Poland J, Huerta-Espino J, Shrestha S, Crossa J et al (2019) Improving grain yield, stress resilience and quality of bread wheat using large-scale genomics. Nat Genet 51:1530–1539. https://doi.org/10.1038/s41588-019-0496-6
Kajimura T, Murai K, Takumi S (2011) Distinct genetic regulation of flowering time and grain-filling period based on empirical study of D-genome diversity in synthetic hexaploid wheat lines. Breed Sci 61:130–141
Khalid M, Afzal F, Gul A, Amir R, Subhani A, Ahmed Z et al (2019) Molecular characterization of 87 functional genes in wheat diversity panel and their association with phenotypes under well-watered and water-limited conditions. Front Plant Sci 10:717
Kirchgessner N, Liebisch F, Yu K, Pfeifer J, Friedli M, Hund A, Walter A (2017) The ETH field phenotyping platform FIP: a cable-suspended multi-sensor system. Funct Plant Biol 44:154–168
Kobayashi F, Takumi S, Handa H (2010) Identification of quantitative trait loci for ABA responsiveness at the seedling stage associated with ABA-related gene expression in common wheat. Theor Appl Genet 121:629–641
Kobayashi F, Tanaka T, Kanamori H, Wu J, Katayose Y, Handa H (2016) Characterization of a mini core collection of Japanese wheat varieties using single-nucleotide polymorphisms generated by genotyping-by-sequencing. Breed Sci 66:213–225
Koyama K, Okumura Y, Okamoto E, Nishijima R, Takumi S (2018) Natural variation in photoperiodic flowering pathway and identification of photoperiod-insensitive accessions in wild wheat, Aegilops tauschii. Euphytica 214:3
Law CN (1966) The location of genetic factors affecting a quantitative character in wheat. Genetics 53:487–498
Li W, Yang B (2017) Translational genomics of grain size regulation in wheat. Theor Appl Genet 130:1765–1771
Li XP, Zhao XQ, He X, Zhao GY, Li B, Liu DC et al (2011) Haplotype analysis of the genes encoding glutamine synthetase plastic isoforms and their association with nitrogen-use- and yield-related traits in bread wheat. New Phytol 189:449–458
Li X, Zhu C, Wang J, Yu J (2012) Computer simulation in plant breeding. Adv Agron 116:219–264
Li H, Singh RP, Braun H-J, Pfeiffer WH, Wang J (2013) Doubled haploids versus conventional breeding in CIMMYT wheat breeding programs. Crop Sci 53:74–83
Li B, Li Q, Mao X, Li A, Wang J, Chang X et al (2016a) Two novel AP2/EREBP transcription factor genes TaPARG have pleiotropic functions on plant architecture and yield-related traits in common wheat. Front Plant Sci 7:1191
Li B, Liu D, Li Q, Mao X, Li A, Wang J, Chang X, Jing R (2016b) Overexpression of wheat gene TaMOR improves root system architecture and grain yield in Oryza sativa. J Exp Bot 67:4155–4167
Li L, Shi X, Zheng F, Li C, Wu D, Bai G, Gao D, Wu J, Li T (2016c) A novel nitrogen-dependent gene associates with the lesion mimic trait in wheat. Theor Appl Genet 129:2075–2084
Li M, Li B, Guo G, Chen Y, Xie J, Lu P et al (2018) Mapping a leaf senescence gene els1 by BSR-Seq in common wheat. Crop J 6:236–243
Li N, Xu R, Duan P, Li Y (2018b) Control of grain size in rice. Plant Reprod 31:237–251
Li G, Zhou J, Jia H, Gao Z, Fan M et al (2019) Mutation of a histidine-rich calcium-binding-protein gene in wheat confers resistance to Fusarium head blight. Nat Genet 51:1106–1112
Liu HJ, Yan J (2019) Crop genome-wide association study: a harvest of biological relevance. Plant J 97:8–18
Liu YN, He ZH, Appels R, Xia XC (2012) Functional markers in wheat: current status and future prospects. Theor Appl Genet 125:1–10
Liu J, He Z, Wu L, Bai B, Wen W, Xie C, Xia X (2016) Genome-wide linkage mapping of QTL for black point reaction in bread wheat (Triticum aestivum L.). Theor Appl Genet 129:2179–2190
Liu J, He Z, Rasheed A, Wen W, Yan J, Zhang P et al (2017) Genome-wide association mapping of black point reaction in common wheat (Triticum aestivum L.). BMC Plant Biol 17:220
Liu H, Li T, Wang Y, Zheng J, Li H, Hao C, Zhang X (2019a) TaZIM-A1 negatively regulates flowering time in common wheat (Triticum aestivum L.). J Integr Plant Biol 61:359–376
Liu J, Rasheed A, He Z, Imtiaz M, Arif A, Mahmood T et al (2019b) Genome-wide variation patterns between landraces and cultivars uncover divergent selection during modern wheat breeding. Theor Appl Genet 132:2509–2523
Liu K, Cao J, Yu K, Liu X, Gao Y, Chen Q et al (2019c) Wheat TaSPL8 modulates leaf angle through auxin and brassinosteroid signaling. Plant Physiol 181:179–194
Long YM, Chao WS, Ma GJ, Xu SS, Qi LL (2016) An innovative SNP genotyping method adapting to multiple platforms and throughputs. Theor Appl Genet 130:597–607
Ma DY, Yan J, He ZH, Wu L, Xia XC (2012) Characterization of a cell wall invertase gene TaCwi-A1 on common wheat chromosome 2A and development of functional markers. Mol Breed 29:43–52
Ma L, Tian L, Hao C, Wang Y, Chen X, Zhang XY (2016) TaGS5-3A, a grain size gene selected during wheat improvement for larger kernel and yield. Plant Biotechnol J 14:1269–1280
Manickavelu A, Jighly A, Ban T (2014) Molecular evaluation of orphan Afghan common wheat (Triticum aestivum L.) landraces collected by Dr. Kihara using single nucleotide polymorphic markers. BMC Plant Biol 14:320
Matsuoka Y, Nasuda S (2004) Durum wheat as a candidate for the unknown female progenitor of bread wheat: an empirical study with a highly fertile F1 hybrid with Aegilops tauschii Coss. Theor Appl Genet 109:1710–1717
Matsuoka Y, Takumi S, Kawahara T (2007) Natural variation for triploid F1 hybrid formation in allohexaploid wheat speciation. Theor Appl Genet 115:509–518
Mayer KFX, Martis M, Hedley PE, Simková H, Liu H et al (2011) Unlocking the barley genome by chromosomal and comparative genomics. Plant Cell 23:1249–1263
McIntosh R (1973) A catalogue of gene symbols for wheat. In: Missouri C (ed) Proceedings of 4th international wheat genetics symposium
Michikawa A, Yoshida K, Okada M, Sato K, Takumi S (2019) Genome-wide polymorphisms from RNA sequencing assembly of leaf transcripts facilitate phylogenetic analysis and molecular marker development in wild einkorn wheat. Mol Genet Genom 294:1327–1341. https://doi.org/10.1007/s00438-019-01581-9
Miki Y, Yoshida K, Mizuno N, Nasuda S, Sato K, Takumi S (2019) Origin of wheat B-genome chromosomes inferred from RNA sequencing analysis of leaf transcripts from section Sitopsis species of Aegilops. DNA Res 26:171–182
Mizuno N, Hosogi N, Park P, Takumi S (2010) Hypersensitive response-like reaction is associated with hybrid necrosis in interspecific crosses between tetraploid wheat and Aegilops tauschii Coss. PLoS ONE 5:e11326
Mizuno N, Shitsukawa N, Hosogi N, Park P, Takumi S (2011) Autoimmune response and repression of mitotic cell division occur in inter-specific crosses between tetraploid wheat and Aegilops tauschii Coss. that show low temperature-induced hybrid necrosis. Plant J 68:114–128
Mizuno N, Nitta M, Sato K, Nasuda S (2012) A wheat homologue of PHYTOCLOCK 1 is a candidate gene conferring early heading phenotypes to einkorn wheat. Genes Genet Syst 87:357–367
Mizuno N, Kinoshita M, Kinoshita S, Nishida H, Fujita M, Kato K et al (2016) Loss-of-function mutations in three homoeologous PHYTOCLOCK 1 genes in common wheat are associated with the extra-early flowering phenotype. PLoS ONE 11:e0165618
Mori M, Uchino N, Chono M, Kato K, Miura H (2005) Mapping QTLs for grain dormancy on wheat chromosome 3A and the group 4 chromosomes, and their combined effect. Theor Appl Genet 110:1315–1323
Nakamura S (2018) Grain dormancy genes responsible for preventing pre-harvest sprouting in barley and wheat. Breed Sci 68:295–304
Nakamura T, Shimbata T, Vrinten P, Saito M, Yonemaru J, Seto Y, Yasuda H, Takahama M (2006) Sweet wheat. Genes Genet Syst 81:361–365
Nakamura S, Abe F, Kawahigashi H, Nakazono K, Tagiri A, Matsumoto T et al (2011) A wheat homolog of MOTHER OF FT AND TFL1 acts in the regulation of germination. Plant Cell 23:3215–3229
Nakano H, Mizuno N, Tosa Y, Yoshida K, Park P, Takumi S (2015) Accelerated senescence and enhanced disease resistance in hybrid chlorosis lines derived from interspecific crosses between tetraploid wheat and Aegilops tauschii. PLoS ONE 10:e0121583
Nguyen AT, Iehisa JCM, Kajimura T, Murai K, Takumi S (2013) Identification of quantitative trait loci for flowering-related traits in the D genome of synthetic hexaploid wheat lines. Euphytica 192:401–412
Ni F, Qi J, Hao Q, Lyu B, Luo MC, Wang Y et al (2017) Wheat Ms2 encodes for an orphan protein that confers male sterility in grass species. Nat Commun 8:15121
Nishijima R, Yoshida K, Motoi Y, Sato K, Takumi S (2016) Genome-wide identification of novel genetic markers from RNA sequencing assembly of diverse Aegilops tauschii accessions. Mol Genet Genom 291:1681–1694
Nishijima R, Okamoto Y, Hatano H, Takumi S (2017) Quantitative trait locus analysis for spikelet shape-related traits in wild wheat progenitor Aegilops tauschii: implications for intraspecific diversification and subspecies differentiation. PLoS ONE 12:e0173210
Nishijima R, Yoshida K, Sakaguchi K, Yoshimura S, Sato K, Takumi S (2018) RNA sequencing-based bulked segregant analysis facilitates efficient D-genome marker development for a specific chromosomal region of synthetic hexaploid wheat. Int J Mol Sci 19:3749
Okada M, Ikeda TM, Yoshida K, Takumi S (2018a) Effect of the U genome on grain hardness in nascent synthetic hexaploids derived from interspecific hybrids between durum wheat and Aegilops umbellulata. J Cereal Sci 83:153–161
Okada M, Yoshida K, Nishijima R, Michikawa A, Motoi Y, Sato K, Takumi S (2018b) RNA-seq analysis reveals considerable genetic diversity and provides genetic marker saturating all chromosomes in the diploid wild wheat relative Aegilops umbellulata. BMC Plant Biol 18:271
Okamoto Y, Nguyen AT, Yoshioka M, Iehisa JCM, Takumi S (2013) Identification of quantitative trait loci controlling grain size and shape in the D genome of synthetic hexaploid wheat lines. Breed Sci 63:423–429
Onishi K, Yamane M, Yamaji N, Tokui M, Kanamori H, Wu J et al (2017) Sequence differences in the seed dormancy gene Qsd1 among various wheat genomes. BMC Genom 18:497
Pont C, Leroy T, Seidel M, Tondelli A, Duchemin W et al (2019) Tracing the ancestry of modern bread wheats. Nat Genet 51:905–911
Qin L, Hao CY, Hou J, Wang YQ, Li T, Wang LF, Ma ZQ, Zhang XY (2014) Homologous haplotypes, expression, genetic effects and geographic distribution of the wheat yield gene TaGW2. BMC Plant Biol 14:107
Ramirez-Gonzalez RH, Segovia V, Bird N, Fenwick P, Holdgate S, Bery S et al (2015) RNA-Seq bulked segregant analysis enables the identification of high-resolution genetic markers for breeding in hexaploid wheat. Plant Biotech J 13:613–624
Rasheed A, Xia X (2019) From markers to genome-based breeding in wheat. Theor Appl Genet 132:767–784
Rasheed A, Wen W, Gao FM, Zhai S, Jin H, Liu JD et al (2016) Development and validation of KASP assays for functional genes underpinning key economic traits in wheat. Theor Appl Genet 129:1843–1860
Rasheed A, Hao Y, Xia X, Khan A, Xu Y, Varshney RK, He Z (2017) crop breeding chips and genotyping platforms: progress, challenges, and perspectives. Mol Plant 10:1047–1064
Rawat N, Pumphrey MO, Liu S, Zhang X, Tiwari VK, Ando K et al (2016) Wheat Fhb1 encodes a chimeric lectin with agglutinin domains and a pore-forming toxin-like domain conferring resistance to Fusarium head blight. Nat Genet 48:1576–1580
Rutkoski J, Poland J, Mondal S, Autrique E, Pérez LG, Crossa J et al (2016) Canopy temperature and vegetation indices from high-throughput phenotyping improve accuracy of pedigree and genomic selection for grain yield in wheat. G3 Genes Genom Genet 6:2799–2808
Sajjad M, Ma X, Habibullah KS, Shoaib M, Song Y, Yang W et al (2017) TaFlo2-A1, an ortholog of rice Flo2, is associated with thousand grain weight in bread wheat (Triticum aestivum L.). BMC Plant Biol 17:164
Sakaguchi K, Nishijima R, Iehisa JCM, Takumi S (2016) Fine mapping and genetic association analysis of Net2, the causative D-genome locus of low temperature-induced hybrid necrosis in interspecific crosses between tetraploid wheat and Aegilops tauschii. Genetica 144:523–533
Sato K, Yamane M, Yamaji N, Kanamori H, Tagiri A, Schwerdt JG et al (2016) Alanine aminotransferase controls seed dormancy in barley. Nat Commun 7:11625
Schmundt D, Stitt M, Jähne B, Schurr U (1998) Quantitative analysis of the local rates of growth of dicot leaves at a high temporal and spatial resolution, using image sequence analysis. Plant J 16:505–514
Sears ER (1954) The aneuploids of common wheat. Univ Missouri Agric Exp Stn Bull 572:1–58
Sears ER, Sears LMS (1978) The telocentric chromosomes of common wheat. In: 5th international wheat genetics symposium, pp 389–407
Sehgal D, Mondal S, Guzman C, Garcia Barrios G, Franco C, Singh R, Dreisigacker S (2019) Validation of candidate gene-based markers and identification of novel loci for thousand-grain weight in spring bread wheat. Front Plant Sci 10:1189
Shavrukov Y, Suchecki R, Eliby S, Abugalieva A, Kenebayev S, Langridge P (2014) Application of next-generation sequencing technology to study genetic diversity and identify unique SNP markers in bread wheat from Kazakhstan. BMC Plant Biol 14:258
Shepherd K (1968) Chromosomal control of endosperm proteins in wheat and rye. In: Proceedings of 3rd international wheat genetics symposium, Australian Academy of Science, pp 86–96
Shi W, Hao C, Zhang Y, Cheng J, Zhang Z, Liu J et al (2017) A combined association mapping and linkage analysis of kernel number per spike in common wheat (Triticum aestivum L.). Front Plant Sci 8:1412
Shimbata T, Ai Y, Fujita M, Inokuma T, Vrinten P, Sunohara A et al (2012) Effects of homoeologous wheat Starch Synthase IIa genes on starch properties. J Agric Food Chem 60:12004–12010
Su ZQ, Hao CY, Wang LF, Dong YC, Zhang XY (2011) Identification and development of a functional marker of TaGW2 associated with grain weight in bread wheat (Triticum aestivum L.). Theor Appl Genet 122:211–223
Su Z, Bernardo A, Tian B, Chen H, Wang S, Ma H et al (2019) A deletion mutation in TaHRC confers Fhb1 resistance to Fusarium head blight in wheat. Nat Genet 51:1099–1105
Sun C, Zhang F, Yan X, Zhang X, Dong Z, Cui D, Chen F (2017) Genome-wide association study for 13 agronomic traits reveals distribution of superior alleles in bread wheat from the Yellow and Huai Valley of China. Plant Biotechnol J 15:953–969
Sun L, Yang W, Li Y, Shan Q, Ye X, Wang D et al (2019) A wheat dominant dwarfing line with Rht12, which reduces stem cell length and affects gibberellic acid synthesis, is a 5AL terminal deletion line. Plant J 97:887–900
Tabiki T, Nishio Z, Ito M, Yamauchi H, Takata K, Kuwabara T et al (2011) A new extra-strong hard red winter wheat variety: ‘Yumechikara’. Res Bull NARO Hokkaido Agric Res Cent 195:1–12
Takamatsu K, Iehisa JCM, Nishijima R, Takumi S (2015) Comparison of gene expression profiles and response to zinc chloride among inter- and intraspecific hybrids with growth abnormalities in wheat and its relatives. Plant Mol Biol 88:487–502
Takumi S, Naka Y, Morihiro H, Matsuoka Y (2009) Expression of morphological and flowering time variation through allopolyploidization: an empirical study with 27 wheat synthetics and their parental Aegilops tauschii accessions. Plant Breed 128:585–590
Tardieu F, Cabrera-Bosquet L, Pridmore T, Bennett M (2017) Plant phenomics, from sensors to knowledge. Curr Biol 27:R770–R783
Tian S, Mao X, Zhang H, Chen S, Zhai C, Yang S, Jing R (2013) Cloning and characterization of TaSnRK2.3, a novel SnRK2 gene in common wheat. J Exp Bot 64:2063–2080
Torada A, Koike M, Ikeuchi S, Tsutsui I (2008) Mapping of a major locus controlling seed dormancy using backcrossed progenies in wheat (Triticum aestivum L.). Genome 51:426–432
Torada A, Koike M, Ogawa T, Takenouchi Y, Tadamura K, Wu J et al (2016) A causal gene for seed dormancy on wheat chromosome 4A encodes a MAP kinase. Curr Biol 26:782–787
Trick M, Adamski NM, Mugford SG, Jiang CC, Febrer M, Uauy C (2012) Combining SNP discovery from next-generation sequencing data with bulked segregant analysis (BSA) to fine-map genes in polyploid wheat. BMC Plant Biol 12:14
Ur Rehman S, Wang J, Chang X, Zhang X, Mao X, Jing R (2019) A wheat protein kinase gene TaSnRK2.9-5A associated with yield contributing traits. Theor Appl Genet 132:907–919
Virlet N, Sabermanesh K, Sadeghi-Tehran P, Hawkesford MJ (2017) Field Scanalyzer: An automated robotic field phenotyping platform for detailed crop monitoring. Funct Plant Biol 44:143–153
Walter A, Schurr U (2005) Dynamics of leaf and root growth: endogenous control versus environmental impact. Ann Bot 95:891–900
Wang J, Van Ginkel M, Podlich D, Ye G, Trethowan R, Pfeiffer W et al (2003) Comparison of two breeding strategies by computer simulation. Crop Sci 43:1764–1773
Wang J, Eagles H, Trethowan R, Van Ginkel M (2005) Using computer simulation of the selection process and known gene information to assist in parental selection in wheat quality breeding. Aust J Agric Res 56:465–473
Wang J, Chapman SC, Bonnett DG, Rebetzke GJ (2009a) Simultaneous selection of major and minor genes: use of QTL to increase selection efficiency of coleoptile length of wheat (Triticum aestivum L.). Theor Appl Genet 119:65–74
Wang J, Singh RP, Braun HJ, Pfeiffer WH (2009b) Investigating the efficiency of the single backcrossing breeding strategy through computer simulation. Theor Appl Genet 118:683–694
Wang J, Wen W, Hanif M, Xia X, Wang H, Liu S et al (2016) TaELF3-1DL, a homolog of ELF3, is associated with heading date in bread wheat. Mol Breed 36:161
Wang Y, Xie J, Zhang H, Guo B, Ning S, Chen Y et al (2017a) Mapping stripe rust resistance gene YrZH22 in Chinese wheat cultivar Zhoumai 22 by bulked segregant RNA-Seq (BSR-Seq) and comparative genomics analyses. Theor Appl Genet 130:2191–2201
Wang Y, Yu H, Tian C, Sajjad M, Gao C, Tong Y, Wang X, Jiao Y (2017b) Transcriptome association identifies regulators of wheat spike architecture. Plant Physiol 175:746
Wang J, Wang R, Mao X, Li L, Chang X, Zhang X, Jing R (2018a) TaARF4 genes are linked to root growth and plant height in wheat. Ann Bot. https://doi.org/10.1093/aob/mcy218
Wang Y, Zhang H, Xie J, Guo B, Chen Y, Zhang H et al (2018b) Mapping stripe rust resistance genes by BSR-Seq: YrMM58 and YrHY1 on chromosome 2AS in Chinese wheat lines Mengmai 58 and Huaiyang 1 are Yr17. Crop J 6:91–98
Wang H, Wang S, Chang X, Hao C, Sun D, Jing R (2019a) Identification of TaPPH-7A haplotypes and development of a molecular marker associated with important agronomic traits in common wheat. BMC Plant Biol 19:296
Wang X, Zhang R, Song W, Han L, Liu X, Sun X et al (2019b) Dynamic plant height QTL revealed in maize through remote sensing phenotyping using a high-throughput unmanned aerial vehicle (UAV). Sci Rep 9:3458
Watanabe K, Guo W, Arai K, Takanashi H, Kajiya-Kanegae H, Kobayashi M et al (2017) High-throughput phenotyping of sorghum plant height using an unmanned aerial vehicle and its application to genomic prediction modeling. Front Plant Sci 8:421–421
Wei J, Geng H, Zhang Y, Liu J, Wen W, Zhang Y, Xia X, Chen X, He Z (2015) Mapping quantitative trait loci for peroxidase activity and developing gene-specific markers for TaPod-A1 on wheat chromosome 3AL. Theor Appl Genet 128:2067–2076
Wen W, Guo X, Wang Y, Zhao C, Liao W (2017) Constructing a three-dimensional resource database of plants using measured in situ morphological data. Appl Eng Agric 33:747–756
White JW, Conley MM (2013) A flexible, low-cost cart for proximal sensing. Crop Sci 53:1646–1649
Wicker T, Mayer KFX, Gundlach H, Martis M, Steuernagel B et al (2011) Frequent gene movement and pseudogene evolution is common to the large and complex genomes of wheat, barley, and their relatives. Plant Cell 23:1707–1718
Worland AJ (1996) The influence of flowering time gene on environmental adaptability in European wheats. Euphytica 89:49–57
Wu J, Zeng Q, Wang Q, Liu S, Yu S, Mu J et al (2018a) SNP-based pool genotyping and haplotype analysis accelerate fine-mapping of the wheat genomic region containing stripe rust resistance gene Yr26. Theor Appl Genet 131:1481–1496
Wu P, Xie J, Hu J, Qiu D, Liu Z, Li J et al (2018b) Development of molecular markers linked to powdery mildew resistance gene Pm4b by combining SNP discovery from transcriptome sequencing data with bulked segregant analysis (BSR-Seq) in wheat. Front Plant Sci 9:95
Würschum T, Langer SM, Longin CFH (2015) Genetic control of plant height in European winter wheat cultivars. Theor Appl Genet 128:865–874
Würschum T, Langer SM, Longin CFH, Tucker MR, Leiser WL (2017) A modern Green Revolution gene for reduced height in wheat. Plant J 92:892–903
Xia C, Zhang L, Zou C, Gu Y, Duan J, Zhao G et al (2017) A TRIM insertion in the promoter of Ms2 causes male sterility in wheat. Nat Commun 8:15407
Xing L, Hu P, Liu J, Witek K, Zhou S, Xu J et al (2018) Pm21 from Haynaldia villosa encodes a CC-NBS-LRR protein conferring powdery mildew resistance in wheat. Mol Plant 11:874–878
Xu Y, Crouch JH (2008) Marker-assisted selection in plant breeding: from publications to practice. Crop Sci 48:391–407
Yan X, Zhao L, Ren Y, Dong Z, Cui D, Chen F (2019) Genome-wide association study revealed that the TaGW8 gene was associated with kernel size in Chinese bread wheat. Sci Rep 9:2702
Yang G, Liu J, Zhao C, Li Z, Huang Y, Yu H et al (2017) Unmanned aerial vehicle remote sensing for field-based crop phenotyping: current status and perspectives. Front Plant Sci 8:1111
Yao J, Zhao D, Chen X, Zhang Y, Wang J (2018) Use of genomic selection and breeding simulation in cross prediction for improvement of yield and quality in wheat (Triticum aestivum L.). Crop J 6:353–365
Yao H, Xie Q, Xue S, Luo J, Lu J, Kong Z et al (2019) HL2 on chromosome 7D of wheat (Triticum aestivum L.) regulates both head length and spikelet number. Theor Appl Genet 132:1789–1797
Yokota H, Iehisa JCM, Nishijima R, Nitta M, Takenaka S, Nasuda S, Takumi S (2016) Variation in abscisic acid responsiveness at the early seedling stage is related to line differences in seed dormancy and in expression of genes involved in abscisic acid responses in common wheat. J Cereal Sci 71:167–176
Yongjun Z, Shenghui Y, Chunjiang Z, Liping C, Lan Y, Yu T (2017) Modelling operation parameters of UAV on spray effects at different growth stages of corns. Int J Agric Biol Eng 10:57–66
Yue J, Yang G, Li C, Li Z, Wang Y, Feng H, Xu B (2017) Estimation of winter wheat above-ground biomass using unmanned aerial vehicle-based snapshot hyperspectral sensor and crop height improved models. Remote Sens 9:708
Zhai S, He Z, Wen W, Jin H, Liu J, Zhang Y, Liu Z, Xia X (2016a) Genome-wide linkage mapping of flour color-related traits and polyphenol oxidase activity in common wheat. Theor Appl Genet 129:377–394
Zhai S, Li G, Sun Y, Song J, Li J, Song G, Li Y, Ling H, He Z, Xia X (2016b) Genetic analysis of phytoene synthase 1 (Psy1) gene function and regulation in common wheat. BMC Plant Biol 16:228
Zhang G, Li YM, Zhang Y, Dong YL, Wang XJ, Wee GR et al (2010) Cloning and characterization of a pathogenesis-related protein gene TaPR10 from wheat induced by stripe rust pathogen. Agric Sci China 9:549–556
Zhang L, Zhao YL, Gao LF, Zhao GY, Zhou RH, Zhang BS, Jia JZ (2012) TaCKX6-D1, the ortholog of rice OsCKX2, is associated with grain weight in hexaploid wheat. New Phytol 195:574–584
Zhang YJ, Liu JD, Xia XC, He ZH (2014a) TaGS-D1, an ortholog of rice OsGS3, is associated with grain weight and grain length in common wheat. Mol Breed 34:1097–1107
Zhang YJ, Miao XL, Xia XC, He ZH (2014b) Cloning of seed dormancy genes (TaSdr) associated with tolerance to pre-harvest sprouting in common wheat and development of a functional marker. Theor Appl Genet 127:855–866
Zhang B, Liu X, Xu W, Chang JZ, Li A, Mao XG, Zhang XY, Jing RL (2015a) Novel function of a putative MOC1 ortholog associated with spikelet number per spike in common wheat. Sci Rep 5:12211
Zhang Y, Guo X, Du J, Zhao C (2015b) Review on characterization of maize phenotypic diversity: from genome and genotyping to phenomics and high-throughput phenotyping. Res Crops 16:351
Zhang W, Zhao G, Gao LF, Kong XY, Guo Z, Wu B, Jia JZ (2016) Functional studies of heading date-related gene TaPRR73, a paralog of Ppd1 in common wheat. Front Plant Sci 7:772
Zhang B, Xu W, Liu X, Mao X, Li A, Wang J, Chang X, Zhang X, Jing R (2017a) Functional conservation and divergence among homoeologs of TaSPL20 and TaSPL21, two SBP-box genes governing yield-related traits in hexaploid wheat. Plant Physiol 174:1177
Zhang P, He Z, Tian X, Gao F, Xu D, Liu J et al (2017b) Cloning of TaTPP-6AL1 associated with grain weight in bread wheat and development of functional marker. Mol Breed 37:78
Zhang Y, Xia X, He Z (2017c) The seed dormancy allele TaSdr-A1a associated with pre-harvest sprouting tolerance is mainly present in Chinese wheat landraces. Theor Appl Genet 130:81–89
Zhang X, Liu G, Zhang L, Xia C, Zhao T, Jia J, Liu X, Kong X (2018) Fine mapping of a novel heading date gene, TaHdm605, in hexaploid wheat. Front Plant Sci 9:772
Zhao G, Zou C, Li K, Wang K, Li T, Gao L et al (2017) The Aegilops tauschii genome reveals multiple impacts of transposons. Nat Plants 3:946–955
Zhao C, Zhang Y, Du J, Guo X, Wen W, Gu S, Wang J, Fan J (2019a) Crop phenomics: current status and perspectives. Front Plant Sci 10:714
Zhao J, Wang Z, Liu H, Zhao J, Li T, Hou J, Zhang X, Hao C (2019b) Global status of 47 major wheat loci controlling yield, quality, adaptation and stress resistance selected over the last century. BMC Plant Biol 19:5
Zheng J, Liu H, Wang Y, Wang L, Chang X, Jing RL, Hao C, Zhang XY (2014) TEF-7A, a transcript elongation factor gene, influences yield-related traits in bread wheat (Triticum aestivum L.). J Exp Bot 65:5351–5365
Zhong S, Toubia-Rahme H, Steffenson BJ, Smith KPJP (2006) Molecular mapping and marker-assisted selection of genes for septoria speckled leaf blotch resistance in barley. Phytopathology 96:993–999
Zhou C, Liang D, Yang X, Xu B, Yang G (2018a) Recognition of wheat spike from field based phenotype platform using multi-sensor fusion and improved maximum entropy segmentation algorithms. Remote Sens 10:246
Zhou Y, Chen Z, Cheng M, Chen J, Zhu T, Wang R et al (2018b) Uncovering the dispersion history, adaptive evolution and selection of wheat in China. Plant Biotechnol J 16:280–291
Zhu XF, Zhang HP, Hu MJ, Wu ZY, Jiang H, Cao JJ, Xia XC, Ma CX, Chang C (2016) Cloning and characterization of Tabas1-B1 gene associated with flag leaf chlorophyll content and thousand-grain weight and development of a gene-specific marker in wheat. Mol Breed 36:142
Zhu Z, Xu D, Cheng S, Gao C, Xia X, Hao Y, He Z (2018) Characterization of fusarium head blight resistance gene Fhb1 and its putative ancestor in chinese wheat germplasm. Acta Agron Sin 44:473–482
Zou C, Wang P, Xu Y (2016) Bulked sample analysis in genetics, genomics and crop improvement. Plant Biotechnol J 14:1941–1955
Acknowledgements
This work was supported by Grant-in-Aids for Scientific Research (B) No. 16H04862 and for Scientific Research on Innovative Areas No. 19H04863 from MEXT to ST. We acknowledge Natural National Science Foundation (NSFC), China for Research Fund of International Young Scientists award (31950410563) and Pak-China (PSF-NSFC) Joint Research Grant. We apologize that due to the space limitations, we could not cite several citations.
Author information
Authors and Affiliations
Contributions
AR, ZH and TM conceived the idea. AR, AM, ST and MI outlined the manuscript. AR, ST, MAH and MA wrote the manuscript. MI, AM, ZH and TM reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Xianchun Xia.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rasheed, A., Takumi, S., Hassan, M.A. et al. Appraisal of wheat genomics for gene discovery and breeding applications: a special emphasis on advances in Asia. Theor Appl Genet 133, 1503–1520 (2020). https://doi.org/10.1007/s00122-019-03523-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-019-03523-w