
Abstract
Key message
A repertoire of the genomic regions involved in quantitative resistance to Leptosphaeria maculans in winter oilseed rape was established from combined linkage-based QTL and genome-wide association (GWA) mapping.
Abstract
Linkage-based mapping of quantitative trait loci (QTL) and genome-wide association studies are complementary approaches for deciphering the genomic architecture of complex agronomical traits. In oilseed rape, quantitative resistance to blackleg disease, caused by L. maculans, is highly polygenic and is greatly influenced by the environment. In this study, we took advantage of multi-year data available on three segregating populations derived from the resistant cv Darmor and multi-year data available on oilseed rape panels to obtain a wide overview of the genomic regions involved in quantitative resistance to this pathogen in oilseed rape. Sixteen QTL regions were common to at least two biparental populations, of which nine were the same as previously detected regions in a multi-parental design derived from different resistant parents. Eight regions were significantly associated with quantitative resistance, of which five on A06, A08, A09, C01 and C04 were located within QTL support intervals. Homoeologous Brassica napus genes were found in eight homoeologous QTL regions, which corresponded to 657 pairs of homoeologous genes. Potential candidate genes underlying this quantitative resistance were identified. Genomic predictions and breeding are also discussed, taking into account the highly polygenic nature of this resistance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Blackleg or stem canker caused by the fungus Leptosphaeria maculans (Desm.) Ces. et de Not. is one of the most devastating diseases of winter and spring oilseed rape (Brassica napus L.) worldwide. The disease is of major economic importance in Europe, North America and Australia (West et al. 2001; Fitt et al. 2006). Different oilseed rape plant developmental stages are affected by L. maculans and in some cases it can completely kill the young susceptible seedlings and cause catastrophic yield loss (Marcroft et al. 2002; Fitt et al. 2006). Different resistance types, such as monogenic or polygenic resistance, usually operate at different plant developmental stages and control the disease at different stages of the fungus life cycle. Plants show resistance either through large effect R genes (monogenic) or moderate to small effect quantitative (polygenic) resistance. In B. napus, blackleg resistance at the seedling stage is governed by race-specific R genes (Rlm genes) active from cotyledon stage onwards. In adult stages, race non-specific QTL limit the development of necrosis at the basis of the stem (Delourme et al. 2006; Rimmer 2006; Huang et al. 2009; Raman et al. 2013; Larkan et al. 2016). Major R genes are often used to effectively control a particular pathogen but their durability is a concern for breeders. Quantitative resistance is generally more durable than major-gene-mediated resistance but its genetic dissection might not be easy due to the influence of R genes in the genetic background or interactions with the environment. However, once QTL are identified, their deployment either in combination with R gene(s) or alone in elite varieties could be a long-term solution for controlling disease infestation for several years in a sustainable way (Pilet-Nayel et al. 2017). This was clearly demonstrated for blackleg of oilseed rape. Varieties whose resistance is based solely on major genes lose their effectiveness only 3–4 years after their release (Brun et al. 2000; Li et al. 2003; Rouxel et al. 2003). Furthermore, quantitative resistance mediated by multiple genetic factors was shown to increase the potential for durability of major resistance genes (Brun et al. 2010; Delourme et al. 2014).
The genetic architecture of oilseed rape resistance to blackleg is quite complex. More than fifteen specific resistance genes were identified in B. napus or introgressed in B. napus from the related Brassica species B. rapa, B. juncea and B. nigra (Rlm1–11; LepR1-4) (Delourme et al. 2006; Rimmer 2006; Balesdent et al. 2013) and mapped on A02, A06, A07 and A10 (Delourme et al. 2004, 2006; Long et al. 2011; Yu et al. 2005, 2008, 2013). QTL regions have been mapped by linkage-based mapping and GWAS. Quantitative resistance is complex in nature, with multiple genes working together to defend the plant against the pathogen and it is often drastically affected by changing environments or genetic backgrounds. Thus, QTL stability is often verified in multiple environments and/or crosses. In oilseed rape, the effect of genetic background and environment on quantitative resistance to blackleg has been shown. In winter oilseed rape, quantitative estimation of blackleg disease led to the identification of more than ten QTL using two doubled haploid (DH) populations derived from the resistant parent ‘Darmor’ (Pilet et al. 1998, 2001; Jestin et al. 2012). In multi-environment trials, six of these QTL were found to be less sensitive to environmental factors and thus appear to be relatively stable QTL (Huang et al. 2016). Jestin et al. (2015) used a multi-parental design to identify QTL common among different genetic backgrounds and background-specific QTL. Larkan et al. (2016) carried out a multi-environment study in Australia and identified and delineated four significant and stable QTL in spring oilseed rape. Quantitative resistance could be mapped effectively only in the absence of R genes that correspond to highly frequent avirulence genes in the field fungus population. Otherwise, the R genes would interfere with leaf lesion development and fungus progression into the plants, which would render quantitative resistance evaluation less effective. Hence, mapping populations and panels that do not segregate for effective R genes in the field conditions used for phenotyping offer advantages in efficiently capturing quantitative resistance. This strategy was employed in the first GWAS studies for blackleg resistance where the selected panel was devoid of highly effective specific resistance genes to L. maculans in field conditions and the identified regions were involved in quantitative resistance (Jestin et al. 2011; Fopa Fomeju et al. 2014; 2015). R genes and QTL for qualitative and quantitative resistances, respectively, have also been reported in B. napus even in the presence of genotypes containing major R genes through the combination of field experiments and cotyledon stage inoculation in controlled conditions (Raman et al. 2012, 2013, 2016). Nevertheless, in B. napus, there is a paucity of blackleg-related association mapping studies which explore natural allelic diversity for quantitative resistance in available germplasms and assess the stability of the associations across environments. The identification of consistent QTL regions among different GWAS would help for breeding and identification of candidate genes for quantitative resistance through combined functional characterization.
Before SNPs, microsatellites were the most commonly used molecular markers to generate genetic maps and QTL studies in B. napus. Since the availability of SNP arrays in Brassica, saturated genetic maps have been developed (Chalhoub et al. 2014; Clarke et al. 2016). These maps as well as the B. napus genome sequence (Chalhoub et al. 2014) are now available for more precise comparisons of linkage and GWAS QTL. In this study, we took advantage of multi-year data available on three segregating populations derived from the resistant cv Darmor and multi-year data available on oilseed rape panels. We integrated these data with previously published data to obtain a complete repertoire of the genomic regions involved in quantitative resistance to L. maculans in winter oilseed rape. A combined study including both linkage-based QTL mapping and GWAS should provide complementary results which can be used to refine the similar QTL genomic regions targeted by both studies. Prospects for breeding and identification of potential causal genes in the associated genomic regions are discussed.
Materials and methods
Plant material
For QTL analysis, the plant material consisted of three populations sharing the same resistant parent and its dwarf version on one hand and three susceptible oilseed rape accessions on the other hand. ‘Darmor’ is a French winter double-low line registered in 1983 and exhibits partial but high level resistance to L. maculans. The mutated dwarfing-gene Bzh was introgressed into ‘Darmor’ by backcrossing and selfing (Foisset et al. 1995) to derive the ‘Darmor-bzh’ isogenic line. The segregating DH population, derived from the ‘Darmor-bzh’ × ‘Yudal’ cross (DY population), was previously described by Foisset et al. (1996). ‘Yudal’, which is susceptible to blackleg, is a Spring Korean line which behaves as an early flowering winter type in temperate climates. More than 900 DH lines were produced either by spontaneous chromosome doubling or colchicine treatment (Foisset et al. 1996). Different sets of DH lines from the DY population were evaluated in France and the UK in seven winter oilseed rape field experiments over different cropping seasons (Table 1). ‘Samouraï’ and ‘Bristol’ are two winter-type double-low lines, released in 1989 and 1993, respectively, and susceptible to blackleg. From ten ‘Darmor’ × ‘Samouraï’ F1 plants, a total of 134 DH lines were derived by in vitro androgenesis (DS population) and evaluated in 2-year field experiments (Table 1; Pilet et al. 2001). A single ‘Darmor’ × ‘Bristol’ F1 plant was self-pollinated to derive the F2:3 population (DB population) of 119 individuals and the F3 families were evaluated in a 2-year field experiment (Table 1; Jestin et al. 2015).
For GWAS analysis, a panel of 166 winter B. napus varieties was used (Supplementary Table 1). The panel was composed of modern winter oilseed rape (WOSR) varieties with low seed erucic acid and glucosinolate content (00 quality) and old varieties with high seed erucic acid and high glucosinolate content (++ quality). The varieties were chosen to represent a wide range of WOSR diversity and derived from different breeding programs. The absence of any highly effective specific resistance genes conferring total resistance in our field trial conditions was verified for the varieties included in the panel so that only the level of quantitative resistance was evaluated. This was deduced from cotyledon tests using isolates carrying known AvrLm genes (Balesdent et al. pers. comm.). Thus, we excluded WOSR varieties known to carry Rlm7, a highly effective gene that was recently introduced into WOSR varieties in France. The panel was evaluated for blackleg resistance in four different years (2006, 2013, 2014 and 2015). In 2006, only 96 genotypes were included for disease resistance analysis; however, in the other years, 166 genotypes were evaluated for blackleg resistance.
Field experiments
Field experiments for segregating populations consisted of α-designs with three replicates and were carried out in 1995, 1996, 2007, 2011 and 2012 at Le Rheu, INRA, France (Jestin et al. 2012) and in 2008 and 2009 at Rothamsted Research (RRES), UK (Huang et al. 2016) for DY population, in 1998 and 1999 at Le Rheu, INRA, France (Pilet et al. 2001) for DS population and in 2008 and 2010 at Le Rheu, INRA, France (Jestin et al. 2015) for DB population. The panel was also evaluated in α-designs with three replicates in 2006, 2013, 2014 and 2015 at Le Rheu, INRA, France (Jestin et al. 2011; Fopa Fomeju et al. 2015). Each replicate included a 5-row plot (2.5 m2) or a 10-row plot (6 m2) of each DH line/F3 family and the parental lines at INRA and RRES, respectively. In each replicate, control resistant and susceptible varieties were replicated in each row × column block to check the homogeneity of the trial. To ensure homogeneous disease infection, contaminated residues collected from the previous season were scattered throughout the plots in each cropping season at a density of one stem (at RRES) or two stems (at INRA) per m2 when the crop was at the two to three leaf growth stage. Stem canker severity was assessed 2–3 weeks before harvest (mid-June). Forty plants per plot were uprooted and stem canker was assessed on a 0–9 disease index scale (DI) (Aubertot et al. 2004). DI increases with crown canker severity, starting from zero for healthy plants to nine for completely lodged plants.
Statistical analysis of the phenotypic data
All statistical analyses were carried out with R software (R Core Team 2013). Normality of the phenotypic data was tested with Shapiro–Wilk test (α = 0.05). To account for different variances and correlation between years, BLUP (Best linear unbiased predictions) was estimated using a mixed-effects model implemented in the lme4 package (Bates et al. 2015). For each mapping population and the panel, the effect of genotype, experiment (year) and block was estimated following the global mixed model: Pijk = µ + Gi + Ej + Bk/j + εijk, where Pijk is the DI of the ith genotype in the kth replicate of the jth experiment, Gi is the random effect of genotype, Ej is the random effect of experiment, Bk/j is the random effect of replicate and εijk is the random residual error effect. Broad-sense heritability was then calculated as h2 = σ2G/[σ2G + σ2ε/N], where σ2G is the genotypic variance, σ2ε is the residual error variance estimated from the selected model, N is the total number of replicates for all experiments (2 or 3 per experiment).
A second mixed linear model was applied to each individual year experiment: Pij = µ + Gi + Bj + εij, where Pij is the DI of the ith genotype in the kth block, Gi is the random effect of genotype, Bj is the random effect of replicate and εij is the random residual error effect. For each experiment, broad-sense heritability was calculated as H2 = σ2G/[σ2G + (σ2ε/n)], where σ2G is the genotypic variance, σ2ε is the residual error variance estimated from the selected model and n is the number of replicates in the considered experiment. Marker-based estimation of narrow-sense heritability (h2) for all the biparental mapping populations was obtained using the heritability package (Kruijer et al. 2015) in R software.
Genetic maps
Genotyping data for the DY population included SNPs from the Brassica 60K Illumina Infinium SNP array (Clarke et al. 2016), the 20K Illumina Infinium SNP array (Chalhoub et al. 2014) and from the 8K Illumina Infinium SNP array (Delourme et al. 2013). Genotyping data for the DB population included SNPs from the 20K Illumina Infinium SNP array (Chalhoub et al. 2014). The DY and DB genetic maps were obtained from Chalhoub et al. (2014) and 60K SNPs were added to the DY map. The DS population was genotyped using the Brassica 60K Illumina Infinium SNP array. Automatic allele calling for each locus was done using GenomeStudio software (Illumina Inc., San Diego, USA). The clusters were manually edited when necessary. Technical replicates and signal intensities were verified and only the most reliable calls were retained. The genetic maps were built with Carthagene software (De Givry et al. 2005) using the group command to assign markers to linkage groups and the annealing, flips and polish commands to refine the marker order. For QTL analysis, SNPs were selected with a custom script to retain one marker per genetic position and as many common markers between populations as possible. Three genetic maps were then used for QTL mapping: the DY, DB and DS maps contained 3767, 2374 and 929 SNPs and covered 2128.2, 1942.9 and 1946.1 cM, respectively (Supplementary Table 2).
Panel genotyping
The association panel was genotyped using the Brassica 60K Illumina Infinium SNP array (Clarke et al. 2016). Chromosomal physical positions for all these genotyped SNPs were retrieved from already mapped SNPs (Clarke et al. 2016) or inferred from their genetic positions on our genetic maps for SNPs anchoring on random chromosomes. Following genotyping, the data were loaded into GenomeStudio V2011.1 (Illumina Inc.) for manual cluster refinement considering QC functions such as GenTrain and GenCall scores. After removing monomorphic and badly clustered SNPs, 37346 high-quality SNPs were retrieved for further analysis. Out of 37346, 8582 SNPs were further removed because of high heterozygosity or minor allele frequencies (< 5%). Finally, 28764 high-quality SNPs were used for GWAS. Missing values among these 28764 SNPs were imputed using the knncatimputeLarge function of the scrime package implemented in R (Schwender and Ickstadt 2008).
QTL mapping
For each population, BLUPs of genotype random effects extracted from the multi-year experiment as well as the different year-wise mean DI were used for QTL detection. A multiple QTL mapping model was tested using the R/qtl package (Broman et al. 2003) for the differently calculated BLUP.
For each variable, a simple interval mapping (SIM) was performed using the scanone function. The number of positions with a LOD score equal to or above 2.5 was used to declare the number of cofactors in the composite interval analysis (CIM) performed with the cim function. Positions with LOD scores equal to or above 2.5 were included as QTL in the first model. An ANOVA was fitted to the multiple QTL model (fitqtl function). QTL were retained in this first model when their effects were significant (α = 0.05). Interactions were investigated using the addint function and were included in the model when significant (α = 0.05). Again, an ANOVA was fitted to the multiple QTL model with added interactions (fitqtl function). The addqtl function was then used to test for further significant QTLs, followed again by an ANOVA fitted to the new model. These tests for new interactions and new QTL were performed iteratively until no further QTL or interactions could be added to the model. We then applied the refineqtl function which used an iterative algorithm to refine the locations of QTL in our multiple QTL model, with the aim of obtaining the maximum likelihood estimates of the QTL positions. A last ANOVA was fitted to this final multiple QTL model. Based on the ANOVA, the percentage of variation explained by the global model and the R2 of each QTL was assessed. The fiqtl function also provided the LOD value for each QTL. The confidence intervals of each QTL were assessed with a LOD drop of one (lodint function). QTL positions and marker names at these positions, confidence intervals, percentage of variation explained by each QTL (R2), allelic effect and favorable alleles were scored.
Population structure, relative kinship analysis and GWAS
For population structure and kinship analysis, a reduced set of SNPs was used to avoid biasness due to linkage disequilibrium (LD). For this, SNPs were pruned using the “indep-pairwise” option in PLINK 1.03 (Purcell et al. 2007) such that all SNPs within a given window size of 100 had pairwise r2 < 0.2. After filtering, only 2661 independent SNPs were retained for principal component analysis (PCA) and kinship analysis. Pairwise LD analysis (r2) was performed considering the whole set of SNPs (28764) using PLINK 1.03. Structure in the diversity panel was deciphered using the SNPRelate package in R employing the whole set as well as a pruned set of SNP markers (Zheng et al. 2012). For each variety, BLUPs extracted from the multi-year experiment as well as the different year-wise mean DI were used for GWAS to detect common and year-specific associations. GWAS was performed using a compressed mixed linear model (CMLM) implemented in the genomic association and prediction integrated tool (GAPIT), which is based on efficient mixed model association (EMMA) developed by Kang et al. (2008). An independent SNP-based matrix of genetic similarity generated through the EMMA function of GAPIT was used to model the variance–covariance matrix of the random effect. For significance of association, a P value threshold of − log10 (3) was used to consider false negatives or type II errors in the study and to rule out the overly conservative nature of type I errors. We also generated LD-based haplotype blocks only around significant SNPs (selected through GAPIT) in PLINK using the default method of Gabriel et al. (2002). Haplotypes < 0.2 frequency were removed while others were tested for association with blackleg BLUP data using PLINK. Narrow-sense heritability (h2), which is the proportion of phenotypic variance explained by the genetic variance, was also estimated as h2 = σ2g/(σ2g + (σ2ε) implemented in GAPIT, where σ2g is the additive genetic variance and σ2ε is the residual variance.
Genome-wide prediction
For genome-wide prediction of blackleg resistance using SNP markers in the current germplasm set, we used ridge regression best linear unbiased prediction, rrBLUP (Endelman 2011) implemented in R. To assess the prediction ability, 20–70% of the whole dataset (166 genotypes) were randomly sampled as training sets to predict trait values of the corresponding validation sets. The prediction ability (r) of the model was assessed using the Pearson’s correlation between the observed and cross-validated (500 iterations) predicted trait values to obtain the prediction accuracy. SNP markers were also tested for their prediction accuracy on the basis of their P value threshold (P < 0.001, P < 0.01, P < 0.1) in GWAS.
Results
Phenotypic analysis of biparental mapping populations
The frequency distribution of phenotypes observed among different environments and locations as well as BLUPs all showed continuous distributions for all three populations (DY, DB and DS) (Table 1; Supplementary Table 2; Supplementary Fig. 1A, 1B & 1C). Phenotypic distributions of the DI for the DY population in 2008 and 2011 were skewed towards low or high disease levels, respectively. The mean disease severity as estimated by the mean DI varied across years but the DI from the different environments and locations were significantly highly correlated within all three populations (Supplementary Fig. 1A, 1B & 1C). The overall frequency distribution of BLUPs for the DS population was very narrow where, except for one DH line, the BLUPs ranged from − 0.90 to 0.84, which is less variable as compared to DY (− 2.74 to 2.10) and DB (− 1.53 to 1.90) populations. Variability in disease scores led to the highest broad-sense heritability in the DY (0.95) population followed by the DB (0.83) and DS (0.68) populations (Table 1).
QTL identification
Using the composite interval mapping (CIM) for QTL identification, three to 12 QTL were detected according to the year in the DY population (Supplementary Table 3A), which individually explained from 2.1 to 17.0% of the phenotypic variation. Overall 16 QTL were detected for the BLUP estimates extracted over years in the DY population. All these QTL were located on 15 linkage groups and cumulatively accounted for 75.9% of the phenotypic variation (Table 2). One QTL on A04 showed the largest LOD score (22.8) and contributed to more than 10% of the phenotypic variation. Most of the DY BLUP QTL were detected in two to five individual years except for the QTL on A03 which was only detected in 2007 and the QTL on C06 which was never detected in individual years. Nine and 11 QTL were detected in 2008 and 2010 in the DB population, respectively (Supplementary Table 3B), with each accounting for 3.1–34.7% of the phenotypic variation. This resulted in 13 QTL detected from the BLUP estimated over years that were located on 11 linkage groups and accounted for 94% of the phenotypic variation (from 2.2 to 17.0% each) (Table 2). The DB BLUP QTL were either detected in one or two individual years or were not detected in individual years. Different genetic effects were observed according to the QTL. Five and one QTL were detected in 1998 and 1999 in the DS population, respectively (Supplementary Table 3C), with each accounting for 4.8–19.7% of the phenotypic variation. This resulted in four QTL detected from the BLUP estimated over years that were located on three linkage groups and accounted for 39.8% of the phenotypic variation (from 7.1 to 10.4% each) (Table 2). The DS BLUP QTL were either detected in one individual year or were not detected in individual years. Narrow-sense heritability (h2) and the QTL number identified in each population were related to the broad-sense heritability (H2) variations. Only one QTL was identified in 1999 for the DS population with H2 = 0.41 and h2 = 0.40. In contrast, the highest numbers of QTL were identified for the DY population in 2007 and 2012 (11 and 12 QTL, respectively), the 2 years with the highest H2 (0.93 and 0.92, respectively).
In each population, the support intervals for the BLUP QTL generally overlapped with those of individual-year QTL but in some cases, their positions were quite different (Supplementary Fig. 2). Some significant epistatic interactions were detected between some QTL in the DY and DB populations (Supplementary Table 3) but these were not consistent between years or with those detected from the BLUP estimates. In the three populations, the resistance allele was mainly inherited from the resistant parent ‘Darmor’ except on A01, A03, C01 where it was from ‘Yudal’ or ‘Bristol’, C06 from ‘Samourai’, C08 from ‘Bristol’ and ‘Samourai’ and C09 from ‘Bristol’.
The support intervals for each individual-year and BLUP QTL were projected on the ‘Darmor-bzh’ sequence using the physical position of the nearest flanking SNP (Supplementary Table 4; Supplementary Fig. 3) in order to compare QTL localization between the three populations. Only one region on C04 showed overlapping support intervals between the three populations. Fifteen regions were detected in two populations on A01, A02 (two regions), A03, A04, A05, A06 (two regions), A08, C02, C04, C05, C06 and C08 (two regions). Most (nine regions) were common in the DY and DB populations. Six of these regions were detected for BLUP estimates in the two populations (on A02, A04, A06, A08, C04, C06) and five regions for BLUP estimates in one population and individual-year QTL in the other population (on A01, A02, A05, A06, C08). The last three regions carried overlapping individual-year QTL.
Population structure and LD patterns in the diversity panel
The germplasm collection used in this study included a total of 166 genotypes collected from different countries and breeders. To look for structure in the germplasm collection, we used the whole set of SNPs (28764) as well as the pruned set of markers (2661). No significant differences in the germplasm structure and variance explained by principal components (PCs) were observed because of LD between the markers. In PC analysis, no major PC was detected and the first three PCs explained only 12.2% of the variance (Supplementary Fig. 4) where the first component explained only 4.7% of the variance. The genotypes were scattered throughout the PC plots without any specific or deep sub-population structure (Supplementary Fig. 4). We also generated a kinship matrix of identity by state (IBS) coefficients across all the genotypes using the pruned set of SNPs. An IBS coefficient-based heatmap implied no clear ancestry in the panel and an average of 0.67 random allele sharing was observed throughout the panel. We observed three sub-groups (reported in Supplementary Table 1) in genotype clustering based on allele-sharing coefficients. Genome-wide pairwise LD between SNPs was calculated across all the genotypes to investigate the potential of the array to capture all significant regions associated with disease. The pairwise r2 values were divided into bins of 10 kb and mapped against physical distance (kb) across the chromosome, and LD decay was observed up to 2 Mb. The maximum average r2 (0.83) was observed between SNPs at a distance of 10 kb distance from each other and it decreased to its half value (0.40) at around 90 kb. However, a sharp decay in LD was noted up to 150 kb after which it decayed relatively more slowly.
Phenotypic variation for blackleg disease in the diversity panel
Differences among year-wise means of the genotypes for disease resistance with continuous phenotypes were observed. The DI of genotypes among different years were highly correlated. The maximum correlation (0.87) was observed between the 2014 and 2015 datasets, while the lowest correlation was between the 2006 and 2013 phenotypic means. The histograms and boxplots of individual-year DI and BLUPs are presented in Supplementary Fig. 5. Disease data from 2006 were normally distributed while a small amount of positive skewness was observed in the 2014 and 2015 data. However, in 2013, the DI was highly positively skewed towards a low level of disease. All sources of variations due to genotype, year and block/year were significant in the global model. The ANOVA showed that DI was significantly variable between different replicates within a year (P value 3.29e−05) with a large F value (10.48) in the case of 2013 followed by 2015 (P value: 0.00327, F value: 5.781). Differences due to replicates within years were non-significant in the case of 2006 and 2014. Thus, by considering years and replicates as major sources of variation, BLUPs were calculated to minimize the errors caused by simple means of all the years for GWAS. BLUPs ranged from − 1.086 to 2.78 (Supplementary Table 1).
Association analysis
After filtering the SNPs with quality criteria, 28764 SNPs were finally retained for association analysis. Compressed MLM was used to examine the association of SNPs with blackleg disease scores. To control for false-positive associations because of population structure and cryptic relatedness among individuals, we used a kinship matrix based on the pruned set of SNPs as a random effect in the mixed model. As discussed earlier, PCs in this study were not informative and did not identify any new clustering over kinship relationship matrix. Significant threshold levels were adjusted according to the background noise of false-positive associations and − log10 (3) was found to be the most appropriate in our study, where only a few SNPs were truly significantly associated with blackleg resistance and where this trait is highly polygenic with individual associations explaining only a small part of the phenotypic variation. We re-checked the significant associations using a linear model in R and by generating haplotypes around the significant SNPs and checking their re-association with blackleg disease scores in PLINK. We also performed conditioning on some of the most significant SNPs to investigate the confounding effects of background loci that may be present throughout the genome due to LD. Using the most significant SNPs as cofactors in the model helped to find some new associations and to increase the power in terms of P values of already captured significant loci with low power. Overall, five regions were detected to be significantly associated with the DI for BLUP on chromosomes A08, A09, C03, C04 and C09 (Fig. 1; Supplementary Table 5). The most significant associations were detected on chromosome A08 followed by C04 with large LD blocks. Most of the significant SNP alleles were common alleles with MAF ranging from 32 to 48%. However, two of the alleles on C03 and C09 had a MAF of 11 and 14%, respectively.

Manhattan and Q–Q plots showing the stem canker resistance-associated regions (pink) from multi-year BLUP estimates in the GWAS panel. A1, A2 are the GWAS results employing CMLM model without conditioning on SNPs. B1, B2 are the GWAS results by conditioning on most significant SNP from A08 chromosome. C1, C2 are the GWAS results by conditioning on most significant SNPs from A08 and C04 chromosomes
A highly significant association on A08 was detected with SNP Bn-A08-p14512862 (12213572 bp), with a P value = 2.37E−06 and explaining almost 9% of the phenotypic variation. We also reviewed the LD haplotype and haplotype frequency around this significant SNP marker. We tracked the physical interval of the LD block by setting the LD decay to r2 = 0.2 from the most significantly associated SNPs and found that the A08 LD region is large (1.7 Mb) and located between 10557498 bp (Bn-A08-p12638473) and 12279616 bp (Bn-A08-p14718347).
The most significant SNP on C04 was located at 30349322 bp (Bn-C4-p33755696) and explained almost 5% of the phenotypic variation with a P value = 0.0002. Significant SNPs from this C04 region clustered in a LD block which was located from 30153297 bp (Bn-C4-p32955161) to 31443717 bp (Bn-C4-p34813310) covering a 1.2 Mb region.
Apart from some major regions on A08 and C04, other less significant regions were also detected on A09, C03 and C09. Significant SNPs on A09 were located at 4769933 bp (Bn-A09-p4862871) and explained almost 4% of the phenotypic variation with a P value = 0.0006. The major LD block was quite small (30337 bp) for this region, but it has an extended association (r2) with several other SNPs of up to several kb. The significant region on C03 showed an R2 value of almost 4% with a P value = 0.0008 and was detected at around 29823812 bp (Bn-C3-p31174419). Significant SNPs from this region were located in a large LD block (~ 7 MB) spanning from 23339613 bp (Bn-C3-p25434294) to 30313720 bp (Bn-C3-p32268237). However, all the significant SNPs from this region were found at the end of this LD block in a little isolated region with high r2. A single SNP (Bn-C9-p4178485) was also detected on C09 with an R2 value of almost 5% and a P value = 0.0004. This SNP had a MAF of only 14% and was linked through high r2 (LD) with only two other SNPs in the same LD block.
We also used the most significant SNP on A08 as a cofactor in the model and a new association was detected on C06. Conditioning on this SNP also improved the significance level of SNPs located on A09 and C04. We also conditioned on the second most significant SNP from C04 in the model which resulted in another significant association on C01 at a significance level of − log10 (3) P value. We reconfirmed the significance of these associations by plotting phenotypic means expressed by the respective haplotype of the SNP and testing the significance of the slope using a linear model in R. Both these newly detected SNPs on C01 and C06 showed an R2 of almost 4% and were, respectively, located at 9059861 (Bn-C1-p9646961) and 28381033 (Bn-C6-p8469110) bp.
Finally, we investigated the cumulative explanatory power by looking at the joint effect of all the significant SNPs. For this, multiple linear regression was fitted in R where all the significant SNPs from A08, A09, C01, C03, C04, C06 and C09 were used as explanatory variables while BLUP was the response variable. Collectively, all these variables explained 55% of the phenotypic variation.
Significant alleles and narrow-sense heritability
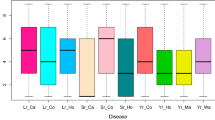
A minor allele haplotype of the most significant SNP on A08 was related to resistance and provided the highest level of resistance (BLUP = − 0.58) with a moderate allelic effect (− 0.31) (Fig. 2). In all other cases, major alleles of all the significantly associated SNPs showed higher resistance with a moderate allelic effect (Fig. 2). High narrow-sense heritability was observed for the year-wise DI as well as for BLUPs estimated from over all the years. A year-wise minimum h2 = 87% was observed for 2014 while the maximum was for 2013 (h2 = 98.7%). As BLUP is a cumulative expression of all types of random effects, it showed the highest h2 (99.4%).

Boxplots of the distribution of stem canker BLUPs within each genotypic class of the seven most significant SNPs in GWAS. Genotypic data are transformed into numeric format where ‘0’ and ‘2’ represent the homozygous state while ‘1’ is for heterozygous state
Genotype × environment effects
Substantial changes in the significant loci were observed between years. None of the GWAS peaks were consistently significant in all the years but BLUP-associated regions on A08, C01, C03 and C04 were significant in more than one year (Supplementary Fig. 6). However, the effects of the most stable GWAS peaks varied greatly across the years. Some of the significant GWAS peaks were detected in only 1 year. For example, two peaks on A03 were only significant in 2013 when the DI was quite positively skewed compared to other years. In 2013, two other GWAS peaks on A05 and C01 were also detected but linear modeling and cofactor analysis later showed that these peaks were associated because of LD with two of the most significant signals on A03. In 2014, some of the loci from the A genome were detected near the significance threshold; however, only one small effect variant (Bn-A06-p15037349) on A06 was significant. A significant SNP (Bn-C6-p14996206) on C06 (21920027 bp) was also detected from 2006 data. It was quite far from the significant SNP (Bn-C6-p8469110: 28381033 bp) detected for BLUP on the same chromosome but plenty of SNPs in these two regions were in LD.
Overlapping QTL in linkage and association mapping
We investigated the combined results of linkage-based mapping and LD-based association mapping to identify the QTL that are significantly associated with blackleg resistance in the B. napus panel. Small differences were evident since different sets of markers were used in the linkage and association mapping. Globally none of the QTL were detected throughout all the populations in linkage-based mapping and GWAS. However, some of the GWAS regions did partially or completely overlapped with QTL detected in one or two linkage-based mapping populations (Fig. 3; Supplementary Fig. 3). Co-localization with linkage-based QTL was observed for both of the most effective BLUP-GWAS QTL (on A08 and C04). The most significant GWAS-BLUP peak (A08:12213572) on A08 completely overlapped with the major BLUP-QTL (DY_BLUP_A08) identified in the DY population. The second most significant GWAS-BLUP peak on C04 overlapped with the BLUP-QTL (DB_BLUP_C04) detected in DB populations. Overlapping was also detected in other GWAS-based less effective QTL but with less stringency. A 2014-specific GWAS region on A06 overlapped with only one QTL detected in 2007 in the DY population, even though the region harbors some other significant year-wise and BLUP QTL. The 2014-specific GWAS peak on A06 is also situated very close to the most significant BLUP-QTL detected in the DB population that explained almost 15.8% of the phenotypic variation. The significant GWAS-BLUP region mapped on A09 overlapped with DY_BLUP_A09, DY_2011_A09 and DY_2012_A09 QTL. However, as the region was quite large because of extended LD, the most significant SNPs from this region were situated a small distance away from the overlapped region. A partial overlap was also detected between a GWAS-BLUP peak on C01 and DB_2008_C01 QTL and between a GWAS peak on C06 and DB_2010_C06 QTL. None of the GWAS-based QTL overlapped with DS QTL except on C04 where DS_BLUP_C04 QTL was located within an uncertain interval.

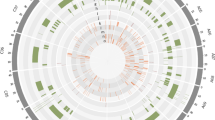
Schematic representation of the physical position of the linkage-based (in orange and red) and LD-based (in blue) QTL identified from individual years and BLUP estimates of DI. The upper curve represents the density of QTL presence along all the chromosomes
To find out the homoeologous relationship between QTL identified from multi-year BLUPs and year-wise data in GWAS and the mapping populations, the co-localized QTL were considered as same ones. We then identified a total of 31 and 29 QTL regions on the A and C genomes, respectively. We inferred the corresponding A. thaliana genomic block for each QTL using homology between B. napus and A. thaliana genes. QTL were found in most genomic blocks and many were localized in homoeologous or paralogous regions (Supplementary Table 6). Homoeologous B napus genes were found in eight pairs of homoeologous regions (Fig. 4), which corresponded to 657 pairs of homoeologous genes. These homoeologous relationships were shared between regions either identified only in GWAS (A09/C09), or in linkage mapping (A01/C01, A03/C07, A06/C06, A06/C05, A06/C07) or between regions identified in GWAS and in linkage mapping (A04/C04, A09/C09). The number of homoeologous genes varied across these regions from 15 on A04/C04 (T genomic block) to 217 (U genomic block) on A01/C01. C04 was identified as the second most important QTL in the GWAS and completely overlapped with a BLUP-QTL identified with the DB and DS populations. It was located only 879 kb away from an important BLUP-QTL in the DY population. Its homoeologous counterpart corresponded to DY_2007_A04 QTL and was very close to DY_BLUP_C04 QTL.

Comparative physical map showing the homoeologous relationship between genes underlying blackleg resistance QTL. Visualized by Circos. The middle ring represents the Arabidopsis thaliana genomic blocks (GB). The inner ring represents the compilation of support intervals of the linkage-based (in red) and the LD-based (in blue) QTL. Dark red and blue indicate the BLUP QTL
Prediction of stem canker quantitative resistance
We found 23 varieties that shared resistance alleles at the most significant BLUP-associated SNPs, especially on A08, A09, C03, C04 and C09 regions (Supplementary Table 7). Collectively, all these 23 varieties showed a BLUP mean of − 0.97. In the most significant regions on A08 and C04, the resistant haplotypes from C04 contributed to a higher level of resistance (− 0.79) than A08 (− 0.56).
The ability to predict blackleg resistance in the B. napus panel was assessed employing various combinations of training sets and SNP markers. The predictive ability varied from 0.53–0.64 for 20–70% training population ratios. However, it changed slightly to 0.63 for a 60% training set. Thus, all the remaining combinations were made using a 60%:40% ratio for training and validation sets. We then selected only the most significant markers identified in GWAS to assess predictive ability, including markers with P < 0.001 (77 SNPs), P < 0.01 (461 SNPs), P < 0.1 (3134 SNPs), and the predictive abilities were 0.68, 0.84 and 0.88, respectively. The predictive ability of year-wise blackleg DI was also assessed using 60%:40% training and validation set ratio and the 3134 significant SNPs at P < 0.1. It was 0.80 for 2014 and 2015 and 0.81 for 2013.
Discussion
With combined multi-year analyses of LD- and linkage-based mapping, we could obtain a wide overview of quantitative resistance factors to L. maculans in winter oilseed rape under field conditions. The continuous distribution of stem canker DI for the biparental populations and the panel in different years and locations clearly confirmed that the resistance tagged in our study is quantitative. None of the genomic regions captured through LD- and linkage-based mapping explained a very high level of disease variation, which supports the fact that this quantitative resistance is highly polygenic and controlled by several low effect loci distributed throughout the genome, as earlier pointed out in our previous studies (Pilet et al. 2001; Jestin et al. 2011, 2012; Fopa Fomeju et al. 2014, 2015). Using high numbers of SNPs and the available B. napus sequence, we were able to project all the identified QTL regions on the physical map of B. napus and to precisely investigate the stability of these QTL over genetic backgrounds and years. Many consistent as well as population/GWAS-specific genomic regions were identified from this wide analysis.
Broad-sense heritability for stem canker resistance in all biparental populations was high and comparable to that found in Pilet et al. (1998), Jestin et al. (2015), Larkan et al. (2016) and Raman et al. (2016), which suggests that a large percentage of phenotypic variation for stem canker DI is due to genotypic components. The DS population was less variable for stem canker disease and the BLUP range was narrower compared to the DY and DB populations. The three populations ranked in the same order for broad-sense heritability. The number of detected QTL was related to disease severity distribution and broad-sense heritability in different years. In the case of the DY population, the highest numbers of QTL (11–12) were identified when disease was normally distributed and with the highest broad-sense heritabilities, e.g., in years 2007 and 2012. In GWAS, there is also an implicit assumption that the phenotypes follow a normal distribution, a skewed phenotype can severely affect the power and type I error (Goh and Yap 2009). In our GWAS panel, phenotypic data from 2013 showed a positively skewed DI. This led to two false-positive associations on A05 and C01, which were detected through allele discriminant and cofactor analysis. Compared to 2014 (mean MAF = 0.34) and 2015 (mean MAF = 0.33), MAFs in the associated regions were also comparatively low (0.22) for 2013-specific associations. BLUP-GWAS results were more similar to 2006, 2014 and 2015 results than those of 2013.
In recent years, BLUP estimates of phenotypic data gained importance in plant breeding to obtain a more precise genetic value for each genotype and has been widely used in multi-environment QTL mapping in many crops for detection of QTL (Gore et al. 2014; Rabbi et al. 2014; Allard et al. 2016; Avia et al. 2017; Zhang et al. 2017). In our study, DI means and variances varied significantly between different years. BLUP offers advantages over means of phenotypes over different years by accounting for non-genetic variations caused by replications and environments. Globally, except for DS_BLUP_C04 QTL, the mean size of the support intervals was lower with BLUP estimates. The BLUP QTL were compared with those obtained in different years to determine which regions are most robust and could be the most reliable for marker-assisted breeding. In the DY population, which was evaluated across many years, clusters of individual-year QTL either overlapped or were scattered around BLUP QTL. Such QTL clusters were observed on linkage groups A01, A02, A04, A05, A06, A07, A09, C01, C04, C08 and C09. The variation in locations of individual-year QTL between years confirms that genotype × environment interactions have a strong impact on QTL detection and location accuracy as shown in Huang et al. (2016) for stem canker. Disease severity is assessed at the end of a long cropping season, which is about 9 months for WOSR, and then results from this long period of interaction between the host plant, L. maculans and the environment. QTL effects might then differ across varying environments due to climatic variation that can impact the level and timing of infection (Evans et al. 2008) or the effect of individual determinants of plant response to infection. Phenotyping through visual assessment of the necrosis on the stem sections may also give rise to slight variations in the assessment of the DI that could affect the QTL location. Sixteen regions were common to at least two populations, mainly between the DY and DB populations. Of these, nine were common to the 15 regions previously identified in a connected multi-parental design on A01, A02, A05, A06, C04, C05, C06 and C08 (Jestin et al. 2015). The six other regions on A09, C01, C03, C04 and C09 from Jestin et al. (2015) were detected in a single population in the present study. These co-localizations highlight the fact that many QTL present in ‘Darmor’ or its parent ‘Jet Neuf’ were introduced into current varieties through breeding.
GWAS from BLUP estimates summarized the associated regions that were detected in the year-wise analyses and discarded the non-significant ones found in 2013. Significance level of some of the BLUP peaks and number of associations within these peaks have been improved in comparison to individual year GWAS results. This current GWAS study with multiple linear regressions, including all the significant loci, overall explained 55% of the phenotypic variation. This ensures that the 60k chip would be sufficient to harness the phenotypic variability of stem canker in B. napus under field conditions. All accessions in the GWAS panel showed a moderate genetic relationship with each other, which limits the probability of false positives in the case of a deep population sub-structure. Thus, only kinship matrices were used to account for sub-structure in our GWAS. Fopa Fomeju et al. (2014, 2015) previously performed GWAS to identify genomic regions underlying blackleg resistance and used PCs to check for false positives. This could have overestimated the structure and lead to false negatives. The current study included many years of disease evaluation with a more stringent significance threshold which led to increased association significance and accuracy. In all, 138 SNPs were significant at -log10 (3), which on a LD (r2 = 0.2) basis can be divided into 11 genomic regions. Two highly significant regions on A08 and C04 contained most of these SNPs (63 and 27, respectively). Both these BLUP-associated significant regions are common with the genomic regions published in Fopa Fomeju et al. (2014, 2015) where both regions were the most significant in a KP-CML model. Our BLUP-associated genomic results from A09 and C09 were also common with the significant results in Fopa Fomeju et al. (2014, 2015). Most of the significant results in different years were consistent with those obtained with the estimated BLUP but some were specific to a year, due to genotype × environment interactions. In 2013, the less severe disease resulted in phenotype skewness and two specific GWAS peaks were observed, specifically on chromosome A03 (Bn-A03-p2891056: 2451234 and Bn-A03-p20550791: 19385492). Both these were also observed in Fopa Fomeju et al. (2015). Indeed, our GWAS results for 2006- and 2013-specific disease index were exactly the same as the GWAS results of Fopa Fomeju et al. (2014, 2015). GWAS results for BLUP associated with C03 (~ 29.8 MB) and C04 (~ 30.5 MB) are new in our study. This could result from a normal non-skewed distribution of the disease index in 2014 and 2015 compared to 2013, which improved the overall BLUP estimates. We also found a 2014-specific association on A06 (Bn-A06-p15037349: 16498797) which is new with regard to the studies of Fopa Fomeju et al. (2014, 2015). Other newly associated genomic regions would have been detected from our BLUP estimates if we had set a low P value threshold (P < 0.05) like Fopa Fomeju et al. (2015) but that may also have resulted in some false-positive associations.
Consistent mapping of BLUP-associated genomic regions makes them a better choice for identifying candidate or causal markers in future studies or for marker-assisted breeding. Stem canker resistance sharing haplotypes across 23 varieties in the panel on chromosomes A08, A09, C03, C04 and C09 conferred a high resistance phenotype. These varieties may also contain some other small effect alleles which were untagged in the current GWAS. All these genotypes were distributed randomly in the three kinship groups; however, most (13) were located in group 2 where all the varieties showed a good level of resistance (BLUP = − 0.73). This set of varieties can further be incorporated in different breeding programs for improvement of stem canker resistance in B. napus.
The identification of overlapping genomic regions in LD- and linkage-based mapping is a two-way validation of stem canker resistance QTL from ‘Darmor’ that are segregating in the WSOR panel. In particular, the markers associated in A08, A09/C09 and C04 genomic regions captured through both analyses and their consistency with previous studies (Fopa Fomeju et al. 2014, 2015) suggest their major role in controlling stem canker quantitative resistance. Thus, these markers could be used to trace introgression of favorable QTL alleles in canola breeding programs. GWAS is basically known to resolve the large LD blocks because of the accumulated recombination events in the germplasm (Mitchell-Olds and Schmitt 2006; Nordborg and Weigel 2010). However, in our study, one of the GWAS peaks from A08 spanned over a distance of 1.7 MB which is either due to the presence of a cluster of genes involved in stem canker resistance in adult plants in this region or because this region is pericentromeric with a low recombination rate. The most significant SNPs from this region showed a scattered LD pattern over a large physical distance that did not show the expected continuous decrease of r2 for LD. This could result from genetic or physical mapping inaccuracy in this region.
Some of our significant genomic regions were very close to or within 100 kb of SNPs identified by Raman et al. (2016) from GWAS and linkage mapping results obtained in ascospore shower tests in controlled conditions, which should be at least partly related to quantitative adult plant resistance (Supplementary Fig. 3). This suggests that these regions may be involved in quantitative resistance both in WOSR and SOSR. BLUP-QTL on A06, A07, A09 and C08 were found in the vicinity of the associations from ascospore tests in Raman et al. (2016) and a few single associated markers from Raman et al. (2016) were localized in or close to our QTL or GWAS regions (on A04, A05, A08, C04, C05, C07). However, most significant GWAS peaks (A08, C03, C04 and C09) from our study were new with regard to Raman et al. (2016). In contrast, the A01 region identified as Rlm12 in Raman et al. (2016) is located between the two QTL regions we detected in the DY and DB populations. However, it does correspond to QTL region found by Larkan et al. (2016) who studied field stem canker resistance in two DH populations derived from SOSR-resistant Australian varieties (AG-Spectrum and AV-Sapphire). This region may thus be specific to SOSR. Raman et al. (2016) also performed GWAS at the cotyledon stage and identified many significant associated markers of which clusters of associated markers at − log10(p) ≥ 4 were located on A05, A07, A10, C03, C06, C09. The A05, C03 and C09 regions were located in the vicinity of QTL detected in our study and the C06 region co-localized with a QTL detected in Larkan et al. (2016) from AV-Sapphire. The region on A07 corresponds to the Rlm3/Rlm4 location that was previously detected (Delourme et al. 2004) and more precisely mapped in Raman et al. (2012) and Larkan et al. (2016). The region on A10 corresponds to the LepR3/Rlm2 location. Whether any Rlm-like R genes account for a proportion of quantitative adult resistance remains to be investigated. Pilet et al. (2001) already raised this hypothesis since a QTL was located at the Rlm2 position in the DS population. As discussed in Pilet et al. (2001), it is not possible to discriminate between the partial effect of overcome R genes or linked gene hypotheses.
The GWAS panel selected for this study was devoid of highly effective race-specific R genes (Rlm) to accurately estimate the genetic architecture of quantitative resistance to stem canker in B. napus. However, R gene analogs are frequent throughout the B. napus genome (Chalhoub et al. 2014; Raman et al. 2016; Alamery et al. 2017), so all the significant genomic regions in our study, 200 kb on either side of the most significant SNPs, were searched for R genes. None of the associations in our study was located on A07 which suggests that this R-gene hotspot was ineffective in providing resistance in adult plants under our field conditions. Two closely located disease resistance genes (BnaA08g14760D [At4g36140] and BnaA08g14770D) were 178 kb away from the most significant SNP (Bn-A08-p14512862: 12213572 bp) on A08 in GWAS. One of these, At4g36140, is a TIR-NBS-LRR disease resistance protein known to provide an innate immunity response in Arabidopsis. However, its role in quantitative resistance needs to be further investigated, for example through an adult stage transcriptomic study of contrasting susceptible and resistant genotypes The potential role of this candidate gene in quantitative resistance meets the question raised above, i.e., partial effect of overcome R genes or linked genes.
The genetic control of a trait for stem canker quantitative resistance could originate in homoeologous regions in B. napus as discussed in Fopa Fomeju et al. (2014, 2015). Our study confirmed that homoeologous and paralogous regions are involved in the control of this trait. These duplicated regions may have retained some common genes with similar functions that could be good candidates for causal genes underlying the QTL. However, the contribution of these common genes to the resistance needs to be further investigated. As known from Brassica species genome comparisons and as discussed in Fopa Fomeju et al. (2014, 2015), fractionation or sequence evolution has occurred in these duplicated regions and led to gene loss or gene sub-/neo-functionalization. The causative genes in these regions may then also be genes with different functions. With the hypothesis that increasing the diversity of genetic factors controlling the resistance would result in an increase in the potential durability of the resistance, knowledge of the level of functional redundancy and allelic diversity of the genes controlling stem canker resistance would help to construct resistant varieties with improved durability.
Based on the completely sequenced and annotated genomes of A. thaliana and B. napus, LD regions surrounding the significant associations from our study were searched for candidate genes for stem canker. In the A08 region (892400 bp, 442 genes), 6.2% of the genes were stress responsive and the most significant SNPs from this region are closely surrounded by important genes such as the mitogen-activated protein kinase kinase kinase 16 (MAPKKK16/At4g26890) and a WRKY transcriptional factor, which play important roles in signal transduction and transcriptional regulation under biotic and abiotic stress (Ichimura et al. 2002). The most significant SNP (Bn-A09-p4862871) from chromosome A09 is located in a mitogen-activated protein kinase 7 (MAPK 7). Elicitors such as chitin produced mainly by hemibiotroph fungi such as L. maculans can activate downstream defense responses through cytosolic protein kinases such as the MAPK kinase kinase (MAPKKK) pathway which in turn activates an array of plant transcription factors such as WRKY to regulate resistance-related metabolites (RRMs) and proteins (RRPs) (Kushalappa et al. 2016). These RRMs and RRPs suppress pathogens due to anti-microbial properties or through further enforcement of cell walls by producing structural barriers (Kushalappa and Gunnaiah 2013) and lead to reduced susceptibility or quantitative resistance (Kushalappa et al. 2016). The LD region from C01 contains two defensin-like proteins (At4g21520 and At4g22115) which are known to be involved in defense against a broad range of fungi (Lay and Anderson 2005; Wong et al. 2007; Thomma et al. 2002) and their activities have been noted in roots, seeds, flowers, stems, and leaves (Nawrot et al. 2014). Other significant regions were also surrounded by several stress responsive proteins but further studies are needed to find out the causal marker/gene and to validate them functionally. Transcriptomic studies of the B. napus/L. maculans pathosystem through high-throughput RNA sequencing have so far only been performed at the cotyledon stage to analyze compatible interactions (Lowe et al. 2014; Haddadi et al. 2016) or the LepR1-AvrLepR1 interaction (Becker et al. 2017). Such studies performed at different time points at later stages on stems would help to identify potential mechanisms underlying quantitative resistance to L. maculans.
Stem canker resistance is definitely a highly polygenic trait with a high number of genomic regions that each accounts for a low proportion of phenotypic variation and that shows high genotype × environment interactions. Hence, breeding for such a trait may be more efficient through genomic selection. We thus investigated the potential for predicting this trait in the studied panel. Due to the high heritability of stem canker in our panel, the predictive ability was less sensitive to the number of markers (Tan et al. 2017) and changed slightly when the whole set of markers (0.63) was reduced to 5000 randomly selected SNPs (0.62) with a slight reduction in predictive ability. However, the incorporation of only 3134 SNPs that were significantly associated with resistance at P < 0.1 in the prediction model was sufficient to get high prediction accuracy (88%). These 3134 SNP markers are distributed throughout the chromosomes but many markers are located in the regions with significant associations in GWAS or in the support intervals of the QTL detected in the biparental populations. The need for this entire set of SNPs to improve the predictive ability shows the low power of GWAS to detect all regions associated with stem canker in our panel, which could result from the highly polygenic genetic nature of this trait (Bian and Holland 2017). This entire set of 3134 SNPs may be used to predict stem canker resistance of elite WOSR lines for breeding purposes but this would need to be validated in an independent panel.
Author contribution statement
VK, SP and BFF carried out the genetic analyses. VK, SP, BFF, MMD and RD wrote the manuscript and assisted its editing. CF, GD, CB and LB were in charge of the genotyping and of the genetic maps construction. PV was in charge of the field experiments and of the phenotypic data collection. RD and MMD conceived and coordinated the study.
References
Alamery S, Tirnaz S, Bayer P, Tollenaere R, Chaloub B, Edwards D, Batley J (2017) Genome-wide identification and comparative analysis of NBS-LRR resistance genes in Brassica napus. Crop Pasture Sci 69:72–93
Allard A, Bink MCAM, Martinez S, Kelner J, Legave J, Guardo M, Di Pierro EA, Laurens F, Van De Weg EW, Costes E (2016) Detecting QTLs and putative candidate genes involved in budbreak and flowering time in an apple multiparental population. J Exp Bot 67:2875–2888
Aubertot JN, Schott JJ, Penaud A, Brun H, Doré T (2004) Methods for sampling and assessment in relation to the spatial pattern of phoma stem canker (Leptosphaeria maculans) in oilseed rape. Eur J Plant Pathol 110:183–192
Avia K, Coelho SM, Montecinos GJ, Cormier A, Lerck F, Mauger S, Faugeron S, Valero M, Cock JM, Boudry P (2017) High-density genetic map and identification of QTLs for responses to temperature and salinity stresses in the model brown alga. Ectocarpus Sci Rep 7:43241
Balesdent MH, Fudal I, Ollivier B, Bally P, Grandaubert J, Eber F, Chèvre AM, Leflon M, Rouxel T (2013) The dispensable chromosome of Leptosphaeria maculans shelters an effector gene conferring avirulence towards Brassica rapa. New Phytol 198:887–898
Bates D, Mächler M, Bolker B, Walker S (2015) Fitting linear mixed-effects models using lme4. J Stat Soft 67:1–48
Becker MG, Zhang X, Walker PL, Wan JC, Millar JL, Khan D et al (2017) Transcriptome analysis of the Brassica napus-Leptosphaeria maculans pathosystem identifies receptor, signalling and structural genes underlying plant resistance. Plant J 90:573–586
Bian Y, Holland JB (2017) Data from: enhancing genomic prediction with genome-wide association studies in multiparental maize populations. Dryad Digit Repos. https://doi.org/10.5061/dryad.cd3hv
Broman KW, Wu H, Sen S, Churchill GA (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19:889–890
Brun H, Levivier S, Somda I, Ruer D, Renard M, Chevre AM (2000) A field method for evaluating the potential durability of new resistance sources: application to the Leptosphaeria maculans-Brassica napus pathosystem. Phytopathol 90:961–966
Brun H, Chèvre AM, Fitt BD, Powers S, Besnard AL, Ermel M, Huteau V, Marquer B, Eber F, Renard M, Andrivon D (2010) Quantitative resistance increases the durability of qualitative resistance to Leptosphaeria maculans in Brassica napus. New Phytol 185:285–299
Chalhoub B, Denoeud F, Liu S, Parkin IAP, Tang H, Wang X et al (2014) Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345:950–953
Clarke WE, Higgins EE, Plieske J, Wieseke R, Sidebottom C et al (2016) A high-density SNP genotyping array for Brassica napus and its ancestral diploid species based on optimised selection of singlelocus markers in the allotetraploid genome. Theor Appl Genet 129:1887–1899
de Givry S, Bouchez M, Chabrier P, Milan D, Schiex T (2005) CARTHAGENE: multipopulation integrated genetic and radiation hybrid mapping. Bioinformatics 21:1703–1704
Delourme R, Pilet-Nayel ML, Archipiano M, Horvais R, Tanguy X, Rouxel T, Brun H, Renard M, Balesdent MH (2004) A cluster of major specific resistance genes to Leptosphaeria maculans in Brassica napus. Phytopathol 94:578–583
Delourme R, Chevre AM, Brun H, Rouxel T, Balesdent MH, Dias JS et al (2006) Major gene and polygenic resistance to Leptosphaeria maculans in oilseed rape (Brassica napus). Eur J Plant Pathol 114:41–52
Delourme R, Falentin C, Fomeju BF, Boillot M, Lassalle G et al (2013) High-density SNP-based genetic map development and linkage disequilibrium assessment in Brassica napus L. BMC Genomics 14:120
Delourme R, Bousset L, Ermel E, Duffé P, Besnard AL, Marquer B, Fudal I, Linglin J, Chadoeuf J, Brun H (2014) Quantitative resistance affects the speed of frequency increase but not the diversity of the virulence alleles overcoming a major resistance gene to Leptosphaeria maculans in oilseed rape. Infect Genet Evol 27:490–499
Endelman JB (2011) Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome 4:250–255
Evans N, Baierl A, Semenov MA, Gladders P, Fitt BDL (2008) Range and severity of a plant disease increased by global warming. J R Soc Interface 5:525–531
Fitt BDL, Brun H, Barbetti MJ, Rimmer SR (2006) World-wide importance of phoma stem canker (Leptosphaeria maculans and L. biglobosa) on oilseed rape (Brassica napus). Eur J Plant Pathol 114:3–15
Foisset N, Delourme R, Barret P, Renard M (1995) Localization of agronomic traits on a B. napus map. In: Proceedings of 9th international Rapeseed congress, vol 4. Cambridge, UK, pp 1199–1201
Foisset N, Delourme R, Barret P, Hubert N, Landry BS, Renard M (1996) Molecular mapping analysis in Brassica napus using isozyme, RAPD and RFLP markers on a doubled haploid progeny. Theor Appl Genet 93:1017–1025
Fopa Fomeju B, Falentin C, Lassalle G, Manzanares-Dauleux M, Delourme R (2014) Homologous duplicated regions are involved in quantitative resistance of Brassica napus to stem canker. BMC Genomics 15:498
Fopa Fomeju B, Falentin C, Lassalle G, Manzanares-Dauleux M, Delourme R (2015) Comparative genomic analysis of duplicated homoeologous regions involved in the resistance of Brassica napus to stem canker. Front Plant Sci 6:772
Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M et al (2002) The structure of haplotype blocks in the human genome. Science 296:2225–2229
Goh L, Yap VB (2009) Effects of normalization on quantitative traits in association test. BMC Bioinform 10:415
Gore MA, Fang DD, Poland JA, Zhang J, Percy RG, Cantrell RG et al (2014) Linkage map construction and quantitative trait locus analysis of agronomic and fiber quality traits in cotton. Plant Genome. https://doi.org/10.3835/plantgenome2013.07.0023
Haddadi P, Ma L, Wang H, Borhan MH (2016) Genome-wide transcriptomic analyses provide insights into the lifestyle transition and effector repertoire of Leptosphaeria maculans during the colonization of Brassica napus seedlings. Mol Plant Pathol 17:1196–1210
Huang YJ, Pirie EJ, Evans N, Delourme R, King GJ, Fitt BDL (2009) Quantitative resistance to symptomless growth of Leptosphaeria maculans (phoma stem canker) in Brassica napus (oilseed rape). Plant Pathol 58:314–323
Huang YJ, Jestin C, Welham SJ, King GJ, Manzanares-Dauleux MJ, Fitt BDL, Delourme R (2016) Identification of environmentally stable QTL for resistance against Leptosphaeria maculans in oilseed rape (Brassica napus). Theor Appl Genet 129:169–180
Ichimura K, Shinozaki K, Tena G, Sheen J, Henry Y, Champion A et al (2002) Mitogen-activated protein kinase cascades in plants: a new nomenclature. Trends Plant Sci 7:301–308
Jestin C, Lodé M, Vallée P, Domin C, Falentin C, Horvais R, Coedel S, Manzanares-Dauleux MJ, Delourme R (2011) Association mapping of quantitative resistance for Leptosphaeria maculans in oilseed rape (Brassica napus L.). Mol Breed 27:271–287
Jestin C, Vallée P, Domin C, Manzanares-Dauleux MJ, Delourme R (2012) Assessment of a new strategy for selective phenotyping applied to complex traits in Brassica napus. Open J Genet 2:190–201
Jestin C, Bardol N, Lodé M, Duffé P, Domin C, Vallée P, Mangin B, Manzanares-Dauleux MJ, Delourme R (2015) Connected populations for detecting quantitative resistance factors to phoma stem canker in oilseed rape (Brassica napus L.). Mol Breed 35:1–16
Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, Eskin E (2008) Efficient control of population structure in model organism association mapping. Genetics 178:1709–1723
Kruijer W, Boer MP, Malosetti M, Flood PJ, Engel B, Kooke R, Keurentjes JJ, van Eeuwijk FA (2015) Marker-based estimation of heritability in immortal populations. Genetics 199:379–398
Kushalappa AC, Gunnaiah R (2013) Metabolo-proteomics to discover plant biotic stress resistance genes. Trends Plant Sci 18:522–531
Kushalappa AC, Yogendra KN, Karre S (2016) Plant innate immune response: qualitative and quantitative resistance. Crit Rev Plant Sci 35:38–55
Larkan NJ, Raman H, Lydiate DJ, Robinson SJ, Yu F, Barbulescu DM, Raman R, Luckett DJ, Burton W, Wratten N, Salisbury PA, Rimmer SR, Borhan MH (2016) Multi-environment QTL studies suggest a role for cysteine-rich protein kinase genes in quantitative resistance to blackleg disease in Brassica napus. BMC Plant Biol 16:183
Lay FT, Anderson MA (2005) Defensins-components of the innate immune system in plants. Curr Protein Pept Sci 6:85–101
Li H, Sivasithamparam K, Barbetti MJ (2003) Breakdown of a Brassica rapa subsp. sylvestris single dominant blackleg resistance gene in B. napus rapeseed by Leptosphaeria maculans field isolates in Australia. Plant Dis 87:752
Long Y, Wang Z, Sun Z, Fernando DWG, McVetty PBE, Li G (2011) Identification of two blackleg resistance genes and fine mapping of one of these two genes in a Brassica napus canola cultivar ‘Surpass 400’. Theor Appl Genet 122:1223–1231
Lowe RG, Cassin A, Grandaubert J, Clark BL, Van De Wouw AP, Rouxel T et al (2014) Genomes and transcriptomes of partners in plant-fungal-interactions between canola (Brassica napus) and two Leptosphaeria species. PLoS ONE 9:e103098
Marcroft SJ, Purwantara A, Salisbury P, Potter TD, Wratter N, Khangura R et al (2002) Reaction of a range of Brassica species under Australian conditions to the fungus, Leptosphaeria maculans, the causal agent of blackleg. Aust J Exp Agric 42:587–594
Mitchell-Olds T, Schmitt J (2006) Genetic mechanisms and evolutionary significance of natural variation in Arabidopsis. Nature 441:947–952
Nawrot R, Barylski J, Nowicki G, Broniarczyk J, Buchwald W, Gózdzicka-Józefiak A (2014) Plant antimicrobial peptides. Folia Microbiol 59:181–196
Nordborg M, Weigel D (2010) Next-generation genetics in plants. Nature 456:10–13
Pilet ML, Delourme R, Foisset N, Renard M (1998) Identification of loci contributing to quantitative field resistance to blackleg disease, causal agent Leptosphaeria maculans (Desm.) Ces. et de Not., in Winter rapeseed (Brassica napus L.). Theor Appl Genet 96:23–30
Pilet ML, Duplan G, Archipiano M, Barret P, Baron C, Horvais R, Tanguy X, Lucas MO, Renard M, Delourme R (2001) Stability of QTL for field resistance to blackleg across two genetic backgrounds in oilseed rape. Crop Sci 41:197–205
Pilet-Nayel ML, Moury B, Caffier V, Montarry J, Kerlan MC, Fournet S, Durel CE, Delourme R (2017) Quantitative resistance to plant pathogens in pyramiding strategies for durable crop protection. Front Plant Sci 8:1838
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575
R Core Team (2013) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/
Rabbi IY, Hamblin MT, Kumar PL, Gedil M, Ikpan AS, Jannink J-L, Kulakow P (2014) High-resolution mapping of resistance to cassava mosaic geminiviruses in cassava using genotyping-by-sequencing and its implications for breeding. Virus Res 186:87–96
Raman R, Taylor B, Marcroft S, Stiller J, Eckermann P, Coombes N et al (2012) Molecular mapping of qualitative and quantitative loci for resistance to Leptosphaeria maculans; causing blackleg disease in canola (Brassica napus L.). Theor Appl Genet 125:405–418
Raman H, Raman R, Larkan N (2013) Genetic dissection of blackleg resistance loci in rapeseed (Brassica napus L). In: Andersen SB (ed) Plant breeding from laboratories to fields. InTech, Luton. https://doi.org/10.5772/53611. ISBN 978-953-51-1090-3
Raman H, Raman R, Coombes N, Song J, Prangnell R, Bandaranayake C et al (2016) Genome-wide association analyses reveal complex genetic architecture underlying natural variation for flowering time in canola. Plant Cell Environ 39:1228–1239
Rimmer SR (2006) Resistance genes to Leptosphaeria maculans in Brassica napus. Can J Plant Pathol 28:S288–S297
Rouxel T, Penaud A, Pinochet X, Brun H, Gout L, Delourme R et al (2003) A 10-year survey of populations of Leptosphaeria maculans in France indicates a rapid adaptation towards the Rlm1 resistance gene of oilseed rape. Eur J Plant Pathol 109:871–881
Schwender H, Ickstadt K (2008) Imputing missing genotypes with k nearest neighbors. Technical report, SFB 475, Department of Statistics, University of Dortmund
Tan B, Grattapaglia D, Martins GS, Zamprogno Ferreira K, Sundberg B, Ingvarsson PK (2017) Evaluating the accuracy of genomic prediction of growth and wood traits in two Eucalyptus species and their F1 hybrids. BMC Plant Biol 17:110
Thomma BP, Cammue BP, Thevissen K (2002) Plant defensins. Planta 216:193–202
West JS, Kharbanda PD, Barbetti MJ, Fitt BDL (2001) Epidemiology and management of Leptosphaeria maculans (phoma stem canker) on oilseed rape in Australia, Canada and Europe. Plant Pathol 50:10–27
Wong JH, Xia L, Ng TB (2007) A review of defensins of diverse origins. Curr Protein Pept Sci 8:446–459
Yu F, Lydiate DJ, Rimmer SR (2005) Identification of two novel genes for blackleg resistance in Brassica napus. Theor Appl Genet 110:969–979
Yu F, Lydiate DJ, Rimmer SR (2008) Identification and mapping of a third blackleg resistance locus in Brassica napus derived from B rapa subsp sylvestris. Genome 51:64–72
Yu F, Gugel RK, Kutcher HR, Peng G, Rimmer SR (2013) Identification and mapping of a novel blackleg resistance locus LepR4 in the progenies from Brassica napus × B rapa subsp sylvestris. Theor Appl Genet 126:307–315
Zhang X, Huang C, Wu D et al (2017) High-throughput phenotyping and QTL mapping reveals the genetic architecture of maize plant growth. Plant Physiol 173:1554–1564
Zheng X, Levine D, Shen J, Gogarten SM, Laurie C, Weir BS (2012) A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28:3326–3328
Acknowledgements
We would like to acknowledge Gilles Lassalle and Anne Laperche for the script that was used to choose the markers for QTL analysis and Mathieu Rousseau-Gueutin, Jérôme Morice for Circos representation of homoeologous relationships between the QTL regions. The authors are grateful to Claude Domin and the INRA Experimental Unit (UE La Motte, Le Rheu) for field experimentations. The authors would like to thank the BrACySol biological resource center (INRA Ploudaniel, France) for providing the seeds used in this study. This work was supported by the French ‘Institut National de la Recherche Agronomique’—Department of ‘Biologie et Amélioration des Plantes’, Terres Inovia and PROMOSOL. VK was funded through the European PLANT-KBBE-IV research program in the French-German Project GEWIDIS (Exploiting genome-wide diversity for disease resistance improvement in oilseed rape; ANR13-KBBE-0004-01).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Heiko C. Becker.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kumar, V., Paillard, S., Fopa-Fomeju, B. et al. Multi-year linkage and association mapping confirm the high number of genomic regions involved in oilseed rape quantitative resistance to blackleg. Theor Appl Genet 131, 1627–1643 (2018). https://doi.org/10.1007/s00122-018-3103-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-018-3103-9