
Abstract
Key message
Aside from the identification of 32 QTL for N metabolism in the seedling leaves of a maize testcross population, alanine aminotransferase was found to be a central enzyme in N assimilation.
Abstract
Excessive application of nitrogen (N) fertilizer to grow commercial crops like maize is a cause of concern because of the runoff of excess N into streams and rivers. Breeding maize with improved N use efficiency (NUE) would reduce environmental pollution as well as input costs for the farmers. An understanding of the genetics underlying N metabolism is key to breeding for NUE. From a set of 176 testcrosses derived from the maize IBMsyn10 population grown in hydroponics, we analyzed the youngest fully expanded leaf at four-leaf stage for enzymes and metabolites related to N metabolism. Three enzymes, along with one metabolite explained 24% of the variation in shoot dry mass. Alanine aminotransferase (AlaAT) stood out as the key enzyme in maintaining the cellular level of glutamate as it alone explained 58% of the variation in this amino acid. Linkage mapping revealed 32 quantitative trait loci (QTL), all trans to the genomic positions of the structural genes for various enzymes of N assimilation. The QTL models for different traits accounted for 7–31% of the genetic variance, whereas epistasis was generally not significant. Five coding regions underlying 1-LOD QTL confidence intervals were identified for further validation studies. Our results provide evidence for the key role of AlaAT in N assimilation likely through homeostatic control of glutamate levels in the leaf cells. The two QTL identified for this enzyme would help to select desirable recombinants for improved N assimilation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nitrogen (N) is the most important mineral nutrients for plant growth and development. N deficiency reduces dry matter accumulation and kernel number, and results in a substantial reduction in grain yield (Uhart and Andrade 1995; DeBruin et al. 2013). Excessive application of N, on the contrary, is detrimental to the environment, as being highly soluble, it can leach into the water table or runoff into the rivers and streams, eventually causing dead zones in the ocean where the rivers drain.
Improving N use efficiency (NUE) of maize would help reduce N losses from the soil. NUE, ratio of grain yield to soil N, can be subdivided into two components: N uptake efficiency (total plant N/soil N) and N utilization efficiency (total grain yield/total plant N) (Moll et al. 1982; Dhugga and Waines 1989). Since N uptake efficiency is derived from multiplying final biomass with N concentration, it should, in fact, be referred to as N acquisition efficiency because of the difficulty in separating the effects of feedback inhibition and a limitation at the cellular level in root uptake. At normal soil N, total plant N is nearly completely explained by biomass, suggesting that the ability of the tissue N to produce dry matter rather than N concentration should be the focus of NUE studies (Dhugga and Waines 1989).
Once absorbed by the roots, nitrate is mostly transported to the leaves for reduction and incorporation into amino acids and other molecules, followed by incorporation into various macromolecules, including enzymes. A limitation at any point in the N metabolism pathway can limit N acquisition and utilization and, consequently, biomass production. We set out to evaluate the enzymes involved in N reduction and incorporation into organic molecules with the goal of identifying the steps that affect N assimilation, and to determine associated quantitative trait loci (QTL). QTL identified henceforth would help in selecting recombinants that combine desirable activities for improved NUE.
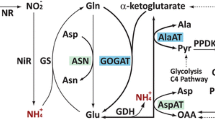
The pathway for N reduction and its incorporation into organic molecules is well documented (Fig. 1) (Yemm and Folkes 1958; Lea 1990a, b; Lea and Azevedo 2007; Lea and Miflin 2010; Plett et al. 2016). Nitrate is reduced to nitrite by nitrate reductase (NR) in the cytoplasm followed by transport of the nitrite into the plastid and reduction by nitrite reductase (NiR) to ammonium, which then becomes the substrate along with glutamate for glutamine synthetase (GS) to form the amino-acid glutamine, the entry point of inorganic N into organic form. Another enzyme, glutamine-2-oxoglutarate aminotransferase (GOGAT), which is also referred to as glutamate synthase, regenerates glutamate from glutamine and α-ketoglutarate. Glutamate further serves as an amino-group donor for other amino acids, a reaction catalyzed by various amino transferases. For instance, alanine aminotransferase (AlaAT) catalyzes the reversible reaction between pyruvate and glutamate on one side and α-ketoglutarate and alanine on the other (Miyashita et al. 2007), while aspartate aminotransferase (AspAT) reversibly forms α-ketoglutarate and aspartate from glutamate and oxaloacetate. Asparagine synthase (ASN) produces asparagine and glutamate from glutamine and aspartate. Following N assimilation, asparagine, glutamate, glutamine, and other amino acids are transported via phloem to the growing organs. Alternatively, they can be stored as vegetative storage proteins, which can aid plant growth during periods of N deficiency (Anderson et al. 1989; Staswick et al. 1991; Tranbarger et al. 1991; Dhugga et al. 2007; Plett et al. 2016).

(adapted from Plett et al. 2016). The AlaAT portion of the pathway is highlighted
Enzymes and proteins involved in N acquisition and assimilation in C4 plants (created with Adobe Illustrator CS2). AlaAT alanine aminotransferase, ASN asparagine synthase, AspAT aspartate aminotransferase, GDH glutmate dehydrogenase, GOGAT glutamate synthase, GS glutamine synthetase, NR nitrate reductase, NiR nitrite reductase, PEPC phosphoenol pyruvate carboxylase, PPDK pyruvate orthophosphate dikinase
Certain metabolites and enzymes perform key roles in C metabolism and are regulated by the status of N in the cell (Sugiharto et al. 1990). Oxaloacetate, one of the carbon skeletons utilized in the synthesis of amino acids, is made from the addition of bicarbonate to phosphoenol pyruvate (PEP) by a reaction catalyzed by phosphoenol pyruvate carboxylase (PEPC). Pyruvate orthophosphate dikinase (PPDK) is responsible for catalyzing the regeneration of PEP. Ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco), considered the most abundant enzyme on Earth, initiates primary carbon fixation by catalyzing the carboxylation of ribulose-1,5-biphosphate (Farquharson 2012). The information on the genetics underlying the regulation of plant C and N metabolism is scarce.
Most of today’s commercial maize germplasm originated from seven progenitor lines, including B73 and Mo17 (Mikel and Dudley 2006). Both these inbred lines are known to differ in their response to soil N (Balko and Russell 1980). They are also the parents of the IBM (intermated B73 × MO17) mapping population (Lee et al. 2002). After ten rounds of random mating, doubled haploid (DH) lines were generated from the IBMSyn10 population (Hussain et al. 2007), which represented a high-resolution mapping population that could be directly associated with the physical map established for the B73 inbred line (http://www.maizesequence.org). This population, especially at the testcross (TC) level, has direct relevance to commercial maize production, because it is the hybrids that are grown in the farmers’ fields.
Several QTL were previously identified for enzymes related to N metabolism (Bertin and Gallais 2000). QTL for yield and its components clustered mainly on chromosome 5. Interestingly, one of the genes encoding cytosolic GS was also identified in the same genomic region. The same research group suggested that NUE could be improved by marker-assisted selection and genetic engineering (Masclaux et al. 2001). Another group identified two nitrate transporters to be associated with the ability of the lines to maintain biomass under reduced N in a hydroponic system (Garnett et al. 2015). Amino-acid profiles of the root and shoot tissues were relatively stable across the lifecycle of Gaspe, a short-cycle maize, but transcript levels fluctuated (Plett et al. 2016).
From a study of the inbred lines of the IBMSyn4 maize population, 73 QTL associated with ten enzymes for C and N metabolism and 8 QTL associated with biomass were identified (Zhang et al. 2010). Most of the QTL for enzyme activities were away from the known genomic locations of the corresponding structural genes, but three cis-QTL were identified for NR, glutamate dehydrogenase and shikimate dehydrogenase. Recently, a QTL analysis was performed for 12 metabolites related to C and N metabolism in the inbred lines of a maize nested association mapping (NAM) population where mapping revealed 101 candidate genes (Zhang et al. 2015). QTL associated with enzymes of N metabolism in hybrid seedlings based on a high-resolution mapping population were not studied. We believe that the identification of QTL in a hybrid background is important in allaying the concern that the ones identified from an inbred population may be masked when constituted into a hybrid and may thus not be commercially relevant.
In this investigation, a mapping population of testcrosses (TC) derived from the crosses of the IBMSyn10-DH lines with an elite inbred line was grown in hydroponics and leaf samples were analyzed to identify QTL associated with enzyme activities and metabolites involved in N assimilation.
Materials and methods
Plant material
A total of 176 TC genotypes derived from the cross between each IBMSyn10-DH line and an elite inbred were used. The IBMSyn10-DH population, developed by Hussain et al. (2007), is a set of DH lines derived from a population after ten generations of random mating from the cross between inbred lines B73 × Mo17. Each DH line was crossed by an elite inbred (PEI), property of DuPont Pioneer (closed pedigree), to generate the TC genotypes.
Experimental design
Kernels from each TC genotype were germinated in autoclaved paper rolls and sterilized water and, subsequently grown under hydroponic conditions. Ten tanks (i.e., sets) containing appropriate growing media were planted with a total of 264 seedlings in each set. In every set, 22 different genotypes were grown, and each genotype was replicated 12 times, randomly distributed with the help of random numbers generated beforehand. Two genotypes (B73 and Mo17 each crossed to the PEI) served as controls, and were included in every set and replication.
The growing media consisted of MgSO4·7H2O 0.5 mM, KH2PO4 0.5 mM, Fe-EDTA 0.1 mM, FeEDDHA 0.1 mM, Ca(NO3)2·4H2O 1.25 mM, KNO3 2.5 mM, Na(OH) 0.1 mM, and 0.4 L of trace elements (25 mM H3BO3, 2 mM MnSO4·H2O, 2 mM ZnSO4·7H2O, 0.5 mM CuSO4·5H2O, 0.5 mM Na2MoO4·2H2O, and 50 mM KCl) in a total of 400 L solution per hydroponic tank. Two weeks after planting, the six most representative plants of each genotype, based on both their root and shoot development, were selected and transplanted into another hydroponic tank with same media.
When plants reached V4 stage (Abendroth et al. 2011), the youngest fully expanded leaf was harvested and stored at −80 °C, while the rest of the plant tissues were dried for 12 days at 48 °C.
Biochemical assays
Activities of eight enzymes related to N metabolism were determined. These enzymes were: NR, NiR, GS, Fd-GOGAT, AlaAT, ASN, AspAT, and PEPC, and specific protocols are described in Plett et al. (2016). GS, Fd-GOGAT, AspAT, and PEPC assay protocols were adapted from Gibon et al. (2004), while NR from Lea et al. (1990a, b), NiR from Bourne and Miflin (1973), ASN assay from Joy and Ireland (1990), and AlaAT protocol were modified from Ashton et al. (1990). In addition, the metabolites nitrate, nitrite, and glutamate were measured as byproducts of enzyme reactions. All measurements were determined by comparing absorbance of each specific biochemical reaction with known standards using a spectrophotometer (Spectramax Plus 384 Microplate Reader, Molecular Devices).
Plant leaf tissues were dried and weighed (SW), and N content was determined by total combustion as previously described in detail (DeBruin et al. 2013).
Statistical analysis
All statistical analyses were implemented in R statistical program (RCoreTeam 2014). The initial data analysis of the raw data was based on the ggplot2 package (Wickham 2010) and GGally (Schloerke et al. 2014). As a first step, a univariate analysis, where a single variable is fitted in a model, followed by a multivariate approach, where multiple variables are analyzed simultaneously, was performed to comprehend the relationship among variables. The determination of outliers present in the data set, based on a jackknife resampling strategy, was applied. As described in Trucillo Silva et al. (2016), a statistical model is fitted n times, systematically omitting one observation from the data set, followed by the prediction of random effects for a subset of most consistent genotypes each of the n times. The aim of the process is to target “real outliers” based on the complete information gathered in the experiment and fine-tune the statistical model, quantified by improvements in log-likelihood, Akaike and Bayesian information criterion values after discarding misleading observations, while keeping informative and true observations for later analysis. The mixed model was fitted with ASReml R package (Butler et al. 2007) and correspondent mixed model equations were solved for prediction of random effects and estimation of fixed effects.
The statistical model can be represented as follows:
where \(y\) denotes a n × 1 vector of observed response values, \(b\) is a p × 1 vector of fixed effects, \(X\) is a n × p design matrix, \(u\) is a q × 1 vector of random effects, \(~Z\) is a n × q design matrix, and \(e\) being the error term.
The following assumptions were used: E (u) = 0, E (e) = 0, Cov (u, e) = 0, and Var (u) = G and, Var (e) = R. The G matrix had a compound symmetry structure on the genotype levels and R matrix is a diagonal matrix with different values for each set, allowing non-constant variance across sets. The response variable was the activity of the enzyme, metabolite concentration, final ELISA determination and N content, respectively. Set, the light replicate and plate were included as fixed effects in the model (where replicate and plate are nested in set), and check genotype effect was included as a continuous covariate. Finally, a random effect for the genotype was included in the linear model. During the process described above, several genotypes were discarded separately for each trait. Extreme cases were traits as AlaAT with eight total genotypes discarded, and nitrate with no genotypes removed from the analysis and sample size totalized 176 genotypes.
Significance of genetic variance was calculated based on log-likelihood ratio test by comparing models with and without the TC random effect. Correlation was calculated, after Bonferroni correction for multiple comparisons, among BLUP values for each pair of traits, and repeatability was derived from the variance estimates from ASReml. As variance components were estimated for each of the different sets, a different value of repeatability was estimated for each set and then partial estimates were averaged correspondingly. Multiple regression analyses were performed using standard procedures.
Path coefficient analysis was performed by the method of Wright (1921) as described in Li (1975).
Genotypic information, and genetic and physical maps
TC genotypes were analyzed with a total of 5306 single-nucleotide polymorphism (SNP) markers generated at Beijing Genomics Institute. Physical and genetic positions of each SNP were determined, and genetic maps were created using R/qtl (Broman et al. 2003). Based on the approach used for the determination of real outliers described previously (Trucillo Silva et al. 2016), different genotypes were omitted from the analysis of each trait. As a result, a different genetic map was determined for each individual trait. Recombination fractions were estimated and Kosambi mapping function was implemented to calculate genetic map distances (Kosambi 1944). Furthermore, as the recombination between linked loci increases every generation, leading to an expansion of the genetic map, mapping distances were adjusted with the purpose of comparison with previous investigations. The expansion factor was determined based on the following equation: \(\propto ~=\frac{j}{2}+(2i - 1)/2i\), where j corresponds to the number of generations of intermating including the two generations for generating the F 2, and i is the number of inbred generations after intermating (Teuscher et al. 2005).
A genetic map of 11,275 cM length was produced. Following QTL mapping, real genetic map distances were reduced by a factor of 6.5 to estimate adjusted F2 map distances (Supplementary Material A) with the goal of comparing the results with the previous QTL studies. Final adjusted map was 1734.65 cM in length (Supplementary Material B). Average spacing between markers was 2.13 cM (0.33 cM adjusted distance), while the maximal spacing between markers was nearly 45 cM (7 cM adjusted distance), located in chromosome 6.
Regarding physical distance, the length of the total genome was 2051.8 Mb, and on average, a marker was positioned every 400 Kb. The widest gaps between markers, 69.8 and 67.4 Mb, were in chromosomes 2 and 9, respectively.
QTL mapping and identification of candidate genes within QTL regions
Associations between phenotypes and genotypes were determined using QTL Cartographer (Basten et al. 2002). Single-marker analysis followed by linear regression analysis and composite interval mapping (CIM) was performed. For CIM, Zmap (model 6) was implemented, using the ten most significant marker cofactors identified by forward and backward regression. In addition, QTL were scanned at intervals of 1 cM and at every marker, while cofactors located within a window of 10 cM of the scanned position were excluded from the analysis. To determine LOD score thresholds of 5%, and to further identify significant QTL, 1000 permutations were performed for every trait. Two nearby QTL were considered as different when LOD peaks were localized 20 cM or greater apart. Furthermore, a multiple interval mapping (MIM) analysis was performed by fitting all previously identified QTL from CIM analysis, and parameters were re-estimated and positions refined. In addition, all pairwise interactions between QTL in every model were studied for each trait. The significance was determined based on the information criterion: IC (k) = −2 (log (L) − kc (n)/2), where the penalty function corresponds to: c (n) = log (n) and a threshold of 0.0 was used (Basten et al. 2002). The proportion of the total phenotypic variance associated with each model was estimated.
In addition, physical genomic regions corresponding to 1-LOD QTL regions were examined, and putative genes related to N metabolism were prioritized based on their annotations at MaizeGDB (Lawrence et al. 2008) and NCBI (http://www.ncbi.nlm.nih.gov), and proposed as targets for further studies.
Results
Statistical analysis for N metabolism related traits
Genetic variance was highly significant for all the traits studied (Table 1). In some TCs, mean values exceeded two standard deviations compared to the parental (Mo17 and B73) performance in their respective TC version. Repeatability, the proportion of total variance in multiple measurements of a trait due to differences among individuals (Dohm 2002), ranged from 0.27 to 0.86. The lowest repeatability was for ASN, while nitrate exhibited the highest value (Table 1).
Relatedness of N metabolism traits
Pairwise correlation coefficients among all the traits are presented in Figs. 2 and 3. From a total of 66 Pearson correlation coefficients, the percentage of pairs that were significant at a p value of <0.001, <0.01, or <0.05 was, respectively, 23, 2, and 9. Significant correlations between enzyme activity and metabolite concentrations were all positive. Negative significant correlations were found between shoot dry weight (SW) and activities of ASN (−0.31), AspAT (−0.33), and GOGAT (−0.27). Strong, positive correlations were observed between AlaAT activity and glutamate (0.79), as well as between AspAT and GS (0.56).

Correlation matrix-heatmap of the N metabolism-related traits in the IBMSyn10-DH TC population of maize (created with corrplot package, R). Significant correlation (p value <0.05) values are colored in blue (positive correlation) and red (negative correlation). (Color figure online)

Graphical depiction of correlation coefficients among enzymes and metabolites related to N metabolism in maize leaves (created with Keynote from Apple)
Three enzymes along with shoot N concentration (N s) explained 24% of SW. Whereas AspAT explained the most variation in shoot biomass, NiR, N s, and ASN sequentially explained additional, but smaller amounts of variation as judged from the partial R 2 values derived from path coefficient analysis.
Variation explained in each trait by the remaining traits is presented in Fig. 4. Glutamate explained the bulk of the variation in AlaAT and vice versa, GOGAT in ASN, GS in AspAT and vice versa, ASN in GOGAT, N s in NiR, AspAT in NR, AlaAT in nitrate, and AspAT in N s. The maximal variation explained for any trait was 62% for AlaAT and the least amount, 7%, was for NR. AspAT had the largest number of variables, 5, explaining variation, whereas GOGAT and NR had only one each, AspAT and AlaAT, respectively.

Prediction of each trait by the rest of the traits (created with Excel graphics). Sequential R-squared (RSQ) values for each of the independent traits are shown in the form of vertical bars. For example, of the 62% total variation explained in AlaAT, 58% was attributed to glutamate concentration and 4% to AspAT, respectively
Identification of quantitative trait loci: composite interval mapping
A total of 32 QTL spread across all the chromosomes were identified for all the traits. The largest number of QTL, seven, was mapped to chromosomes 6 and the least number, two, to chromosomes 8 and 9 each (Table 2; Fig. 5).

a Genetic map and distribution of QTL associated with N metabolism-related traits in the IBMSyn10-DH TC population of maize (created with Mapchart 2.2). b QTL positions shown at left of chromosomes (in cM) and QTL lengths of bars are determined by 2-LOD confidence intervals. Only selected markers are displayed in the figure (right of chromosomes). QTL for enzyme activity are depicted in blue, QTL for SW are in green, and QTL for metabolites are in brown. Figure created with MapChart 2.2 (Voorrips 2002). (Color figure online)
The number of QTL detected per trait ranged from one for NR to five for ASN (Table 2). Individual QTL explained on average 8.8% of the variance with some explaining as much as 16.2% or as low as 6.0% (both QTL for SW). More than 80% of the QTL accounted for less than 10% of the variance. Positive allele effects across traits, 18 for B73 and 14 for Mo17, were similarly derived from each parent.
Confidence intervals (CI 1-LOD) for QTL localization ranged from 3 to 28 cM (0.46–4.26 cM adjusted distance) in length, with an average of 8.81 cM. Those CI are equivalent to 0.2–12 Mb in physical distance, with an average CI length of 2.1 Mb.
A hotspot QTL region was localized on the short arm of chromosome 6, comprising QTL associated with five different phenotypes including enzyme activities (ASN, GOGAT, and PEPC) and metabolites (glutamate and nitrate). Furthermore, QTL for ASN and GOGAT (Chr. 10) were co-localized on the genetic map.
Multiple interval mapping: epistasis
In most traits, epistatic interactions did not significantly improve the fit of the models, and thus, epistatic effects were excluded from genetic models. Epistatic effects were retained for PEPC and nitrate, where they explained 5 and 2.5% variation, respectively. MIM models explained a significant portion of the total variance in ASN and nitrate (over 30%) and, just over 7% for other traits (AlaAT and NR) (Table 3).
Candidate genes
Candidate genes were identified within 1-LOD QTL regions across the maize genome. However, only a subset of the putative genes could be associated to N metabolism. Most promising candidate genes ID are GRMZM2G008714, GRMZM2G045171, GRMZM2G082780, GRMZM2G088235, and GRMZM2G180625 (Table 4).
All the QTL for enzyme activities and metabolites identified in this study are in trans configuration from the known genomic locations of their corresponding structural genes. For example, QTL for GS were identified on chromosomes 1, 5, 6, and 8 at physical positions 80.15, 83.95, 150.20, and 2.55 Mb, respectively. The structural genes for GS are located on chromosomes 1 (between 27.07 and 27.40 Mb), 2, 4, 5 (between 203.21 and 203.62 Mb), 9, and 10, based on the following nearest loci on the IBM2 2008 Neighbors map, respectively. Thus, the putative N metabolism-related genes identified under the QTL regions might be involved in regulating the activity of the respective enzymes through alteration of the metabolite pools via regulatory enzymes.
Discussion
Four variables, AspAT, NiR, and ASN along with N s, explained approximately a quarter of the variation in shoot biomass (Fig. 6). Only one of these variables, NiR, was positively correlated and the remaining three were each negatively correlated with the biomass (Fig. 2). Nevertheless, these variables could be focused on for further studies for their ability to make shoot biomass.

Variables predictive of shoot biomass in corn seedlings grown in hydroponics (created with Excel graphics). AspAT explained the most variation (11%) in the shoot biomass with NiR, N s, and ASN sequentially explaining incremental variation for a maximal of 24%
Interrelationship among the enzymes and metabolites is represented in Fig. 1. The strongest correlation was between glutamate and AlaAT. Glutamate is a key amino acid in N metabolism as it constitutes the substrate for the incorporation of inorganic N into glutamine and then cycles N from glutamine into other amino acids. AlaAT has previously been reported to improve NUE in plants by several laboratories (Son et al. 1992; Good et al. 2007; Miyashita et al. 2007; Shrawat et al. 2008; Rocha et al. 2010). Although originally identified as conferring upon plants the ability to overcome hypoxia through conversion of pyruvate to alanine pool and thus keeping the energy-producing part of glycolysis functioning, its transgenic overexpression has been shown to improve NUE in several crop plants (Good et al. 2007). We have highlighted the relative importance of various enzymes and glutamate based on their interrelationships in Fig. 1. Its high correlation with glutamate points to the role AlaAT plays in maintaining metabolic homeostasis in primary N assimilation: removal of glutamate from the pool for continued N uptake and assimilation and the conversion of alanine into glutamate for incorporation of N into other amino acids when N availability drops below the threshold of limitation (Fig. 2). Likewise, AspAT could contribute to C4 metabolism by maintaining the concentrations of oxaloacetate and alpha-ketoglutarate in the cells (Fig. 1).
In this investigation, 32 QTL associated with N metabolism were identified in a high-resolution maize TC mapping population. In addition, QTL models explaining even greater than 30% of the genetic variance were identified for certain phenotypes, such as ASN and nitrate.
Consistent with the previous studies, all significant correlations among enzyme activities were positive, suggesting that the enzymes were co-regulated to varying extents (Zhang et al. 2010). Indeed, significant correlations between enzyme activities and metabolites were positive as well (Figs. 2, 3). With an increase in nitrate concentration, an increase in the activities of NR, NiR GOGAT, and GS would be expected. A positive correlation between nitrate and glutamate further supports this viewpoint (Fig. 2). In addition, a negative correlation was found between enzyme activities and biomass (Zhang et al. 2010). Except for NiR, biomass was negatively correlated with most of the enzymes in our study (Fig. 2), which was most likely because of dilution effect, that is, the fast-growing TCs had diluter cell contents than the slow-growing ones as would be expected.
QTL were associated with the activities of eight enzymes, two metabolites, and N s phenotypes. In agreement with a previous investigation (Zhang et al. 2010), the same QTL were detected for NR, GS, and AspAT localized on chromosomes 4, 5, and 9, respectively. Nevertheless, most of the QTL reported in other maize studies (Agrama et al. 1999; Hirel et al. 2001) did not co-localize with the QTL identified in this analysis, whereas a single QTL associated with NR was found on chromosome 4 in this investigation and by Zhang et al. (2010).
A few QTL co-localized with QTL for different agronomic traits in the previous studies. For instance, GOGAT-1 QTL were exactly at the same location as a QTL previously associated with the determination of ear·plant−1 under high soil N, and a QTL associated with number of kernels per ear (Agrama et al. 1999). Hence, some genomic regions seem to be affecting more than one trait, indicative of the presence of pleiotropy.
Three genetic regions (1-LOD confidence interval around the LOD peak) on chromosomes 1, 6, and 7 containing QTL for several enzymes were identified by Zhang et al. (2010); however, no QTL signal was detected under those exact regions in our QTL analysis. The previous investigation was based on the inbred lines of the IBM-Syn4 population, which had fewer recombinants than our population, and genotyped with 2200 DNA marker loci. The co-location of some QTL could be attributed to a lack of genetic resolution (Zhang et al. 2010). The TCs used in our work would be less prone to this bias because of the additional rounds of intermating as well as the use of a higher marker density.
A lower number of QTL identified per trait our study as compared to a preceding investigation could be attributed to an increased recombination because of additional cycles of recombination (Zhang et al. 2010).
Prior investigations found little evidence of common QTL detection between inbred per se and TC progeny, suggesting that marker-assisted selection strategies based on QTL identified at the inbred level, which was the case with the previous study, would not assure the selection of hybrids with superior performance (Beavis et al. 1994; Schon et al. 1994). Genetic studies on a TC mapping populations are thus preferable if the objective is to select superior hybrids based on these QTL.
Several annotated protein-coding genes were identified under QTL intervals determined in this study. One of the maize genes, a cell wall invertase 3 (GRMZM2G123633) located on chromosome 10 under the GOGAT-4 QTL in our study, was also identified in a previous meta-QTL investigation aiming to discover candidate genes for NUE in maize (Liu et al. 2012). In addition, five candidate genes revealed important putative functions related to N metabolism (Table 4). Three of them were also proposed as candidate genes in a recent study from the maize NAM population (Zhang et al. 2015). Those genes were GRMZM2G008714, GRMZM2G045171, and GRMZM2G180625, coding, respectively, for pyruvate kinase, sucrose synthase, and glyceraldehyde-3-phosphate dehydrogenase. Of these three enzymes, sucrose synthase assumes significance, because it catalyzes the reversible reaction between sucrose and uridine diphosphate glucose (UDPG) to mobilize sucrose into multiple pathways that utilize activated sugars, for example, cell wall formation, starch formation, sucrose synthesis, and glycosylation of proteins and other molecules (Subbaiah et al. 2007). Furthermore, sucrose synthase uses the glycosidic bond energy of the sucrose molecule to make UDPG using UDP as the other substrate, sparing a mole of ATP for each mole of sucrose catalyzed by sucrose synthase (Barreiro and Dhugga 2007). The alternative pathway requires phosphorylation of UDP to UTP, which is then used by UDPG-pyrophosphorylase along with glucose-1-phosphate to make UDPG. The additional energy from the sucrose synthase-mediated pathway could favor the formation of additional biomass.
Two other genes were associated with the QTL. One of these, GRMZM2G082780, had been annotated as a PEPC putative gene, involved in primary carbon fixation in C4 plants. In addition, GRMZM2G088235 is a urease accessory protein, which is associated with the urease protein to activate it for urea metabolism into carbon dioxide and ammonium, where the latter is then utilized by the GS/GOGAT cycle to incorporate into glutamine/glutamate amino acids (Witte et al. 2005).
The five coding regions underlying QTL identified in this study constitute promising candidates for validation studies. Furthermore, the two QTL for AlaAT constitute candidates for selection to improve NUE in maize.
Conclusions
One of the enzymes in N assimilation, AlaAT, stands out among others in maintaining metabolic homeostasis of glutamate, apparently for optimal N assimilation. QTL associated with the physiological traits related to N metabolism provide tools to improve NUE in maize. Growing these TCs in the field and assaying the enzymes at flowering time when the canopy is fully developed would make it possible to determine whether and how much of the variation in grain yield they explain. The field-grown TCs will also help determine whether the QTL identified from the seedlings in the growth chamber relate to the ultimate trait: grain yield.
Author contribution statement
KSD, ML, and ITS designed the experiments, analyzed data, and wrote the manuscript. ITS, HKRA, and LPF performed laboratory analyses, HL provided molecular marker information. All authors revised and approved the final manuscript.
References
Abendroth LJ, Elmore RW, Boyer MJ, Marlay SK (2011) Corn growth and development. In: Extension Publication #PMR-1009, Iowa State University
Agrama HAS, Zakaria AG, Said FB, Tuinstra M (1999) Identification of quantitative trait loci for nitrogen use efficiency in maize. Mol Breed 5:187–195
Anderson JM, Spilatro SR, Klauer SF, Franceschi VR (1989) Jasmonic acid-dependent increase in the level of vegetative storage proteins in soybean. Plant Sci 62:45–52
Ashton AR, Burnell JN, Furbank RT, Jenkins CLD, Hatch MD (1990) Enzymes of C4 photosynthesis. In: Lea PJ (ed) Methods in plant biochemistry. Academic Press, London, pp 39–72
Balko LG, Russell WA (1980) Response of maize inbred lines to N-fertilizer. Agron J 72:723–728
Barreiro R, Dhugga KS (2007) From Cellulose to Mechanical Strength: Relationship of the Cellulose Synthase Genes to Dry Matter Accumulation in Maize. In: Brown RM, Saxena IM (eds) Cellulose: Molecular and Structural Biology: Selected Articles on the Synthesis, Structure, and Applications of Cellulose. Springer Netherlands, Dordrecht, pp 63–83
Basten CJ, Weir BS, Zeng ZB (2002) QTL Cartographer, Version 1.16. Department of Statistics, North Carolina State University, Raleigh, NC
Beavis WD, Smith OS, Grant D, Fincher R (1994) Identification of quantitative trait loci using a small sample of topcrossed and F4 progeny from maize. Crop Sci 34:882–896
Bertin P, Gallais A (2000) Genetic variation for nitrogen use efficiency in a set of recombinant maize inbred lines I. Agrophysiological results. Maydica 45(1):53–66
Bourne WF, Miflin BJ (1973) Studies on nitrite reductase in barley. Planta 111:47–56
Broman KW, Wu H, Sen Ś, Churchill GA (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19:2990–2992
Butler D, Cullis BR, Gilmour AR, Gogel BJ (2007) Analysis of mixed models for s language environments. ASReml-R reference manual, Technical support, Queensland Department of Primary Industries
DeBruin J, Messina CD, Munaro E, Thompson K, Conlon-Beckner C, Fallis L, Sevenich DM, Gupta R, Dhugga KS (2013) N distribution in maize plant as a marker for grain yield and limits on its remobilization after flowering. Plant Breeding 132:500–505
Dhugga KS, Waines JG (1989) Analysis of nitrogen accumulation and use in bread and durum-wheat. Crop Sci 29:1232–1239
Dhugga KS, Appenzeller LM, Gupta R, Abbaraju R (2007) Methods and compostions for increasing the nitrogen storage capacity of a plant. World Patent WO2007075557A3
Dohm MR (2002) Repeatability estimates do not always set an upper limit to heritability. Fuctional. Ecology 16:273–280
Farquharson KL (2012) Insight into Ribulose 1,5-Bis-Phosphate Carboxylase/Oxygenase Assembly in Maize. Plant Cell 24:3171–3171
Garnett T, Plett D, Conn V, Conn S, Rabie H, Rafalski JA, Dhugga K, Tester MA, Kaiser BN (2015) Variation for N uptake system in maize: genotypic response to N supply. Front Plant Sci 6:936. doi:10.3389/fpls.2015.00936
Gibon Y, Blaesing OE, Hannemann J, Carillo P, Hohne M, Hendriks JHM, Palacios N, Cross J, Selbig J, Stitt M (2004) A robot-based platform to measure multiple enzyme activities in Arabidopsis using a set of cycling assays: Comparison of changes of enzyme activities and transcript levels during diurnal cycles and in prolonged darkness. Plant Cell 16:3304–3325
Good AG, Johnson SJ, De Pauw M, Carroll RT, Savidov N, Vidmar J, Lu Z, Taylor G, Stroeher V (2007) Engineering nitrogen use efficiency with alanine aminotransferase. Botany 85:252–262
Hirel B, Bertin P, Quillere I, Bourdoncle W, Attagnant C, Dellay C, Gouy A, Cadiou S, Retailliau C, Falque M, Gallais A (2001) Towards a better understanding of the genetic and physiological basis for nitrogen use efficiency in maize. Plant Physiol 125:1258–1270
Hussain T, Tausend P, Graham G, Ho J (2007) Registration of IBM2 SYN10 doubled haploid mapping population of maize. J Plant Regist 1:81–81
Joy KW, Ireland RJ (1990) Methods in plant biochemistry. Academic Press, London, pp 228–295
Kosambi DD (1944) The estimation of map distances from recombination values. Ann Eugenics 12:172–175
Lawrence CJ, Harper LC, Schaeffer ML, Sen TZ, Seigfried TE, Campbell DA (2008) MaizeGDB: the maize model organism database for basic, translational, and applied research. Int J Plant Genomics 2008:496957. doi:10.1155/2008/496957
Lea PJ (1990a) Enzymes of ammonia assimilation. Methods Plant Biochem 3:257–276
Lea PJ (1990b) Methods in plant biochemistry. In, vol 3. Academic Press Limited, San Diego
Lea PJ, Azevedo RA (2007) Nitrogen use efficiency. 2. Amino acid metabolism. Ann Appl Biol 151:269–275
Lea PJ, Miflin BJ (2010) Nitrogen assimilation and its relevance to crop improvement. Annual Plant Reviews 42:1–40
Lee M, Sharopova N, Beavis WD, Grant D, Katt M, Blair D, Hallauer A (2002) Expanding the genetic map of maize with the intermated B73 × Mo17 (IBM) population. Plant Mol Biol 48:453–461
Li CC (1975) Path analysis-a primer. The Boxwood Press
Liu R, Zhang H, Zhao P, Zhang Z, Liang W, Tian Z, Zheng Y (2012) Mining of candidate maize genes for nitrogen use efficiency by integrating gene expression and QTL data. Plant Mol Biol Report 30:297–308
Masclaux C, Quillere I, Gallais A, Hirel B (2001) The challenge of remobilisation in plant nitrogen economy. A survey of physio-agronomic and molecular approaches. Ann Appl Biol 138:69–81
Mikel MA, Dudley JW (2006) Evolution of north American dent corn from public to proprietary germplasm. Crop Sci 46:1193–1205
Miyashita Y, Dolferus R, Ismond KP, Good AG (2007) Alanine aminotransferase catalyses the breakdown of alanine after hypoxia in Arabidopsis thaliana. Plant J 49:1108–1121
Moll RH, Kamprath EJ, Jackson WA (1982) Analysis and interpretation of factors which contribute to efficiency of nitrogen-utilization. Agron J 74:562–564
Plett D, Holthan L, Baumann U, Kalashyan E, Francis K, Enju A, Toubia J, Roessner U, Bacic A, Rafalski A, Dhugga KS, Tester M, Garnett T, Kaiser BN (2016) Nitrogen assimilation system in maize is regulated by developmental and tissue-specific mechanisms. Plant Mol Biol 92:293–312
RCoreTeam (2014) R: A language and environment for statistical computing. In. R Foundation for Statistical Computing, Vienna, Austria
Rocha M, Licausi F, Araújo WL, Nunes-Nesi A, Sodek L, Fernie AR, van Dongen JT (2010) Glycolysis and the tricarboxylic acid cycle are linked by alanine aminotransferase during hypoxia induced by waterlogging of Lotus japonicus. Plant Physiol 152:1501–1513
Schloerke B, Crowley J, Cook D, Hofmann H, Wickham H, Briatte F, Marbach M, Thoen E (2014) GGally: extension to ggplot2. In. R package version 0.4.8, Vienna, Austria
Schon CC, Melchinger AE, Boppenmaier J, Brunklausjung E, Herrmann RG, Seitzer JF (1994) RFLP Mapping in maize—quantitative trait loci affecting testcross performance of elite European flint lines. Crop Sci 34:378–389
Shrawat AK, Carroll RT, DePauw M, Taylor GJ, Good AG (2008) Genetic engineering of improved nitrogen use efficiency in rice by the tissue-specific expression of alanine aminotransferase. Plant Biotechnol J 6:722–732
Son D, Kobe A, Sugiyama T (1992) Nitrogen-dependent regulation of the gene for alanine aminotransferase which is involved in the C4 pathway of Panicum miliaceum. Plant Cell Physiol 33:507–509
Staswick PE, Huang JF, Rhee Y (1991) Nitrogen and Methyl Jasmonate Induction of Soybean Vegetative Storage Protein Genes. Plant Physiol 96:130–136
Subbaiah CC, Huber SC, Sachs MM, Rhoads D (2007) Sucrose synthase: expanding protein function. Plant Signal Behav 2:28–29
Sugiharto B, Miyata K, Nakamoto H, Sasakawa H, Sugiyama T (1990) Regulation of expression of carbon-assimilating enzymes by nitrogen in maize leaf. Plant Physiol 92:963–969
Teuscher F, Guiard V, Rudolph PE, Brockmann GA (2005) The map expansion obtained with recombinant inbred strains and intermated recombinant inbred populations for finite generation designs. Genetics 170:875–879
Tranbarger TJ, Franceschi VR, Hildebrand DF, Grimes HD (1991) The Soybean 94-Kilodalton Vegetative Storage Protein Is a Lipoxygenase That Is Localized in Paraveinal Mesophyll Cell Vacuoles. Plant Cell 3:973–988
Trucillo Silva I, Cook D, Lee M (2016) Outlier detection in a complex biological dataset. Int J of Agron Crop Sci 1:1–13
Uhart SA, Andrade FH (1995) Nitrogen deficiency in maize: I. Effects on crop growth, development, dry matter partitioning, and kernel set. Crop Sci 35:1376–1383
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J heredity 93(1):77–78
Wickham H (2010) A layered grammar of graphics. J Comput Graph Stat 19:3–28
Witte CP, Rosso MG, Romeis T (2005) Identification of three urease accessory proteins that are required for urease activation in Arabidopsis. Plant Physiol 139:1155–1162
Wright S (1921) Correlation and causation. J Agric Res 20:557–585
Yemm W, Folkes BF (1958) The metabolism of aminoacids and proteins in plants. Annu Rev Plant Physiol 9:245–280
Zhang N, Gibon Y, Gur A, Chen C, Lepak N, Hoehne M, Zhang Z, Kroon D, Tschoep H, Stitt M, Buckler E (2010) Fine quantitative trait loci mapping of carbon and nitrogen metabolism enzyme activities and seedling biomass in the maize IBM mapping population. Plant Physiol 154:1753–1765
Zhang N, Gibon Y, Wallace JG, Kruger Lepak N, Li P, Dedow L (2015) Genome-wide association of carbon and nitrogen metabolism in the maize nested association mapping population. Plant Physiol 168:575–583
Acknowledgments
The authors would like to thank R.F. Baker for Plant Breeding—Department of Agronomy—Iowa State University and DuPont Pioneer for making this research possible and Iowa State University undergraduate students (especially Guan Yi Lai) who assisted with planting and harvesting in the hydroponic experiments. Requests for testcross materials and IBMSyn10-DH lines may be directed to DuPont Pioneer and Dr. Michael Lee, respectively.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Natalia de Leon.
Electronic supplementary material
Below is the link to the electronic supplementary material.
122_2017_2900_MOESM1_ESM.tif
Adjustment of real genetic map and final adjusted F2 map (created with R\qtl package, R). (A) For each chromosome, the line located on the left represents the real map estimated from the actual data, while the line on the right corresponds to the adjusted genetic distance (cM). (B) Final adjusted genetic map. (TIF 766 KB)
Rights and permissions
About this article

Cite this article
Trucillo Silva, I., Abbaraju, H.K.R., Fallis, L.P. et al. Biochemical and genetic analyses of N metabolism in maize testcross seedlings: 1. Leaves. Theor Appl Genet 130, 1453–1466 (2017). https://doi.org/10.1007/s00122-017-2900-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-017-2900-x