
Abstract
The red blood cells (RBCs) are essential to transport oxygen (O2) and nutrients throughout the human body. Changes in the structure or functioning of the erythrocytes can lead to several deficiencies, such as hemolytic anemias, in which an increase in reactive oxidative species generation is involved in the pathophysiological process, playing a significant role in the severity of several clinical manifestations. There are important lines of defense against the damage caused by oxidizing molecules. Among the antioxidant molecules, the enzyme peroxiredoxin (Prx) has the higher decomposition power of hydrogen peroxide, especially in RBCs, standing out because of its abundance. This review aimed to present the recent findings that broke some paradigms regarding the three isoforms of Prxs found in RBC (Prx1, Prx2, and Prx6), showing that in addition to their antioxidant activity, these enzymes may have supplementary roles in transducing peroxide signals, as molecular chaperones, protecting from membrane damage, and maintenance of iron homeostasis, thus contributing to the overall survival of human RBCs, roles that seen to be disrupted in hemolytic anemia conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The red blood cell
Approximately two million red blood cells (RBCs) are produced in healthy adults every second through a coordinated process that starts with multipotent hematopoietic stem and progenitor cells (HSPCs) [1, 2] stimulated by the decrease of oxygen in the circulation detected by the kidney. The kidney then secretes the hormone erythropoietin (EPO), which enables the proliferation and differentiation of erythrocyte precursors in hematopoietic tissues, giving rise to a process known as erythropoiesis (Fig. 1) [1]. The size of the cell and the nucleus gradually decreases in the precursors, and the mature RBC are anucleate and devoid of ribosomes and mitochondria. Despite these drawbacks, RBCs can effectively transport oxygen to peripheral tissues for 100 to 120 days while still in circulation [2, 3].

Scheme of the erythropoiesis. Erythropoietin (EPO) stimulates stem cells to differentiate into proerythroblasts, which undergo various modifications that result in the formation of basophilic erythroblasts, polychromatophilic erythroblasts, and orthochromatic/acidophilic erythroblasts, successively. Subsequently, the nucleus is eliminated from the cell, originating the reticulocytes, which are released into the circulation and complete their maturation process, thus becoming erythrocytes cells (The authors created this figure adapting images from Servier Medical Art Commons Attribution 3.0 Unported License (http://smart.servier.com))
The precise structural organization of the RBC membrane is crucial for allowing the human RBCs to experience significant, quick, and reversible changes as they move through the microvasculature and travel through the human spleen. Ankyrin and protein 4.1-based transmembrane protein complexes embedded in the lipid bilayer serve as anchors for the membrane, which comprises a lipid bilayer attached to a two-dimensional elastic network of skeletal proteins. The membrane skeleton and the lipid bilayer are held together by vertical anchoring links [4, 5].
Moreover, to exert its function, RBCs significantly increase the accumulation of hemoglobin (Hb), the oxygen-carrying pigment of erythrocytes, during erythropoiesis [2]. Through Hb switches, mainly mediated by changes in the expression of the β-globin genes, the sequential expression of embryonic, fetal, and adult globin genes occurs during development [2]. The following globulins are connected to the Hb molecule, both throughout the embryonic stage and after birth: α-, β-, γ-, δ-, ε-, and ζ-polypeptide chains. Hb F (α2γ2) is the standard form of Hb in fetal life. After 6 months, just a trace amount of Hb F (less than 1%) and Hb A2 (α2δ2; less than 3.5%) are present in the circulation, and Hb A (α2β2) is the main one, associated with an iron-containing porphyrin ring called heme attached to each subunit to promote efficient transportation of oxygen [6].
In terms of metabolism, due to the loss of mitochondria during reticulocyte maturation and enucleation, glucose is the primary metabolic substrate for RBCs. The main byproducts of glycolysis are 2,3-diphosphoglycerate (2,3-DPG; a crucial intermediate that regulates hemoglobin-oxygen affinity), NADH (an essential cofactor used by methemoglobin (metHb) reductase for the reduction of metHb), and ATP (the source of energy for numerous RBC membrane and metabolic reactions) [3, 7]. Meanwhile, the production of NADPH by the pentose phosphate route (PPP), often referred to as the hexose monophosphate (HMP) shunt, is essential to provide reduction equivalents in RBC, maintaining reduced glutathione (GSH) levels, which helps to limit oxidative damage and assure the survival of healthy RBCs (Fig. 2) [8, 9].

Schematic erythrocyte metabolism. (1) The Embden-Meyerhof pathway, (2) pentose phosphate pathway, (3) redox metabolism, (4) methemoglobin pathway, (5) Luebering-Rapoport pathway, and (6) nucleotide metabolism (elaborated by the authors)
Understanding the different types of hemolytic anemias
The molecular structure, metabolic pathways, and a proper Hb are fundamental to maintaining RBC function and viability. Any defects or failures in these erythrocyte components may result in function/structural issues or/and protection against oxidative stress commitment, leading to several disorders. Hereditary hemolytic diseases are caused by defects in one or more components of erythrocytes, which encompasses a heterogeneous group of anemias [8].
Changes in DNA that result in Hb disorders (such as alterations within globin chains or Hb defects), erythrocyte membrane disorders (such as structural and permeability defects), RBCs’ enzymopathies (that impact the intraerythrocytic metabolism), and also other rare genetic disorders causing ineffective erythropoiesis (causing impaired maturation of erythroid precursors) that lead to hereditary hemolytic anemia are examples of intrinsic defects that results in decreased RBC survival and increased oxidative stress, among other manifestations [1, 8]. Early RBC degradation can occur intravascularly or extravascularly through the reticuloendothelial system’s macrophages, mainly in the liver or spleen [1, 8].
Membranopathies
Other cases of erythrocyte disorder that may lead to hemolytic anemia are the ones involving the membrane, which are heritable conditions caused by mutations in the genes encoding for membrane or cytoskeletal proteins (for example, hereditary spherocytosis [HS]), as well as for transmembrane transporters or channels, which affect volume and ion homeostasis, resulting in altered cell permeability and deformability (for instance, Southeast Asian ovalocytosis [SAO]). The erythrocytes are prematurely removed from the bloodstream and have a shorter half-life due to these changes (reviewed in [5, 8]). A reduction in membrane surface area is seen when the problem is a lack of adherence between the membrane skeleton and the lipid bilayer. Problems in membrane structural stability and further cell rupture are observed when damaged horizontal linkages appear in the junctional complex’s horizontal linkages, which connect spectrin-spectrin dimers and spectrin-actin-protein 4.1R, which offers control of membrane flexibility and mechanical coherence [4].
Hereditary spherocytosis (HS)
With an incidence of 1:2000–5000 in the Caucasian population, HS is the most prevalent inherited membrane condition and causes an architecturally abnormal red cell membrane. HS is caused by a genetic inheritance of dominant, recessive, or de novo mutations in the genes encoding the proteins which compose up the red cell membrane, ankyrin, band 3, α-spectrin, and β-spectrin, as well as protein 4.2. A primary shortfall in one of these components may result in secondary deficiencies in other proteins that interact with that protein [4, 5, 10]. Clinically, mild to severe hemolytic anemia occurs due to an ongoing decrease of membrane surface area caused by reduced cohesion of the lipid bilayer to the spectrin-based membrane skeleton, which leads to the formation of an increasing number of spherical cells that have diminished cellular elasticity that is more prone to removal in the spleen [4, 5].
Southeast Asian ovalocytosis (SAO)
Papua New Guinea, Indonesia, Malaysia, the Philippines, and southern Thailand populations are particularly predisposed to SAO. The 27 nucleotides deletion in the SLC4A1 gene (encoding erythrocyte anion exchanger 1 [AE1, band 3]), which results in the loss of protein band 3’s amino acids 400–408, is the cause of SAO, an autosomal dominant disorder. Patients show distinctive rounded elliptocytes (ovalocytes) and present lower in vitro deformability [5, 11]. The red cell membrane contains band 3 as dimers and tetramers; however, SAO band 3 is thought to generate greater oligomers and connect to the basal erythrocyte cytoskeleton more easily than the wild-type form, explaining the reported rise in membrane rigidity in cells with this mutation [11]. Although they are mostly asymptomatic, hemolytic anemia could appear, especially in babies. Additionally, when exposed to low temperatures, SAO erythrocytes exhibit a slight loss of monovalent cations along with a decrease in anions flux, leading to a more rapid depletion of ATP; therefore, being categorized as an RBC membrane’s permeability hereditary disease [5, 11].
Enzymopathies
To date, at least 16 genetic diseases impact intraerythrocytic metabolism and are classified as RBC enzymopathies. They are all rare except for glucose-6-phosphate dehydrogenase (G6PD) deficiency, which is extremely frequent. Nearly all these enzymopathies exhibit chronic hemolytic anemias, with a beginning usually in the early stages of life, due to the lack of a nucleus alongside other organelles in the RBC, which limits these cells’ ability to endure metabolic impairment [3, 7].
Glucose-6-phosphate dehydrogenase (G6PD) deficiency
According to estimates, more than 500 million individuals globally are thought to be affected with G6PD deficiency, the most prevalent RBC metabolic disease [3, 7]. It is noteworthy to mention that anemia is rare in the stable state of an individual with G6PD deficiency. As a result of oxidizing compounds like divicine, a fava bean product (favism phenomenon), after taking specific medications, such as primaquine, an anti-malarial treatment, or during infection-related oxidative stress, it might nevertheless result in an acute hemolytic crisis [3, 9]. Additionally, a significant contributing element to the disease is the subtype, which varies in severity and, in some cases, can result in a sharp fall in activity, with subtypes that present a half-life of hours (G6PD-Mediterranean). Acute hemolytic events in G6PD-deficient individuals result in both extravascular and intravascular hemolysis since oxidative injury creates inflexible, nondeformable erythrocytes, which are prone to degradation [3, 7].
A vital component of defending RBC against oxidative damage is the HMP/PPP shunt, which metabolizes 5 to 10% of the glucose to produce NADPH [3]. Given that catalase (CAT) is shielded from inactivation by bound NADPH in a state of oxidative stress, G6PD deficiency not only coincides with a decline in NADPH, which lowers red cell GSH, but also results in a concurrent loss of CAT activity [3, 9]. Moreover, thioredoxin levels are maintained by thioredoxin reductases (TrxR) which uses NADPH as the electron donor [9]. This may help further clarify why G6PD-deficient subjects have higher lipid peroxidation levels than carriers and controls [12]. As a result, the activity of all significant scavenging mechanisms is decreased, and hemolysis only ceases when young RBC enters the circulation, and exhibits increased G6PD activity and NADPH concentration [3, 9].
Pyruvate kinase (PK) deficiency
PK deficiency is the most prevalent and well-studied of the enzyme impairments impacting glycolysis. This deficiency occurs due to mutations in the PKLR gene; more than 350 have been described. The incidence is estimated to be 3:1,000,000 to 1:20,000 [13]. One of the two glycolytic reactions that lead to ATP synthesis is the conversion of phosphoenolpyruvate to pyruvate, which PK catalyzes. Decreased utilization of glucose and a reduced ability to produce ATP are the outcomes of this deficiency [3, 13]. As an alternative, ATP is produced by mitochondrial oxidative phosphorylation in PK-deficient reticulocytes, which presents the benefit of generating ATP independently of glycolysis. As the cell finalizes its differentiation, mitochondria, alongside other organelles, are removed from the RBC, and consequently, oxidative phosphorylation stops and ATP levels decrease [3].
Moreover, the Rapoport-Luebering pathway’s 2,3-DPG, a crucial intermediate that is known to improve oxygen unloading from Hb, presents a rise in the concentration up to three times [3, 13] in RBC due to the metabolic block brought on by PK deficit, which leads to inhibition of 6-phosphogluconate dehydrogenase (6PGD). Because of the reduced NADPH production caused by the low HMP/PPP activity and the reduction in ATP that promotes membrane abnormalities, PK-deficient RBC may be more susceptible to oxidative damage and potential hemolysis [7, 9, 13].
Hemoglobinopathies
According to estimates, 7% of the world’s population has a DNA mutation that might cause hemoglobinopathies (300,000 to 400,000 affected infants), making this group of diseases-genetic disorders related to the manufacture of Hb-the most prevalent monogenic disorders globally. DNA variations in or close to the globin genes, which code for the globin chains of the tetrameric Hb protein, are the hereditary cause of this illness. Most of the over 1000 hemoglobinopathies that have been reported are asymptomatic. Major clinical abnormalities, however, were seen in some of these diseases [6, 14]. These DNA mutations could modify the synthesis process of the β-globin, for example, or they could disrupt the basic structure of Hb, resulting in diseases like beta (β)-thalassemia and sickle cell disease (SCD), respectively [14].
Beta-thalassemia
To various extents, over 300 mutations that prevent the synthesis of β-globin chains are known to cause β-thalassemia. More than 95% of such mutations are nucleotide variations, while only about 5% are deletions of the β-globin gene [14, 15]. Ineffective erythropoiesis results from damage and destruction of erythroid precursors caused by free α-globin molecules’ formation of insoluble aggregates. These free α-globins engage with several protein quality control mechanisms, such as ubiquitin-mediated proteolysis and autophagy, which remove some excess chains [1, 15].
Aside from that, it has been proposed that too many unstable α-globin peptide chains bind to and sequester the molecular chaperone HSP70 in the cytoplasm, destabilizing GATA1 (an essential regulator of erythroid cell gene expression and maturation) and affecting gene expression, subsequently resulting in inefficient erythropoiesis. Approximately 65% of erythroblasts in individuals with β-thalassemia die before developing into mature RBCs, according to erythrokinetic and ferrokinetic studies [1, 15]. Chronic anemia is triggered by a shortage of circulating RBCs brought on by ineffective erythropoiesis. This ineffective erythropoiesis stimulates the synthesis of EPO, promoting more ineffective erythropoiesis. This never-ending cycle significantly increases erythroblast proliferation and increased erythroblast cell death [1, 15]. Therefore, extramedullary hematopoiesis may threaten organ functioning, resulting in additional problems such as hepatosplenomegaly and metabolic abnormalities. In contrast, extensive erythropoiesis in the bone marrow may trigger bone alterations that result in osteoporosis. Additionally, mature circulating RBCs hemolyze from free α-globin [1, 14, 15].
In addition, the free heme released from the excess denatured α-globin chains could catalyze the Fenton reaction, increasing ROS in the thalassemic erythroblast. Cell death and loss of function can result from DNA, protein, and lipids modification or even degradation due to increased ROS levels in these cells [15, 16]. Moreover, non-transferrin-bound iron (NTBI) and labile plasma iron (LPI) are products of the disease’s iron overload, brought on by the ongoing hemolytic process. This labile iron pool plays a crucial part in the onset of cellular impairment, apoptosis, and ferroptosis, which catalyzes Fenton and Haber-Weiss reactions and further produces ROS [16]. Thus, β-thalassemia results in decreased production and rapid degradation of RBCs, necessitating both chelation therapy and routine blood transfusions.
Sickle cell disease (SCD)
All manifestations of the erratic hemoglobin S (Hb S), including sickle cell trait (Hb AS), homozygous sickle cell disease (Hb SS—named sickle cell anemia, SCA), and a variety of heterozygous hemoglobinopathies such as Hb SC, SD, and HbS-β-thalassemia, are together referred to as SCD. The Hb S allele is created by just one nucleotide modification in the HBB gene. The mutant protein has an amino acid substitution that, in certain circumstances, such as deoxygenation, causes it to polymerize and give rise to the sickle-shaped erythrocytes, which provide the disease’s name [1, 17].
Individuals with sickle cell trait are often asymptomatic and do not need medical treatment. However, SCA is marked by chronic hemolytic anemia, severe pain events, and extensive damage to various organs [6, 17]. The increased autoxidation of Hb S and the overactivation of several oxidases in the vascular system of SCD patients results in an increased production of ROS, which may directly oxidize several subcellular components and result in the loss of membrane lipid asymmetry. In addition to creating a condition of chronic anemia and endothelial dysfunction, this decreases the deformability of RBCs and increases their fragility and hemolysis [16, 17]. Furthermore, sickled RBCs adhere more readily to one another and the interior walls of small blood vessels as a result of intrinsic rheological changes, secondary impacts on endothelium, inflammatory cells, and plasma proteins, and this leads to vaso-occlusion, tissue hypoxia, pain crisis, and numerous other complications. The lifespan of those who receive the best care maybe 50 to 60 years [1, 16, 17].
Congenital dyserythropoietic anemias (CDA)
The term CDAs refers to a class of uncommon heterogeneous genetic illnesses that impair erythropoiesis and have various morphologic alterations affecting late erythroblasts in the bone marrow and erythroid hyperplasia leading to hemolysis and the formation of frail RBCs [18]. The manifestations are vague since they are similar to other more widespread hemolytic illnesses, including thalassemia and RBC membrane disorders (e.g., spherocytosis) [8, 18].
In addition to X-linked dominant mutations in the heme biosynthetic enzyme gene, ALAS2, this group of diseases has also been linked to mutations in the CDAN1, SEC23B, KIF23, C15ORF41, LPIN2, KLF1, or GATA1 genes [2, 18]. CDA’s categorization has been mainly determined according to bone marrow pathology results utilizing the morphologic classification, in which three primary forms of CDA were discovered and categorized according to structure similarities ([8]; extensively reviewed in [18]).
Redox metabolism in RBC: the most noticeable roles of Prxs so far
The cell’s oxidative status is of utmost importance to cell survival. Signal transmission, gene transcription, and cell proliferation are only a few examples of how oxidative status affects numerous regular physiological cell processes, known as redox biology [19]. When the ratio of pro-oxidants to intracellular antioxidant defenses shifts in favor of the former, oxidative stress occurs. Pro-oxidants are categorized as reactive oxygen species (ROS) and nitrogen species (RNS) and include superoxide radicals (O2−•), hydroxyl radical (OH•), singlet oxygen (1O2), hydrogen peroxide (H2O2), peroxynitrite (ONOO−), and many others. These oxidants are hazardous to cells and cause organ damage and cell death by oxidizing proteins, lipids, and DNA [20, 21]. As a result, it has been proposed that oxidative stress impacts cell aging and apoptosis, aggravating the symptoms of numerous disorders such as hemoglobinopathies (SCA and thalassemia), G6PD deficiency, HS, and CDA. Therefore, although each of these anemias does not have oxidative stress as the root cause, it facilitates numerous of their pathologies, including hemolysis, which exacerbates their symptoms [20].
In hemolytic anemias, RBCs are the primary target of oxidative stress, and Hb is a prominent generator of ROS in these cells. It is estimated that 3% of Hb undergoes auto-oxidation, forming metHb and serving as a constant source of O2•− generation. In turn, this production leads to H2O2 and oxygen as outcomes of dismutation or O2•− may interact with nitric oxide (NO), leading to the formation of ONOO-, the most reactive RNS that can increase lipid peroxidation in addition to the oxidation of Hb to MetHb [20, 22]. Auto-oxidation of unstable Hb can also lead to changes in the RBC membrane, producing hemicromes (byproducts of the oxidative denaturation of Hb), dissociation of cytoskeletal proteins, and also the generation of microparticles (phospholipid microvesicles derived from the cytoplasmic membrane) [23]. Moreover, in addition to ROS production, oxidative denaturation of Hb also results in the accumulation of free heme (which triggers oxidation processes and has a pro-inflammatory effect by activating NF-kB) and iron (which functions as a Fenton reagent in the Haber-Weiss reaction for the synthesis of OH•) [23, 24].
Since redox homeostasis is considered one of the most critical aspects of human physiology, RBCs have been provided with an incredibly efficient redox regulation apparatus exclusively dedicated to maintaining its structural integrity and capacities [21, 22]. Superoxide dismutase (SOD) speeds up the process of turning O2−• into H2O2, which CAT or glutathione peroxidase (GPX) then reduces to water. The reduced thiol GSH, which keeps sulfhydryl buffering capability, is present in millimolar amounts in cells. The PPP’s oxidative metabolism of glucose-6-phosphate (G6P) converts oxidized NADP+ back to NADPH, substantially supporting RBC redox equilibrium by donating an electron to reductases to reduce back oxidized glutathione (GSSG) and metHb to its active form (glutathione reductase and methemoglobin reductase, respectively). Thioredoxin (Trx), an additional antioxidant enzyme found in RBCs, reduces oxidized proteins and is an electron donor for most peroxiredoxins (Prx). This latter one works by reducing organic hydroperoxides (ROOH), peroxidized phospholipids, and peroxides (such as H2O2) [20, 22, 23] and will be the focus of this review.
Investigating which genes are involved in the normal process of erythropoiesis has been an essential goal in the scientific community [25, 26]. Understanding how the metabolic pathways triggered during the development of hemolytic anemia differ from healthy ones and the relationship between the expression of specific proteins and the severity of these diseases remains a puzzling fact. Identifying these proteins could result in new targets for managing anemic patients. In RBCs, Prxs are one of the most important proteins involved in the defense against ROS, and their role in erythrocytes has been shown to go far beyond their antioxidant activity [9, 27].
Mature RBC expresses three Prx isoforms (Prx1, Prx2, and Prx6). In addition to being anemic, Prx1 knockout animals have increased oxidative stress and ROS in their RBC, which leads to protein oxidation, Hb instability, the formation of Heinz bodies, and a reduction in the lifespan of erythrocytes as a result of membrane damage [28]. Prx2 −/− characteristic features include Heinz body formation, aberrant RBC morphology, noticeably increased ROS levels, hemolytic anemia, and several severely oxidized RBC proteins [29, 30]. The other enzymatic H2O2 scavengers, such as GPX and CAT, maintained their activity and expression levels in Prx2−/− RBCs, indicating a non-redundant function [29]. As for Prx6, according to several studies, it plays a significant role in the oxidative stress response [31,32,33,34]. At the same time, the absence of Prx6 makes cells more vulnerable to oxidative stress, severe tissue damage, elevated levels of protein oxidation, and cell death [35]. Furthermore, different antioxidants’ mRNA expression levels showed no differences, pointing to a non-redundant role for this protein [35]. Thus, the aim of this review was present the recent findings that broke some paradigms about these three Prxs present in the mature erythrocyte (Prx1, Prx2, and Prx6), showing that in addition to peroxidase activity, these enzymes may have supplementary roles in the regulation of cellular adaptation mechanisms for redox homeostasis and survival of human RBCs disrupted in hemolytic anemia conditions.
Peroxiredoxins: an overview
Prxs are antioxidant enzymes that exhibit very high peroxide reduction rates (106 to 108 M−1 s−1), using a highly conserved specialized active site architecture to reduce peroxides [36, 37]. The high catalytic efficiency and the abundance of these proteins in the cells make them necessary peroxide scavengers. In humans, six isoforms of Prxs have been described, being detected in different cellular environments, such as the cytosol (Prx1, Prx2, and Prx6), mitochondria (Prx3 and Prx5), peroxisome (Prx5), endoplasmic reticulum (Prx1 and Prx4), and even in the nuclear environment (Prx1) [36, 38, 39].
The activity of the Prx depends on a cysteine residue contained in an active site of the catalytic motif PxxxTxxC (where P designates Proline, T is Threonine, C is Cysteine, and x denotes any amino acid) [36, 40]. Studies have shown that a second cysteine residue is generally present and conserved near the C-terminal portion, defining Prx classes containing 1 or 2 cysteines [45]. Several classifications have already been proposed for Prx, considering different aspects of these enzymes [41,42,43,44,45]. Several studies use a classification based on the number of cysteines involved in the catalysis (1-Cys Prx and 2-Cys Prx) and by the type of disulfide that is formed during the catalysis: inter or intramolecular (typical or atypical 2-Cys Prx, respectively) [36, 46]. Prx cysteines can also be classified according to their role in the catalytic cycle as peroxidase cysteine (CP) or resolution cysteine (CR) [36] (Fig. 3).

Reaction mechanisms of Prx. The reaction mechanisms of typical 2-Cys Prx (1), atypical 2-Cys Prx (2), and 1-Cys Prx (3) enzymes are shown (elaborated by the authors, adapted from [46])
The catalytic cycle of the Prxs is initiated by an attack of peroxidase cysteine CP-SH to the H2O2, cleaving the OO bond of the peroxide and forming sulfenic acid in the CP residue (CP-SOH). This leads to the release of H2O when H2O2 is reduced or the release of alcohol in the case of lipid peroxides reduction [47]. The reduction of the sulfenic acid determines whether the enzyme is a 1-Cys Prx or 2-Cys Prx. 1-Cys Prxs have only CP, and their oxidized form (CP-SOH) can be reduced by ascorbic acid or Trx [48, 49]. In the case of 2-Cys Prx, the sulfenic acid is attacked by a second cysteine, the CR one, releasing H2O and forming an inter- or intramolecular disulfide bond [48] (Fig. 3).
In mammals, Prxs are found in the conformation of antiparallel homodimers, as seen in Fig. 3 [48, 50]. When typical 2-Cys Prx are exposed to high concentrations of peroxide, CP is over-oxidized, taking the form of cysteine sulfinic acid (CP-SO2H) or sulfonic acid (CP-SO3H) after the attack of sulfenic acid (CP-SOH) by other peroxide molecules, avoiding disulfide formation with CR [36, 51, 52]. Overoxidation results in the loss of peroxidase function in these enzymes since Trx cannot reduce the intermediates. However, another low molecular weight enzyme called sulfiredoxin (Srx) can reactivate the peroxidase activity of Prxs. Srx can reduce only the cysteine sulfinic acid (CP-SO2H); no biological reducer for the CP-SO3H form is known to date [53].
Recent findings show that in addition to its pivotal importance in the RBC antioxidant defense, Prxs are also implicated in many different processes, such as cell signaling, proliferation, apoptosis, and mitochondrial permeability [54,55,56]. Thus, the widespread role of Prxs is discussed below, considering the newest discoveries regarding the known (and suggested) roles of these enzymes in erythroid cells.
Multifaceted roles of Prxs: beyond their antioxidant functions
As aforementioned, Prxs play an undeniably important role in protecting cells against oxidative stress due to their antioxidant activity. However, their function is more comprehensive than this role, as these proteins participate in a cascade of peroxide-mediated signaling and interact with a wide variety of proteins and processes of proliferation, differentiation, migration, and regulation of gene expression [57, 58]. The image below (Fig. 4) was generated using databanks of GeneMANIA on Cytoscape; the interactions are made using available data from different databases and demonstrate that the Prxs participate in several pathways interacting with other proteins.

Prx interactome. The image generated by GeneMANIA correlates the interactions among genes and proteins, according to the query, with only the input genes/proteins being represented. The connections among the subjects represent the kind of interactions shown in the legend. Subjects abbreviation: p66SHC (SHC adaptor protein 1); GSS (glutathione synthetase); JNK1 (c-Jun N-terminal kinase); PTEN (phosphatase and tensin homologue); HBA1 (hemoglobin subunit alpha 1); ASK1 (Apoptotic signal-regulating kinase 1); Spectrin (Spectrin); GPX1 (glutathione peroxidase 1); GSTP1 (glutathione S-transferase pi 1); NFKB1 (nuclear factor kappa B subunit 1); Band3 (Band 3); ANK1 (Ankyrin 1); CAT (catalase); PRX4 (Peroxiredoxin-4); SOD1 (Superoxide Dismutase 1); TRX1 (Thioredoxin-1); RHAG (Rh Associated Glycoprotein); PRX5 (Peroxiredoxin-5); PRX2 (Peroxiredoxin-2); PRX1 (Peroxiredoxin-1); SRX1 (Sulfiredoxin 1); TRXR1 (Thioredoxin Reductase 1); PRX3 (Peroxiredoxin-3); PRX6 (Peroxiredoxin-6); APEX1 (Apurinic/Apyrimidinic Endodeoxyribonuclease 1) (Elaborated by the authors)
In addition to peroxidase activity, Prxs interacts with several proteins in many cellular compartments. In different cell types, Prxs were reported to act as modulators of the action of several transcription factors. For example, Prx1 interacts with c-Myc, NF-κB, and AR (androgen receptor) to modulate their activities, thus controlling gene expression [59,60,61,62,63]. Complementarily, Prx1, 2, and 6 appear to play essential roles in the erythrocyte through interacting with proteins such as APEX1, PTEN, ASK1, P66Shc, JNK, HBA1, and Band 3, which are evidenced in Fig. 4. These interactions appear to play an essential role in erythrocyte survival, especially when this cell is exposed to oxidative stress. The details of such interactions and their importance for the erythrocyte will be discussed below.
Prx1
Prx1 plays an essential role in acting as an H2O2 signal transducer [64]. This role is associated with the ability to interact directly with signaling molecules, regulating signaling in the cell temporally and spatially via oxidizing or phosphorylated (i.e., at the Thr90 site, which results in inactivation of peroxidase activity) forms of Prx1 [65, 66]. This role involves their subcellular localization, the availability of Prx1 reducing systems such as thioredoxin Trx and Srx, and even the regulation of transcription factors activity through direct interaction with these factors (most often via oxidation of cysteine thiols) [53]. The mechanistic switch of Prx1 from peroxidase to chaperone function has been discussed in various articles (and previously in topic 2). Briefly, the conserved N-terminal Cys51 (or CP) can be oxidized by H2O2 to cysteine-sulfenic acid (Cys51-SOH) and react with Cys172-SH of the other subunit, thus producing an intermolecular disulfide that can only be reduced by Trx [67].
These Prx1s can become easily over-oxidized on their catalytically active cysteine (i.e., can react with a second H2O2 molecule and becomes hyperoxidized to the cysteine sulfinic acid, CP-SO2H) in conditions of high H2O2 [68, 69], and are eventually recycled through ATP-dependent reduction by Srx [70] (the same occurs with another isoform, the Prx2 [71]). This hyperoxidation of CysP, at first, may seem to limit their role, but in fact demonstrate an additional function of acting as molecular chaperones and their involvement in propagating peroxide-mediated signals in a spatially confined location. The first results from structural changes in which the low molecular weight complexes convert into high molecular complexes (e.g., oligomeric or decameric conformation), enabling them to act as chaperones, which we will explain in more detail below) [69, 70].
The latter is exercised by a small fraction of phosphorylated Prx1 associated with lipid rafts, which results in the inactivation of peroxidase activity. This spatially confined inactivation provides a way to generate H2O2 gradients around sites where signaling proteins are concentrated, minimizing the overall H2O2 accumulation to levels that would disrupt the cell’s global redox potential and optimizes cellular signaling by generating H2O2 gradients around sites where signaling proteins are concentrated (known as the “floodgate mechanism”), thus facilitating H2O2-dependent signaling [72, 73]. Moreover, a slower rate of reactivation of hyperoxidized Prxs (via Srx-dependent reduction) is proposed as a built-in mechanism, allowing sufficient time for H2O2 accumulation and signal propagation [50].
There are different examples of how Prx1 modulates cell signaling when the cell redox status is altered. For instance, although the relationship between ROS and tumorigenesis remains unclear, Neumann et al. [28] were the first to demonstrate that mice with a deficiency in Prx1 production (knockout) showed viability but had a high incidence of anemia and increased malignant cancers. Later, Cao et al. [74] postulated that this relationship was related to the ability of Prx1 to bind to PTEN (phosphatase and tensin homolog), thus protecting against oxidative degradation. PTEN deficiency is a hallmark of several tumors [75] since it is a phosphatase with an essential role in apoptotic regulation and participating in cell migration, adhesion, differentiation, and angiogenesis [74]. This was the first phosphatase associated with tumor suppression, having negative control on one of the most critical pathways: the PI3K-AKT [76]. H2O2 inhibits PTEN activity [77], so the interaction between Prx1 and PTEN is essential for preventing the latter loss of function, therefore maintaining the regulation of the AKT pathway. It is worth mentioning that Huo et al. [78] found a relationship between the absence of PTEN and the consequent imbalance in the expression of several antioxidants, suggesting this protein’s important role in maintaining the overall redox state of cells.
Another exciting example is observed in the hypothesis elaborated by Nassour et al. [79], in which the authors proposed that Prx1 can partake in the inflammatory response through its interaction with APEX1. The APEX1 gene is related to a multifunctional enzyme containing an endonuclease domain that plays a central role in the Base Excision Repair and a redox site. This gene is responsible for activating several transcription factors, including transcription factor nuclear factor kappa B (NF-kB), tumor-suppressor protein p53 (P53), and other genes involved in cellular processes, ranging from cell survival and growth signaling to inflammatory pathways [80,81,82,83,84]. As postulated by Nassour et al. [79], the interaction of Prx1/APEX1 keeps APEX1 in the cytoplasm of the cell instead of in the nucleus, preventing the activation of inflammatory markers such as NF-kB and IL-8 (a pro-inflammatory chemokine involved in the invasion and metastasis of tumors) [85, 86]. Therefore, Prx1 interacts with and blocks the redox factor APEX1 from activating IL-8 expression, demonstrating a novel anti-inflammatory function of Prx1.
Recent studies by our group observed a similar Prx1/APEX1 interaction [87, 88]. Our data showed diminished expression of Prx1 in peripheral blood samples of patients with β-thalassemia major, as well as the protein levels of APEX1, as opposed to the high levels of its mRNA. We hypothesize that with the decrease of Prx1 observed in patients with beta-thalassemia major, the produced APEX1 protein would migrate to the nucleus during RCB cell formation (still in the bone marrow), where new mRNA molecules were being formed. However, with the extrusion of the nucleus at the end of the differentiation process, APEX1 proteins would go along with the nucleus, evidencing this imbalance between the APEX1 protein and its mRNA.
More recently, our group also proposed an exciting role of Prx1 involving the propagation of the peroxide-mediated signal in K562 cells induced to erythroid differentiation submitted to H2O2-oxidative stress. Our hypothesis consists of an interaction between Prx1 with the transcription factor FoxO3. This role is suggested to be a part of a more complex regulatory mechanism that influences the subcellular location of this transcription factor as a part of an adaptive response to low-intensity oxidative stress [89]. Thus, despite being unable to verify this hypothesis, Bernardo et al.’s proposed role of Prx1 is of great importance since FoxO3 is a known key regulator of distinct stages of erythroid cell differentiation and maturation [90, 91]. Hopkins et al. previously described the association of FoxO3 and Prx1 in other cell types [92], and different authors described a modulatory mechanism by which Prx1 is involved in cell signaling via interacting signaling molecules and transcription factors [61, 65, 93, 94], which reinforces the need for further studies in this field, demonstrating that Prx1’s role as a chaperone may be more crucial for the erythroid redox signaling than previously thought.
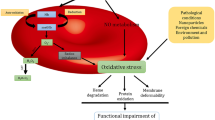
Several studies in different cell types have demonstrated that in parallel to the peroxidase activity, Prx1 and its physiological reductant, Trx1, have anti-apoptotic functions in the cytoplasm through direct or indirect interaction with important apoptotic regulators induced by oxidative stress, such as ASK1 (Apoptosis Signal-Regulating Kinase 1), JNK (Jun N-terminal Kinase), and p66shc [95,96,97]. Under normal conditions, Prx1 assists in suppressing ASK1-JNK signaling, controlling ROS levels and other antioxidant enzymes, while ASK1 remains linked to its physiological inhibitor, Trx1. However, under oxidative stress conditions, Trx1 disconnects from ASK1 and is replaced by Prx1 in its oxidized form. This exchange is a mechanism that aims to keep ASK1 inactive and thereby prevent it from triggering the activation of apoptotic signaling pathways [96]. This enzyme also inhibits the activation of JNK through an indirect interaction mediated by GSTpi (glutathione-S-transferase pi) [98, 99]. Prx1 and Trx1 have been identified as negative modulators of p66Shc activation, which plays a prominent role in apoptosis caused by oxidative stress, inducing respiratory collapse and mitochondrial rupture in many cell types and erythroid precursors (Fig. 5) [99,100,101].

The role of Prx1 in apoptosis suppression under oxidative stress induced through two apoptosis signal regulators, ASK1 and p66Shc. Trx inhibits ASK1 activation during low-stress conditions, while under high oxidative stress conditions; Trx is replaced by oxidized Prx1 to suppress ASK1 activation. Trx and Prx1 maintain p66Shc inactive at the cytoplasm under low oxidative stress conditions since p66Shc is separated from the complex under high oxidative stress due to Prx1 decamerization. Then, p66Shc translocates to the mitochondria, producing ROS and inducing apoptosis (The authors created this figure by adapting images from Servier Medical Art Commons Attribution 3.0 Unported License (http://smart.servier.com))
A recent study by our research group showed significantly elevated levels of Prx1 and Trx1 in reticulocytes and erythrocytes of β-thalassemia intermedia patients, which suggests a possible cellular mechanism to maintain the pathways mentioned above inactive, thus increasing cell survival and consequently reducing the impacts of ROS-mediated ineffective erythropoiesis [87]. The increased apoptosis of the erythroid precursors is one factor contributing to the inefficient erythropoiesis found in β-thalassemia; nevertheless, the degree of apoptosis is unexpectedly varied and has been shown both ex vivo in bone marrow samples as well as in vitro in erythroblast culture techniques [102,103,104]. Centis et al. further elucidated the extent of the ineffective erythropoiesis, demonstrating that thalassemic bone marrow contains about 5- to sixfold more erythroid precursors, which resulted in an absolute rise in erythroid precursor apoptosis of roughly 15-fold above healthy controls [102].
Another disease with high rates of apoptosis is CDA. Bone marrow flow cytometry studies identified that erythroid precursors were arrested in the S phase of the cell cycle, and ultrastructural morphological evaluation revealed alterations in cells going through apoptosis, despite the increased erythropoiesis, which was indicated by an elevated amount of bone marrow erythroid burst-forming units and also by serum EPO [105]. Additionally, in SEC23B-deficient animals, CDA type II caused by germline homozygous or compound-heterozygous SEC23B mutations result in secretory organ deterioration due to endoplasmic reticulum (ER)-stress-associated apoptosis. The extent and duration of the stress stimuli have a significant role in determining the cell’s fate, and failing to respond to ER stress will eventually stimulate apoptotic pathways [106]. Prx1 has recently been shown to be a key regulator of ER stress-induced UPR signaling and apoptosis. The fraction of apoptotic cells was 23% lower in Prx1-overexpressed cells than in control following 24 h of treatment with thapsigargin (Tg), a recognized ER stress inducer. Additionally, Prx1-overexpressed cells had a 40% reduction in ER stress-induced ROS production, demonstrating that Prx1 plays a role in the regulation of ROS production during ER stress, which helped prevent ER stress-induced apoptosis [107]. Therefore, Prx1 might be an interesting target for new studies on diseases with pronounced apoptosis.
Prx2
Prx2 is the most studied isoform among those described in this review, probably because this is the third most abundant erythrocyte protein [108]. As previously mentioned, this is a vital antioxidant in the RBC, mainly responsible for removing H2O2 formed endogenously by Hb autoxidation. Many authors have identified Prx2 oligomers associated with the erythrocyte membrane [109,110,111,112] through binding to band 3 without losing its peroxidase activity [113, 114]. It has been speculated that these membrane-bound Prx2 may be involved in the protection against membrane lipid peroxidation and the membrane proteins from oxidative damage (e.g., band 3) [115, 116]. In SCD human and mouse erythrocytes, phosphorylation of Prx2 by activated Syk kinase was reported, with the latter being associated with membrane translocation of not only Tyr-phosphorylated Prx2 but also of active Syk, which favors band 3 clusterization, erythrocyte membrane rearrangement, and even the release of erythroid microparticles [117].
A significant correlation was found between the binding level of Prx2 to the membrane and the potassium efflux, with a binding rate below 1% capable of initiating the efflux [110]. The fraction of erythrocyte membrane-associated Prx2 was shown to be altered in hemolytic anemias, with most of the studies reporting an augmented Prx2 binding to the membrane, such as SCA [118, 119], HS [10, 120], and SAO [11], all in response to oxidative stress conditions. Concerning the hemolytic anemias for SCD, another study reported increased binding of Prx2 to the membrane during dense cell formation through activating the Ca2+-dependent K+ channel [121]. Additionally, Biondani et al. suggested that the Prx2 increased membrane binding indicates that the severe membrane damage promotes Prx2 dimerization to protect radically sensitive proteins [119]. For the HS, it is suggested that the increased Prx2 membrane binding could be a protection mechanism to maintain membrane integrity by regulating membrane lipid peroxidation [10]. In SAO RBCs, the increase in Prx2 membrane-biding and the large amounts of accumulated misfolded band 3 may cause oxidative damage in these cells [11].
However, other authors reported that the amount of Prx2 bound to the membrane was reduced in some conditions, such as (i) in mouse models of β-thalassemia, in which the authors suggest that the Prx2 membrane displacement possibly occurs because of the membrane-bound hemichromes that masks the Prx2 docking sites. This hypothesis provides a possible mechanism for the increased membrane damage in β-thalassemia RBCs [122]; (ii) in sickle cell SAD mice exposed to hypoxia, which the authors suggests is a reflection that either more damaged red cells are already removed from circulation or that a higher amount of Prx2 is required as free form in red cell cytoplasm [119]; and (iii) when the cells were treated with H2O2, Bayer et al. found a decrease in Prx2 binding to the erythrocyte membrane in oxidative stress conditions. Similar to the previous study, their result suggested an association between the hemichromes and Prx2. The authors propose that the hemichromes competed with Prx2 and decreased their binding to the membrane [123].
Prx2 can also stabilize the Hb by acting as a chaperone (reduced Prx2 in its decameric form binds to Hb through a domain in its N-terminal region [30]). Prx2 interacts with the Hb α, β, and γ subunits [124], thus protecting Hb against H2O2-induced aggregation (i.e., may act as a chaperone for the denatured metHb [120, 125]). Additionally, in hemolysates from Prx2−/− mice, Hb H2O2-induced aggregation and Heinz body formation were more prone to occur, and the addition of recombinant human Prx2 (rhPrx2) restored the aggregation level to wild-type hemolysates [30, 125, 126]. All these findings suggest that Prx2 plays an important role in preventing hemolytic anemia (through its performance in stabilizing Hb under high oxidative stress conditions), with their interaction being severely impaired in RBCs of patients with thalassemia and SCA [30].
Several studies demonstrated that Prx2 is modulated throughout normal erythroid development, especially in the stages of synthesizes of Hb, and that it is overexpressed during erythropoiesis in β-thalassemia [9, 122, 127, 128]. De Franceschi et al. demonstrated that in early β-thal erythroid precursors characterized by high levels of ROS and heme, Prx2 targets both ROS and heme to reduce oxidative stress suggesting a novel multifunctional cytoprotective role of Prx2 [129]. Matte et al. aiming to better understand the role of this enzyme in the context described above, evaluated β-thalassemic mice knockout for Prx2, showed a worsening in the hematological phenotype of the animals, amplification of ineffective erythropoiesis and increased levels of ROS in erythroid precursors when compared to healthy or β-thalassemic mice with preserved Prx2. In addition, the absence of Prx2 resulted in greater activation of Nrf2. In contrast, administration of recombinant Prx2 in mice reduced ineffective erythropoiesis, ROS levels, and apoptosis. These findings suggest that Prx2 and Nrf2 collaborate to reduce oxidative cell damage during pathological erythropoiesis in β-thalassemia [130]. Recently, Prx2 deficiency was associated with dyserythropoiesis and decreased erythrocyte lifespan in patients with CDA. The authors were to describe a case of anemia in humans related to PRX2 mutation, with the variant causing a loss of enzyme function and increased oxidative stress [131].
Interestingly, Prx2 was found to be resistant to hyperoxidation [132], likely because the Srx level in these cells is much lower than that in other cell types [133]. Thus, it has been hypothesized that hyperoxidation does not occur easily in erythrocytes because it is unlikely that the RBC utilizes the “floodgate mechanism” for signal transduction [14]. Nevertheless, transcription-independent self-sustained oscillations of hyperoxidized Prx2 were detected in human erythrocytes, indicating that H2O2 produced from Hb autoxidation is the primary source of Prx2 hyperoxidation [134]. These oscillations were hypothesized to be connected with the peroxidation-enhanced chaperone function [133].
Additionally, it has been reported that hyperoxidation of Prx2 occurs more easily in more oxidizing conditions. For example, it was augmented by the inhibition of CAT, indicating that CAT protects Prx2 against higher levels of H2O2 [71] and in pathological conditions, such as β-thalassemia [87]. The situation is aggravated in G6PD deficiency since it is a condition where inefficient recycling of Prx2 in erythrocytes is reported due to the compromised NADPH production (limited via the pentose phosphate pathway). Cheah et al. reported that G6PD-deficient erythrocytes were severely compromised in their ability to recycle Prx2 oxidized, and its accumulation in the erythrocytes suggests that impaired antioxidant activity of Prx2 could contribute to the hemolysis and other complications associated with the condition [69].
Lastly, Prx2 also regulates iron homeostasis, which is highly relevant in hemolytic anemia since iron overload is an important factor aggravating the clinical outcome of β-thalassemia [135, 136]. Iron homeostasis is a complex process regulated by the interaction of hepcidin, a hormone produced in the liver that the presence of high levels of iron can induce. Ferroportin is a protein responsible for iron absorption, storage, and concentration in the plasma. Hypoxia, a common factor in hemolytic anemias, stimulates the beginning of new erythropoiesis cycles to account for the loss of red cells, which suppress hepcidin, leading to an iron overload that aggravates the clinical outcome of patients [137].
Transcription factors sensitive to redox changes present in the liver, such as STAT3 (signal transducers and activators of transcription), can activate hepcidin expression in response to iron overload and oxidative stress. Not long ago, STAT3 activation was related to Prx2 (i.e., Prx2-dependent STAT3 oxidation as part of a redox relay of a highly organized membrane signaling domain), establishing a possible connection between a transcription factor and a protein responsible for protection against oxidative stress [138,139,140]. A recent study evaluated the effects of iron overload in Prx2−/− mice to explore the role of Prx2 in iron homeostasis. The obtained data showed that the absence of this enzyme failed hepcidin induction in response to the administration of high iron levels. This effect was due to the loss of STAT3 activation. This demonstrated that Prx2 acts as an essential regulator of iron homeostasis through the activation of STAT3 and the consequent induction of hepcidin and can be explored as a future therapeutic strategy [139].
Prx6
The deleterious effects found on erythrocytes of mice genetically deleted for Prx1 and 2 were not described for Prx6 mutants; these biological models remain viable and fertile and display no gross morphological defects [35, 141]. However, it is noteworthy that among all the isoforms of human peroxiredoxins, Prx6 is the most differentiated since it is the only 1-Cys Prx and has phospholipase A2 activity. Cysteine oxidation does not affect this activity and contributes to the repair of oxidized phospholipids by cleavage of oxidized fatty acids [142].
Despite this phospholipase A2 activity never being reported in red cells, the importance of Prx6 to RBC is suggested in a study by Cho et al. [143]. The authors analyzed the abundance of some antioxidant enzymes in the erythrocyte lysate of individuals with SCA, both treated and not treated with hydroxyurea. They reported a significant reduction in Prx6 levels in both sickle groups compared to healthy individuals. This finding suggests that, due to its unique phospholipase A2 activity, the Prx6 may have a nonredundant role in the erythrocyte associated with the ability to repair cell membranes, which may be impaired in the sickle cells, contributing to their greater susceptibility to hemolysis. Nonetheless, the mechanisms underlying the reduction of Prx6 in erythrocytes of individuals with SCA remain unknown [143].
Possibly due to its lower concentration in the erythrocyte, there are very few studies involving this isoform. However, its presence in the RBC may be more important than the scientific community believes. In other cell types, that similarly to the erythrocytes is highly submitted to oxidative stress, a decline in Prx6 expression was linked to a progressive increase in oxidative load, and Prx6 was even a prerequisite for sulforaphane (SFN)-mediated cytoprotection [144]. This phytochemical compound is a known Nrf2 agonist that ameliorated SCD pathology in a murine model by promoting the elimination of heme released by hemolysis from lysed sickle cells [145]. Additionally, another study reported that Prx6 is a bona fide negative regulator of ferroptotic cell death [146].
Thus, by demonstrating the essential role of Prx6 in protecting cells against ferroptosis, the authors provided a potential target for novel studies in the RBC since ferroptosis was revealed to be linked with the pathogenesis of thalassemia-associated bone diseases (i.e., iron overload significantly increases the death of osteoblasts, resulting in a decrease in mineralization capacity. Thus there is increased bone resorption due to ferroptosis) [147]. Additionally, Stuhlmeyer et al. showed that Prx6 binds to the conserved cysteine 47 of heme, having even a higher affinity to it than for Hb. This binding prevented spontaneous and ascorbic acid-induced metHb formation. These results allowed the authors to propose that since heme is the prosthetic group of several proteins and enzymes, Prx6 may also protect these other proteins [148].
Lastly, in other cell types, the loss of Prx6 was associated with down cell division, metabolism alteration, and mitochondrial function, suggesting that Prx6 is a key factor that connects redox homeostasis, proliferation, and apoptosis (i.e., Prx6 interacts with multiple signaling pathways) [149, 150], which could be another promising field of study, mainly when dealing with hemolytic diseases with ineffective erythropoiesis such as thalassemias.
Figure 6 summarizes some of the additional Prx1, 2, and 6 roles covered in this article. Prx1 has an essential role in cellular redox signaling, evidenced by interaction with proteins such as JNK, GSTpi, p66Shc, and ASK1, which inhibits apoptotic signaling pathways. Meanwhile, the interaction of Prx1 with APEX1 prevents the activation of the inflammatory cascade and, with PTEN, leads to cell survival and inhibition of tumorigenesis. Regarding Prx2, we reported the important interaction with the cytoplasmatic portion of band 3 protein that prevents its degradation. Prx2 also binds to free heme preventing oxidative damage caused by its accumulation. More importantly, Prx2 can also form decamers or oligomers of high molecular weight and interact with Hb, inhibiting its denaturation and aggregation. Lastly, Prx6 is hypothesized to repair oxidized phospholipids, consequently preventing membrane damage through phospholipase A2 activity. These additional activities are essential to cell survival, especially in hemolytic anemia, which is frequently exposed to high doses of ROS.

Schematic representation of Prx1, Prx2, and Prx6 roles and peroxidase activity in anemic erythrocytes/precursors. The events shown in the figure are as follows: 1, Binding of Prx2 to protein band 3 preventing its oxidation; 2, Prx2 binding to free heme group to prevent its oxidative actions to the membrane and other proteins, and participating to iron homeostasis; 3, Oligomerization of Prx2 forming decamer that act as chaperone, binding to hemoglobin, preventing denaturation and aggregation of hemoglobin; 4, Prx6 phospholipase A2 activity repairing oxidized phospholipids; 5, Interaction of Prx1 with JNK, GSTpi, p66Shc and ASK1 inhibiting apoptotic signal pathways; 6, Interaction of Prx1 with PTEN, inhibiting the interaction of PL3K and AKT which prevent tumorigenesis; 7, Prx1 regulation of phagocytosis and removal of damaged RBC from peripheral circulation; 8, Prx1 interaction with APEX1 which inhibits its interaction with NF-Kb thus inhibiting inflammatory pathways (The authors created this figure adapting images from Servier Medical Art Commons Attribution 3.0 Unported License (http://smart.servier.com))
Conclusions and future perspectives
This review aimed to demonstrate that the framework for Prxs biology implicates a widespread role that goes far beyond their antioxidant activity. For that, we discussed the newest discoveries regarding the known (and suggested) roles of these enzymes in the erythroid cells, such as (i) selectively transducing peroxide signals to generate appropriate signaling responses, (ii) molecular chaperone; (iii) contribution to cell survival, (iv) protection of membrane damage, and (v) maintenance of iron homeostasis. We also highlighted the role of these enzymes in the context of hemolytic anemias, in which they may represent redox sensors able to differentiate systems operating in a healthy state from pathological conditions where cells are affected by oxidative stress induced by ROS production. Therefore, comprehending the different mechanisms involved in erythrocyte protection by these three proteins can better understand disease pathologies and strategies for developing future therapeutic targets. However, as demonstrated by this review, the knowledge regarding Prxs in the RBC is still evolving, with many unanswered questions, especially concerning Prx6, the lesser studied among the isoforms. These gaps within Prxs’ role in healthy erythroid cells and pathological conditions, such as the hemolytic anemias addressed in this review, comprise a promising field for further research studies.
Availability of data and material
Not applicable.
References
Sankaran VG, Weiss MJ (2015) Anemia: progress in molecular mechanisms and therapies. Nat Med 21:221–230. https://doi.org/10.1038/nm.3814.Anemia
Nandakumar SK, Ulirsch JC, Sankaran VG (2016) Advances in understanding erythropoiesis: evolving perspectives. Br J Haematol 173:206–218. https://doi.org/10.1111/bjh.13938
Grace RF, Glader B (2018) Red blood cell enzyme disorders. Pediatr Clin North Am 65:579–595. https://doi.org/10.1016/j.pcl.2018.02.005
Mohandas N (2018) Inherited hemolytic anemia: a possessive beginner’s guide. Hematol Am Soc Hematol Educ Progr 2018:377–381. https://doi.org/10.1182/asheducation-2018.1.377
Andolfo I, Russo R, Gambale A, Iolascon A (2016) New insights on hereditary erythrocyte membrane defects. Haematologica 101:1284–1294. https://doi.org/10.3324/haematol.2016.142463
Wahed A, Quesada A, Dasgupta A (2020) Hemoglobinopathies and thalassemias. In: Hematology and Coagulation (Second Edition), 2nd ed. Academic Press. 51–75
Luzzatto L (2021) Diagnosis and clinical management of enzymopathies. Hematol Am Soc Hematol Educ Progr 2021:341–352. https://doi.org/10.1182/hematology.2021000266
Risinger M, Emberesh M, Kalfa TA (2019) Rare hereditary hemolytic anemias management: diagnostic approach and considerations in management. Hematol Oncol Clin North Am 33:373–392. https://doi.org/10.1016/j.hoc.2019.01.002
van Zwieten R, Verhoeven AJ, Roos D (2013) Inborn defects in the anti-oxidant systems of human red blood cells. Free Radic Biol Med 67:377–386. https://doi.org/10.1016/j.freeradbiomed.2013.11.022
Rocha S, Rocha-Pereira P, Cleto E et al (2020) Linkage of typically cytosolic peroxidases to erythrocyte membrane – a possible mechanism of protection in Hereditary Spherocytosis. BBA - Biomembr 1862:183172. https://doi.org/10.1016/j.bbamem.2019.183172
Flatt JF, Stevens-hernandez CJ, Cogan NM et al (2020) Expression of South East Asian ovalocytic band 3 disrupts erythroblast cytokinesis and reticulocyte maturation. Front Physiol 11:357. https://doi.org/10.3389/fphys.2020.00357
Gurbuz N, Yalcin O, Aksu TA, Baskurt OK (2004) The relationship between the enzyme activity, lipid peroxidation and red blood cells deformability in hemizygous and heterozygous glucose-6-phosphate dehydrogenase deficient individuals. Clin Hemorheol Microcirc 31:235–242
Fattizzo B, Cavallaro F, Marcello APML et al (2022) Pyruvate kinase deficiency: current challenges and future prospects. J Blood Med 13:461–471. https://doi.org/10.2147/JBM.S353907
Harteveld CL, Achour A, Arkesteijn SJG et al (2022) The hemoglobinopathies, molecular disease mechanisms and diagnostics. Int J Lab Hematol 44:28–36. https://doi.org/10.1111/ijlh.13885
Chaichompoo P, Svasti S, Smith DR (2022) The roles of mitophagy and autophagy in ineffective erythropoiesis in β-thalassemia. Int J Mol Sci 23:10811. https://doi.org/10.3390/ijms23181081
Bou-Fakhredin R, De Franceschi L, Motta I et al (2022) Redox balance in β-thalassemia and sickle cell disease: a love and hate relationship. Antioxidants (Basel) 11:967. https://doi.org/10.3390/antiox11050967
Kato GJ, Piel FB, Reid CD et al (2018) Sickle cell disease. Nat Rev Dis Prim 4:1–22. https://doi.org/10.1038/nrdp.2018.10
Iolascon A, Andolfo I, Russo R (2020) Congenital dyserythropoietic anemias. Blood 136:1274–1283. https://doi.org/10.1182/blood.2019000948
Schieber M, Chandel NS (2014) ROS function in redox signaling and oxidative stress. Curr Biol 24:453–462. https://doi.org/10.1016/j.cub.2014.03.034
Fibach E, Rachmilewitz E (2008) The role of oxidative stress in hemolytic anemia. Curr Mol Med 8:609–619. https://doi.org/10.2174/156652408786241384
Massaccesi L, Galliera E, Romanelli MMC (2020) Erythrocytes as markers of oxidative stress related pathologies. Mech Ageing Dev 191:111333. https://doi.org/10.1016/j.mad.2020.111333
Fujii J, Homma T, Kobayashi S et al (2021) Erythrocytes as a preferential target of oxidative stress in blood. Free Radic Res 55:562–580. https://doi.org/10.1080/10715762.2021.1873318
Voskou S, Aslan M, Fanis P et al (2015) Oxidative stress in β-thalassaemia and sickle cell disease. Redox Biol 6:226–239. https://doi.org/10.1016/j.redox.2015.07.018
Rifkind JM, Mohanty JG, Nagababu E (2015) The pathophysiology of extracellular hemoglobin associated with enhanced oxidative reactions. Front Physiol 5:1–7. https://doi.org/10.3389/fphys.2014.00500
Kingsley PD, Greenfest-Allen E, Frame JM et al (2013) Ontogeny of erythroid gene expression. Blood 121:e5–e13. https://doi.org/10.1182/blood-2012-04-422394
Da Cunha AF, Brugnerotto AF, Duarte AS et al (2010) Global gene expression reveals a set of new genes involved in the modification of cells during erythroid differentiation. Cell Prolif 43:297–309. https://doi.org/10.1111/j.1365-2184.2010.00679.x
Möller MN, Orrico F, Villar SF et al (2023) Oxidants and antioxidants in the redox biochemistry of human red blood cells. ACS Omega 8:147–168. https://doi.org/10.1021/acsomega.2c06768
Neumann CA, Krause DS, Carman CV et al (2003) Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature 424:561–565. https://doi.org/10.1038/nature01819
Lee T, Kim S, Yu S et al (2003) Peroxiredoxin II is essential for sustaining life span of erythrocytes in mice. Blood 101:5033–5038. https://doi.org/10.1182/blood-2002-08-2548.Supported
Han YH, Kim SU, Kwon TH et al (2012) Peroxiredoxin II is essential for preventing hemolytic anemia from oxidative stress through maintaining hemoglobin stability. Biochem Biophys Res Commun 426:427–432. https://doi.org/10.1016/j.bbrc.2012.08.113
Manevich Y, Sweitzer T, Pak JH et al (2002) 1-Cys peroxiredoxin overexpression protects cells against phospholipid peroxidation-mediated membrane damage. Proc Natl Acad Sci USA 99:11599–11604. https://doi.org/10.1073/pnas.182384499
Phelan SA, Wang X, Wallbrandt P et al (2003) Overexpression of Prdx6 reduces H2O2 but does not prevent diet-induced atherosclerosis in the aortic root. Free Radic Biol Med 35:1110–1120. https://doi.org/10.1016/S0891-5849(03)00462-3
Kümin A, Huber C, Rülicke T et al (2006) Peroxiredoxin 6 is a potent cytoprotective enzyme in the epidermis. Am J Pathol 169:1194–1205. https://doi.org/10.2353/ajpath.2006.060119
Walsh B, Pearl A, Suchy S et al (2013) Overexpression of Prdx6 and resistance to peroxide-induced death in Hepa1-6 cells: Prdx suppression increases apoptosis. Redox Rep 14:275–284. https://doi.org/10.1179/135100009X12525712409652
Wang X, Phelan SA, Forsman-semb K et al (2003) Mice with targeted mutation of peroxiredoxin 6 develop normally but are susceptible to oxidative stress *. J Biol Chem 278:25179–25190. https://doi.org/10.1074/jbc.M302706200
Sue GR, Ho ZC, Kim K (2005) Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med 38:1543–1552. https://doi.org/10.1016/j.freeradbiomed.2005.02.026
Zhang B, Wang Y, Su Y (2009) Peroxiredoxins, a novel target in cancer radiotherapy. Cancer Lett 286:154–160. https://doi.org/10.1016/j.canlet.2009.04.043
Szabó C, Ischiropoulos H, Radi R (2007) Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov 6:662–680. https://doi.org/10.1038/nrd2222
Oliveira MA, Discola KF, Alves SV et al (2010) Insights into the specificity of thioredoxin reductase-thioredoxin interactions. A structural and functional investigation of the yeast thioredoxin system. Biochemistry 49:3317–3326. https://doi.org/10.1021/bi901962p
Poole LB, Hall A, Nelson KJ (2012) Overview of peroxiredoxins in oxidant defense and redox regulation. Curr Protoc Toxicol 1–20. https://doi.org/10.1002/0471140856.tx0709s49
Hofmann B, Hecht H, Flohé L (2002) Peroxiredoxins. Biol Chem 383:347–364. https://doi.org/10.1515/BC.2002.040
Copley SD, Novak WRP, Babbitt PC (2004) Divergence of function in the thioredoxin fold suprafamily: evidence for evolution of peroxiredoxins from a thioredoxin-like ancestor. Biochemistry 3:13981–13995. https://doi.org/10.1021/bi048947r
Trivelli X, Krimm I, Ebel C et al (2003) Characterization of the yeast peroxiredoxin Ahp1 in its reduced active and overoxidized inactive forms using NMR. Biochemistry 42:14139–14149. https://doi.org/10.1021/bi035551r
Wood ZA (2003) Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science (80) 300:650–3. https://doi.org/10.1126/science.1080405
Soito L, Williamson C, Knutson ST et al (2011) PREX: PeroxiRedoxin classification indEX, a database of subfamily assignments across the diverse peroxiredoxin family. Nucleic Acids Res 39:332–337. https://doi.org/10.1093/nar/gkq1060
Barranco-medina S, Lázaro J, Dietz K (2009) The oligomeric conformation of peroxiredoxins links redox state to function. FEBS Lett 583:1809–1816. https://doi.org/10.1016/j.febslet.2009.05.029
Poole LB (2007) The catalytic mechanism of peroxiredoxins. Subcell Biochem 44:61–81. https://doi.org/10.1007/978-1-4020-6051-9_4
Rhee SG, Woo HA, Kil IS, Bae SH (2012) Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J Biol Chem 287:4403–4410. https://doi.org/10.1074/jbc.R111.283432
Monteiro G, Horta BB, Pimenta DC et al (2007) Reduction of 1-Cys peroxiredoxins by ascorbate changes the thiol-specific antioxidant paradigm, revealing another function of vitamin C. Proc Natl Acad Sci USA 104:4886–4891. https://doi.org/10.1073/pnas.0700481104
Rhee SG, Woo HA (2011) Multiple functions of peroxiredoxins: peroxidases, sensors and regulators of the intracellular messenger H2O2, and protein chaperones. Antioxidants Redox Signal 15:781–794. https://doi.org/10.1089/ars.2010.3393
Jang HH, Lee KO, Chi YH et al (2004) Two enzymes in one: two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell 117:625–635. https://doi.org/10.1016/j.cell.2004.05.002
Vivancos AP, Castillo EA, Biteau B et al (2005) A cysteine-sulfinic acid in peroxiredoxin regulates H2O2-sensing by the antioxidant Pap1 pathway. Proc Natl Acad Sci U S A 102:1–6. https://doi.org/10.1073/pnas.0503251102
Hopkins BL, Neumann CA (2019) Redoxins as gatekeepers of the transcriptional oxidative stress response. Redox Biol 21:2213–2317. https://doi.org/10.1016/j.redox.2019.101104
Demasi APD, Pereira GAG, Netto LES (2001) Cytosolic thioredoxin peroxidase I is essential for the antioxidant defense of yeast with dysfunctional mitochondria. FEBS Lett 509:430–434. https://doi.org/10.1016/s0014-5793(01)03215-x
Hall A, Karplus PA, Poole LB (2009) Typical 2-Cys peroxiredoxins – structures, mechanisms and functions. FEBS J 276:2469–2477. https://doi.org/10.1111/j.1742-4658.2009.06985.x
Santos MC, Breyer CA, Schultz L et al (2017) Saccharomyces cerevisiae peroxiredoxins in biological processes: antioxidant defense, signal transduction, circadian rhythm, and more. In: Lucas C, Pais C (eds) Old Yeasts - New Questions. InTech. 119–37
Fujii J, Ikeda Y (2002) Advances in our understanding of peroxiredoxin, a multifunctional, mammalian redox protein. Redox Rep 7:123–130. https://doi.org/10.1179/135100002125000352
Kumsta C, Jakob U (2009) Redox-regulated chaperones. Biochemistry 48:4666–4676. https://doi.org/10.1021/bi9003556
Mu ZM, Yin XY, Prochownik EV (2002) Pag, a putative tumor suppressor, interacts with the Myc Box II domain of c-Myc and selectively alters its biological function and target gene expression. J Biol Chem 277:43175–43184. https://doi.org/10.1074/jbc.M206066200
Graves JA, Metukuri M, Scott D et al (2009) Regulation of reactive oxygen species homeostasis by peroxiredoxins and c-Myc. J Biol Chem 284:6520–6529. https://doi.org/10.1074/jbc.M807564200
Hansen JM, Moriarty-Craige S, Jones DP (2007) Nuclear and cytoplasmic peroxiredoxin-1 differentially regulate NF-κB activities. Free Radic Biol Med 43:282–288. https://doi.org/10.1016/j.freeradbiomed.2007.04.029
Wang X, He S, Sun J et al (2010) Selective association of peroxiredoxin 1 with genomic DNA and COX-2 upstream promoter elements in estrogen receptor negative breast cancer cells. Mol Biol Cell 21:2987–2995. https://doi.org/10.1091/mbc.E10
Park S, Yu X, Ip C et al (2007) Peroxiredoxin 1 interacts with androgen receptor and enhances its transactivation. Cancer Res 67:9294–9304. https://doi.org/10.1158/0008-5472.CAN-07-0651
Rhee SG, Kil IS (2017) Multiple functions and regulation of mammalian peroxiredoxins. Annu Rev Biochem 86:749–775. https://doi.org/10.1146/annurev-biochem-060815-014431
Neumann CA, Cao J, Manevich Y (2009) Peroxiredoxin 1 and its role in cell signaling. Cell Cycle 8:4072–4078. https://doi.org/10.4161/cc.8.24.10242
Chang TS, Jeong W, Choi SY et al (2002) Regulation of peroxiredoxin I activity by Cdc2-mediated phosphorylation. J Biol Chem 277:25370–25376. https://doi.org/10.1074/jbc.M110432200
Chae HZ, Uhm TB, Rhee SG (1994) Dimerization of thiol-specific antioxidant and the essential role of cysteine 47. Proc Natl Acad Sci U S A 91:7022–7026. https://doi.org/10.1073/pnas.91.15.7022
Yang KS, Kang SW, Woo HA et al (2002) Inactivation of human peroxiredoxin I during catalysis as the result of the oxidation of the catalytic site cysteine to cysteine-sulfinic acid. J Biol Chem 277:38029–38036. https://doi.org/10.1074/jbc.M206626200
Jung CL, Choi HI, Yu SP et al (2008) Irreversible oxidation of the active-site cysteine of peroxiredoxin to cysteine sulfonic acid for enhanced molecular chaperone activity. J Biol Chem 283:28873–28880. https://doi.org/10.1074/jbc.M804087200
Rhee SG, Jeong W, Chang TS, Woo HA (2007) Sulfiredoxin, the cysteine sulfinic acid reductase specific to 2-Cys peroxiredoxin: its discovery, mechanism of action, and biological significance. Kidney Int 72:3–8. https://doi.org/10.1038/sj.ki.5002380
Sadowska-Bartosz I, Bartosz G (2023) Peroxiredoxin 2: an important element of the antioxidant defense of the erythrocyte. Antioxidants 12:1–30. https://doi.org/10.3390/antiox12051012
Lei XG, Zhu JH, Cheng WH et al (2015) Paradoxical roles of antioxidant enzymes: basic mechanisms and health implications. Physiol Rev 96:307–364. https://doi.org/10.1152/physrev.00010.2014
Woo HA, Yim SH, Shin DH et al (2010) Inactivation of peroxiredoxin I by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell 140:517–528. https://doi.org/10.1016/j.cell.2010.01.009
Cao J, Schulte J, Knight A et al (2009) Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. EMBO J 28:1505–1517. https://doi.org/10.1038/emboj.2009.101
Ding C, Fan X, Wu G (2017) Peroxiredoxin 1 – an antioxidant enzyme in cancer. J Cell Mol Med 21:193–202. https://doi.org/10.1111/jcmm.12955
Stambolic V, Suzuki A, De la Pompa JL et al (1998) Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95:29–39. https://doi.org/10.1016/S0092-8674(00)81780-8
Lee SR, Yang KS, Kwon J et al (2002) Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem 277:20336–20342. https://doi.org/10.1074/jbc.M111899200
Huo YY, Li G, Duan RF et al (2008) PTEN deletion leads to deregulation of antioxidants and increased oxidative damage in mouse embryonic fibroblasts. Free Radic Biol Med 44:1578–1591. https://doi.org/10.1016/j.freeradbiomed.2008.01.013
Nassour H, Wang Z, Saad A et al (2016) Peroxiredoxin 1 interacts with and blocks the redox factor APE1 from activating interleukin-8 expression. Sci Rep 6:1–17. https://doi.org/10.1038/srep29389
Xanthoudakis S, Miao G, Wang F et al (1992) Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J 11:3323–3335. https://doi.org/10.1002/j.1460-2075.1992.tb05411.x
Ema M (1999) Molecular mechanisms of transcription activation by HLF and HIF1alpha in response to hypoxia: their stabilization and redox signal-induced interaction with CBP/p300. EMBO J 18:1905–1914. https://doi.org/10.1093/emboj/18.7.1905
Tell G, Pellizzari L, Cimarosti D et al (1998) Ref-1 controls Pax-8 DNA-binding activity. Biochem Biophys Res Commun 252:178–183. https://doi.org/10.1006/bbrc.1998.9548
Gaiddon C, Moorthy NC, Prives C (1999) Ref-1 regulates the transactivation and pro-apoptotic functions of p53 in vivo. EMBO J 18:5609–5621. https://doi.org/10.1093/emboj/18.20.5609
Bapat A, Fishel ML, Kelley MR (2009) Going ape as an approach to cancer therapeutics. Antioxidants Redox Signal 11:651–667. https://doi.org/10.1089/ars.2008.2218
De Larco JE, Wuertz BRK, Rosner KA et al (2001) A potential role for interleukin-8 in the metastatic phenotype of breast carcinoma cells. Am J Pathol 158:639–646. https://doi.org/10.1016/S0002-9440(10)64005-9
Chen WT, Ebelt ND, Stracker TH et al (2015) ATM regulation of IL-8 links oxidative stress to cancer cell migration and invasion. Elife 4:e07270. https://doi.org/10.7554/eLife.07270
Romanello KS, Teixeira KKL, Silva JPMO et al (2018) Global analysis of erythroid cells redox status reveals the involvement of Prdx1 and Prdx2 in the severity of beta thalassemia. PLoS ONE 13:1–19. https://doi.org/10.1371/journal.pone.0208316
Maia de Oliveira da Silva JP, Brugnerotto AF, S. Romanello K et al (2019) Global gene expression reveals an increase of HMGB1 and APEX1 proteins and their involvement in oxidative stress, apoptosis and inflammation pathways among beta-thalassaemia intermedia and major phenotypes. Br J Haematol 186:608–619. https://doi.org/10.1111/bjh.16062
Bernardo VS, Torres FF, Paula CP de et al (2022) Potential cytoprotective and regulatory effects of ergothioneine on gene expression of proteins involved in erythroid adaptation mechanisms and redox pathways in. Genes (Basel) 13:2368. https://doi.org/10.3390/genes13122368
Menon V, Ghaffari S (2021) Erythroid enucleation: a gateway into a “bloody” world. Exp Hematol 95:13–22. https://doi.org/10.1016/j.exphem.2021.01.001
Menon V, Ghaffari S (2018) Transcription factors FOXO in the regulation of homeostatic hematopoiesis. Curr Opin Hematol 25:290–298. https://doi.org/10.1097/MOH.0000000000000441
Hopkins BL, Nadler M, Skoko JJ et al (2018) A Peroxidase Peroxiredoxin 1-Specific Redox Regulation of the Novel FOXO3 microRNA Target let-7. Antioxidants Redox Signal 28:62–77. https://doi.org/10.1089/ars.2016.6871
Troussicot L, Burmann BM, Molin M (2021) Structural determinants of multimerization and dissociation in 2-Cys peroxiredoxin chaperone function. Structure 29:640–654. https://doi.org/10.1016/j.str.2021.04.007
Jarvis RM, Hughes SM, Ledgerwood EC (2012) Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radic Biol Med 53:1522–1530. https://doi.org/10.1016/j.freeradbiomed.2012.08.001
Gertz M, Fischer F, Leipelt M et al (2009) Identification of Peroxiredoxin 1 as a novel interaction partner for the lifespan regulator protein p66Shc. Aging (Albany NY) 1:254–265. https://doi.org/10.18632/aging.100017
Kim SY, Kim TJ, Lee K-Y (2008) A novel function of peroxiredoxin 1 Prx-1 in apoptosis signal-regulating kinase 1 ASK1. FEBS Lett 582(582):1913–1918. https://doi.org/10.1016/j.febslet.2008.05.015
Saitoh M, Nishitoh H, Fujii M et al (1998) Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J 17:2596–2606. https://doi.org/10.1039/DT9890001143
Kim YJ, Lee WS, Ip C et al (2006) Prx1 suppresses radiation-induced c-Jun NH2-terminal kinase signaling in lung cancer cells through interaction with the glutathione S-transferase Pi/c-Jun NH2-terminal kinase complex. Cancer Res 66:7136–7142. https://doi.org/10.1158/0008-5472.CAN-05-4446
Ishii T, Warabi E, Yanagawa T (2012) Novel roles of peroxiredoxins in inflammation, cancer and innate immunity. J Clin Biochem Nutr 50:91–105. https://doi.org/10.3164/jcbn.11-109
Finetti F, Savino MT, Baldari CT (2009) Positive and negative regulation of antigen receptor signaling by the Shc family of protein adapters. Immunol Rev 232:115–134. https://doi.org/10.1111/j.1600-065X.2009.00826.x
Giorgio M, Migliaccio E, Orsini F et al (2005) Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 122:221–233. https://doi.org/10.1016/j.cell.2005.05.011
Centis F, Tabellini L, Lucarelli G et al (2000) The importance of erythroid expansion in determining the extent of apoptosis in erythroid precursors in patients with β-thalassemia major. Blood 96:3624–3629. https://doi.org/10.1182/blood.V96.10.3624
Pootrakul P, Sirankapracha P, Hemsorach S et al (2000) A correlation of erythrokinetics, ineffective erythropoiesis, and erythroid precursor apoptosis in Thai patients with thalassemia. Blood 96:2606–2612. https://doi.org/10.1182/blood.V96.7.2606
Mathias LA, Fisher TC, Zeng L et al (2000) Ineffective erythropoiesis in β-thalassemia major is due to apoptosis at the polychromatophilic normoblast stage. Exp Hematol 28:1343–1353. https://doi.org/10.1016/s0301-472x(00)00555-5
Tamary BH, Shalev H, Luria D et al (1996) Clinical Features and studies of erythropoiesis in Israeli Bedouins with congenital dyserythropoietic anemia type I. Blood 87:1763–1770. https://doi.org/10.1182/blood.V87.5.1763.1763
Yehia L, Niazi F, Ni Y et al (2015) Germline heterozygous variants in SEC23B are associated with Cowden syndrome and enriched in apparently sporadic thyroid cancer. Am J Hum Genet 97:661–676. https://doi.org/10.1016/j.ajhg.2015.10.001
Kim E, Kim Y, Yang JY, Jang HH (2022) Prx1 regulates thapsigargin-mediated UPR activation and apoptosis. Genes (Basel) 13:2033. https://doi.org/10.3390/genes13112033
Schröder E, Littlechild JA, Lebedev AA et al (2000) Crystal structure of decameric 2-Cys peroxiredoxin from human erythrocytes at 1.7 Å resolution. Structure 8:605–615. https://doi.org/10.1016/S0969-2126(00)00147-7
Plishker GA, Chevalier D, Seinsoth L, Moore RB (1992) Calcium-activated potassium transport and high molecular weight forms of calpromotin. J Biol Chem 267:21839–21843. https://doi.org/10.1016/s0021-9258(19)36688-8
Moore RB, Mankad MV, Shriver SK et al (1991) Reconstitution of Ca2+-dependent K+ transport in erythrocyte membrane vesicles requires a cytoplasmic protein. J Biol Chem 266:18964–18968. https://doi.org/10.1016/s0021-9258(18)55157-7
Harris JR (1969) Some negative contrast staining features of a protein from erythrocite ghosts. J Mol Biol 46:329–335. https://doi.org/10.1016/0022-2836(69)90425-2
Harris JR, Naeem I (1981) Further studies on the characterization of cylindrin and torin, two extrinsic proteins of the erythrocyte membrane. BBA - Protein Struct 670:285–290. https://doi.org/10.1016/0005-2795(81)90021-0
Matte A, Bertoldi M, Mohandas N et al (2013) Membrane association of peroxiredoxin-2 in red cells is mediated by the N-terminal cytoplasmic domain of band 3. Free Radic Biol Med 55:27–35. https://doi.org/10.1016/j.freeradbiomed.2012.10.543
Chu H, Breite A, Ciraolo P et al (2008) Characterization of the deoxyhemoglobin binding site on human erythrocyte band 3: implications for O2 regulation of erythrocyte properties. Blood 111:932–938. https://doi.org/10.1182/blood-2007-07-100180
Cha MK, Yun CH, Kim IH (2000) Interaction of human thiol-specific antioxidant protein 1 with erythrocyte plasma membrane. Biochemistry 39:6944–6950. https://doi.org/10.1021/bi000034j
Cha MK, Kim IH (1996) Purification and characterization of thiol-specific antioxidant protein from human liver: a Mer5-like human isoenzyme. J Biochem Mol Biol 29:236–240
Mattè A, Federti E, Tibaldi E et al (2021) Tyrosine phosphorylation modulates peroxiredoxin-2 activity in normal and diseased red cells. Antioxidants 10:1–16. https://doi.org/10.3390/antiox10020206
Rocha S, Costa E, Coimbra S et al (2009) Linkage of cytosolic peroxiredoxin 2 to erythrocyte membrane imposed by hydrogen peroxide-induced oxidative stress. Blood Cells Mol Dis 43:68–73. https://doi.org/10.1016/j.bcmd.2009.03.002
Biondani A, Turrini F, Carta F et al (2008) Heat-shock protein-27, -70 and peroxiredoxin-II show molecular chaperone function in sickle red cells: Evidence from transgenic sickle cell mouse model. Proteomics Clin Appl 2:706–719. https://doi.org/10.1002/prca.200780058
Rocha S, Vitorino RMP, Lemos-Amado FM et al (2008) Presence of cytosolic peroxiredoxin 2 in the erythrocyte membrane of patients with hereditary spherocytosis. Blood Cells, Mol Dis 41:5–9. https://doi.org/10.1016/j.bcmd.2008.02.008
Moore RB, Shriver SK, Jenkins LD et al (1997) Calpromotin, a cytoplasmic protein, is associated with the formation of dense cells in sickle cell anemia. Am J Hematol 56:100–106. https://doi.org/10.1002/(SICI)1096-8652(199710)56:2%3c100::AID-AJH5%3e3.0.CO;2-2
Matte A, Low PS, Turrini F et al (2010) Peroxiredoxin-2 expression is increased in β-thalassemic mouse red cells but is displaced from the membrane as a marker of oxidative stress. Free Radic Biol Med 49:457–466. https://doi.org/10.1016/j.freeradbiomed.2010.05.003
Bayer SB, Low FM, Hampton MB, Winterbourn CC (2016) Interactions between peroxiredoxin 2, hemichrome and the erythrocyte membrane. Free Radic Res 50:1329–1339. https://doi.org/10.1080/10715762.2016.1241995
Ma Q, An L, Tian H et al (2019) Interactions between human hemoglobin subunits and peroxiredoxin 2. Front Biosci - Landmark Ed 24:1085–1096. https://doi.org/10.2741/4770
Basu A, Chakrabarti A (2015) Hemoglobin interacting proteins and implications of spectrin hemoglobin interaction. J Proteomics 128:469–475. https://doi.org/10.1016/j.jprot.2015.06.014
Rinalducci S, D’Amici GM, Blasi B, Zolla L (2011) Oxidative stress-dependent oligomeric status of erythrocyte peroxiredoxin II (PrxII) during storage under standard blood banking conditions. Biochimie 93:845–853. https://doi.org/10.1016/j.biochi.2011.02.005
Matte A, De Franceschi L (2019) Oxidation and erythropoiesis. Curr Opin Hematol 26:145–151. https://doi.org/10.1097/MOH.0000000000000495
Johnson RM, Ho Y-S, Yu D-Y et al (2010) The effect of disruption of genes for peroxiredoxin-2, glutathione peroxidase-1 and catalase on erythrocyte oxidative metabolism. Free Radic Biol Med 48:1–19. https://doi.org/10.1016/j.freeradbiomed.2009.11.021.The
De FL, Bertoldi M, De FL et al (2011) Oxidative stress modulates heme synthesis and induces peroxiredoxin-2 as a novel cytoprotective response in β-thalassemic erythropoiesis. Haematologica 96:1595–1604. https://doi.org/10.3324/haematol.2011.043612
Matte A, De Falco L, Iolascon A et al (2015) The interplay between peroxiredoxin-2 and nuclear factor-erythroid 2 is important in limiting oxidative mediated dysfunction in β-thalassemic erythropoiesis. Antioxidants Redox Signal 23:1284–1297. https://doi.org/10.1089/ars.2014.6237
Emberesh M, Giger Seu K, Emberesh S et al (2018) Peroxiredoxin II (PRDX2) is a novel candidate gene for congenital dyserythropoietic anemia. Blood 132:3605–3605. https://doi.org/10.1182/blood-2018-99-120056
Ghashghaeinia M, Toulany M, Saki M et al (2012) Roles of the NFκB and glutathione pathways in mature human erythrocytes. Cell Mol Biol Lett 17:11–20. https://doi.org/10.2478/s11658-011-0032-x
Cho C, Yoon HJ, Kim JY et al (2014) Circadian rhythm of hyperoxidized peroxiredoxin II is determined by hemoglobin autoxidation and the 20S proteasome in red blood cells. PNAS 111:12043–12048. https://doi.org/10.1073/pnas.1401100111
O-Neill JS, Reddy AB (2011) Circadian clocks in human red blood cells. Nature 469:498–504. https://doi.org/10.1038/nature09702
Urbinati F, Madigan C, Malik P (2006) Pathophysiology and therapy for haemoglobinopathies. Part II: Thalassaemias. Expert Rev Mol Med 8:1–26. https://doi.org/10.1017/S1462399406010805
Dunn LL, Rahmanto YS, Richardson DR (2007) Iron uptake and metabolism in the new millennium. Trends Cell Biol 17:93–100. https://doi.org/10.1016/j.tcb.2006.12.003
Muckenthaler MU, Rivella S, Hentze MW, Galy B (2017) A red carpet for iron metabolism. Cell 168:344–361. https://doi.org/10.1016/j.cell.2016.12.034
Sobotta MC, Liou W, Stöcker S et al (2015) Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat Chem Biol 11:64–70. https://doi.org/10.1038/nchembio.1695
Matte A, De Falco L, Federti E et al (2018) Peroxiredoxin-2: a novel regulator of iron homeostasis in ineffective erythropoiesis
Talwar D, Messens J, Dick TP (2020) A role for annexin A2 in scaffolding the peroxiredoxin 2–STAT3 redox relay complex. Nat Commun 11:1–11. https://doi.org/10.1038/s41467-020-18324-9
Mo Y, Feinstein SI, Manevich Y et al (2003) 1-Cys peroxiredoxin knock-out mice express mRNA but not protein for a highly related intronless. FEBSLetters 555:192–198. https://doi.org/10.1016/S0014-5793(03)01199-2
Fisher AB (2018) The phospholipase A2 activity of peroxiredoxin 6. J Lipid Res 59:1132–1147. https://doi.org/10.1194/jlr.R082578
Cho C, Lee JS, Gladwin MT, Rhee SG (2010) Hydroxyurea-induced expression of glutathione peroxidase 1 in red blood cells of individuals with sickle cell anemia. Antioxid Redox Signal 13:1–11
Kubo E, Chhunchha B, Singh P et al (2017) Sulforaphane reactivates cellular antioxidant defense by inducing Nrf2/ARE/Prdx6 activity during aging and oxidative stress. Sci Rep 7:1–17. https://doi.org/10.1038/s41598-017-14520-8
Panda H, Keleku-Lukwete N, Kuga A et al (2019) Dietary supplementation with sulforaphane attenuates liver damage and heme overload in a sickle cell disease murine model. Exp Hematol 77:51-60.e1. https://doi.org/10.1016/j.exphem.2019.08.001
Lu B, Chen X, bing, Hong Y cai, et al (2019) Identification of PRDX6 as a regulator of ferroptosis. Acta Pharmacol Sin 40:1334–1342. https://doi.org/10.1038/s41401-019-0233-9
Saki N, Abroun S, Salari F et al (2015) Molecular aspects of bone resorption in β-thalassemia major. Cell J 17:193–200
Stuhlmeier KM, Kao JJ, Wallbrandt P et al (2003) Antioxidant protein 2 prevents methemoglobin formation in erythrocyte hemolysates. Eur J Biochem 270:334–341. https://doi.org/10.1046/j.1432-1033.2003.03393.x
López-Grueso MJ, Lagal DJ, García-Jiménez ÁF, et al (2020) Knockout of PRDX6 induces mitochondrial dysfunction and cell cycle arrest at G2/M in HepG2 hepatocarcinoma cells. Redox Biol 37. https://doi.org/10.1016/j.redox.2020.101737
Jia W, Dong C, Li B (2023) Anti-oxidant and pro-oxidant effects of peroxiredoxin 6: a potential target in respiratory diseases. 1–14
Acknowledgements
We thank the Servier Medical Art Commons Attribution 3.0 Unported License (http://smart.servier.com) for the image used to production of the figures presented in this article.
Funding
This work was supported by FAPESP (2011/50358-3) and CAPES. Author JPMOS has received research support from CAPES.
Author information
Authors and Affiliations
Contributions
All authors contributed to the review conception and design. Carla Peres de Paula wrote the first draft of the manuscript. CPP, KSR, JPMOS, VSB, and FFT reviewed the literature. VSB, FFT, AFC, and DGHS critically revised the work. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors concur with the submission.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
de Paula, C.P., de Oliveira da Silva, J.P.M., Romanello, K.S. et al. Peroxiredoxins in erythrocytes: far beyond the antioxidant role. J Mol Med 101, 1335–1353 (2023). https://doi.org/10.1007/s00109-023-02368-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-023-02368-7