
Abstract
Elevated levels of the anti-apoptotic BCL2 protein associate with favourable outcome in breast cancer. We investigated whether executioner caspase activation downstream of mitochondrial apoptosis was associated with, or independent, of BCL2’s prognostic signature in breast cancer. Levels of pro- and anti-apoptotic BCL2 family proteins were quantified in triple negative breast cancer (TNBC) samples and utilised to calculate BCL2 profiles of 845 breast cancer patients. Biomarkers including single apoptosis proteins and network-enriched apoptosis system signatures were evaluated using uni- and multi-variate Cox-models. In both TNBC and non-TNBC breast cancer, the anti-apoptotic BCL2 protein was particularly abundant when compared to other solid tumours. High BCL2 protein levels were prognostic of favourable outcome across all breast cancers (HR 0.4, 95% CI 0.2–0.6, Wald p < 0.0001). Although BCL2 and cleaved caspase-7 levels were negatively correlated, levels of cleaved caspase-7 were also associated with favourable outcome (HR 0.4, 95% CI 0.3–0.7, Wald p = 0.001). A combination of low BCL2 and low cleaved caspase-7 protein levels was highly prognostic of unfavourable outcome across all breast cancers (HR 11.29, 95% CI 2.20–58.23, Wald p = 0.01). A combination of BCL2 and cleaved caspase-7 levels is a promising prognostic biomarker in breast cancer patients.
Key message
-
BCL2 levels are elevated in breast cancer where they are marker of good prognosis.
-
BCL2 and active caspase levels correlate negatively; yet, active caspases indicate good outcome.
-
Low BCL2 and low caspase-7 are highly prognostic of unfavourable outcome across all breast cancers.
-
BCL2 levels indicate molecular subtype and tumour proliferation status in breast cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The ability of cancer cells to evade apoptosis is believed to play a fundamental role in carcinogenesis [1] and resistance to chemo- and radiotherapy [2]. The BCL-2 family of proteins is gatekeepers of the so-called mitochondrial apoptosis pathway and either promote or inhibit apoptosis [3]. Caspase-3 and caspase-7 represent the main executioners of mitochondrial apoptosis [4]. Cancer cells overexpressing anti-apoptotic BCL2 family proteins including BCL2, BCL(X)L and MCL1 have a reduced ability to activate the mitochondrial apoptosis pathway and show an increased resistance to chemo- and radiotherapy [5]. Due to the complexity of BCL2 protein interactions and redundancies in the BCL-2 signalling pathway, individual BCL-2 family proteins only have limited prognostic power in most cancers [6, 7]. For this reason, we recently developed the computational model, DR_MOMP, that describes BCL-2 signalling quantitatively and holistically [8]. Both preclinical and clinical studies demonstrated that such systems analysis was capable of delivering a powerful prognostic tool, superior to single or combinatorial biomarker approaches [8,9,10]. DR_MOMP is an ordinary differential equation-based computational model of BAX- and BAK-controlled mitochondrial outer membrane permeabilisation (MOMP) during apoptosis. It uses levels of pro-apoptotic BAK and BAX and anti-apoptotic BCL2, BCL(X)L and MCL1 as input, and delivers a single value (‘stress’ dose) that indicates the estimated sensitivity of cancer cells to activate the mitochondrial apoptosis pathway [8]. Previous studies employing quantitative Western blotting [8, 11] or reverse protein phase array (RPPA)-based protein quantification [9] demonstrated a highly prognostic association of high stress doses with unfavourable clinical outcome in patients with colorectal cancer, independent of other (histo)pathological or molecular risk factors including TNM staging [9].
Expression levels of the anti-apoptotic BCL2 protein, as a single biomarker or as part of transcriptome-based risk scores, have been repeatedly identified as a prognostic biomarker in breast cancer, with high levels of BCL2 counter-intuitively associated with favourable prognosis in patients [12, 13]. High BCL2 expression is particularly observed in oestrogen- and progesterone receptor (ER/PR)-positive breast cancer [14] that shows better overall survival (OS) compared to ER/PR-negative or triple negative breast cancer (TNBC) [15]. BCL-2 has been identified as a transcriptional target of oestrogen signalling [16]. Of note, BCL2 signalling is increasingly linked to other non-apoptotic processes, including regulation of bioenergetics, cell proliferation and gene expression [17,18,19,20]. The aim of this study was therefore to quantitatively explore whether individual BCL2 proteins or a systems analysis of BCL-2 family proteins delivers prognostic signatures in breast cancer, whether such signatures are breast cancer subtype-specific, and whether these signatures are related to executioner caspase activation as a read-out of the mitochondrial apoptosis pathway.
Materials and methods
Irish Cancer Society BREAST-PREDICT/National Breast Cancer Resource cohort
Fresh frozen primary cancer samples (n = 25) of non-metastatic female TNBC patients (5-year disease-related survival 76% and recurrence rate 32%) were obtained from Beaumont Hospital, Dublin, Ireland, between 2004 and 2013. Nineteen samples remained after quality control. Pathological stage was determined using the American Joint Committee on Cancer (AJCC) TNM classification system. Clinical follow-up [disease-free survival (DFS) and breast cancer-related overall survival (OS)] was obtained through medical record review. Informed consents were collected following ethical approval from Beaumont Hospital Medical Research Ethics Committee. Patients’ clinical characteristics are provided in supplementary tables S1 and S2.
The Cancer Genome Atlas (TCGA) cohort
The data for protein levels (reverse phase protein array, RPPA), mRNA expressions (mRNA SeqV2), somatic mutations (IlluminaHiSeq) and clinical follow-up of n = 845 patients were accessed from The Cancer Genome Atlas (TCGA) project (20 May 2016). Seventy-four patients were removed from the survival analysis due to history of another malignancy (n = 53), follow-up time≤ 0 days (n = 10) or insufficient information on tumour staging (n = 13). Detailed patient characteristics are provided in supplementary tables S2 and S3.
BCL2 profiling by quantitative Western blotting
Quantitative Western blotting of fresh-frozen tissue protein lysates was performed as previously described using HeLa cells as a standard. Lysates of patient samples were loaded in parallel to HeLa cell lysates. Absolute concentrations of BAK (1109 nM), BAX (519 nM), BCL2 (239 nM), BCL(X)L (110 nM) and MCL1 (83 nM) were previously determined by comparing protein levels in HeLa cells with calibration curves generated using purified proteins [8]. Protein levels of patient tissue were normalised to the known protein concentrations in HeLa cells after correcting with loading-controls (actin and tubulin) as previously described [8]. The HeLa cells were purchased from the American Type Culture Collection (LGC Standards). HeLa cells were collected and lysed in RIPA buffer (150 mM NaCl, 1.0% IGEPAL® CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0, Sigma protease and phosphatase inhibitors mix 1:100). Patient tissue was lysed in 500 μL ice-cold buffer [50 mmol/L HEPES (pH 7.5), 150 mmol/L NaCl, 5 mmol/L Na-EDTA] and protease inhibitor cocktail and homogenised on ice. Samples were centrifuged at 14,000×g for 10 min, supernatant collected and stored at − 80 °C. All densitometry analyses were performed with ImageJ [8, 11]. Primary antibodies to MCL1 (1:1000; BD Biosciences), BCL2 (1:100; Santa Cruz Biotechnology), BCL(X)L (1:250; Santa Cruz Biotechnology), anti-β-actin (A1978, Sigma Aldrich, St. Louis, MO, USA, 1 in 7500) and α-tubulin (1:5000; Sigma Aldrich) were mouse monoclonal. Antibodies to BAK (1:250; Santa Cruz Biotechnology) and BAX (1:1000; Upstate Biotechnology) were rabbit polyclonal. The horseradish peroxidase (HRP)-conjugated secondary antibodies were from Jackson ImmunoResearch (1:5000). Detection of protein bands was carried out using chemiluminescence (EMD Millipore) on a LAS-3000 Imager (FUJIFILM UK Ltd. System).
Immunohistochemistry
Immunohistochemistry (IHC) was carried out on 5-μm-thick paraffin sections using EnVisionTM Kit (Dako, #K4006) as described previously [21]. Briefly, following EDTA-based heat antigen retrieval (pH 9.0, 20 min) and peroxidase (Dako) block step, sections were incubated for 1 h at 25 °C using recommended dilution of BCL2 antibody (1:15; mouse monoclonal, Invitrogen, #MA1-26233). The IHC protein expression was visualised at × 20 magnification under an Olympus light microscope. At least 5–8 random images were taken per tumour (at least 500 cells were assessed in all cases where possible). For the purposes of correlating BCL2 protein score and BCL2 IHC scores, the quantification of positive staining was measured using the Aperio Image Scope digital pathology imaging software (Supplementary Table S4). Results are expressed as mean percentage positive cells as determined by the positive pixel count algorithm using the software.
IHC was performed for five of the TNBC samples (1, 3, 4, 11 and 14) of the Irish Cancer Society BREAST-PREDICT cohort. These were selected to reflect low, mean and high BCL2 protein levels. Five colorectal cancer (CRC) samples, unrelated to this study, were randomly selected from a cohort used by Flanagan et al. [22].
Calculation of TCGA protein profiles
BCL-2 protein profiles (BAK, BAX, BCL2 and BCL(X)L) were calculated for the 845 TCGA patients by using simple linear regression to fit the 25th and 75th percentile of the protein levels of the TCGA patients (RPPA) to the 25th and 75th percentile of the protein levels of our BREAST-PREDICT cohort after excluding extreme outliers (see statistics for outlier calculations). In a first step, protein profiles of TNBC TCGA patients were matched to the TNBC profiles of the BREAST-PREDICT cohort. The fitted linear functions were then applied to calculate the protein profiles of the remaining non-TNBC patients of the TCGA cohort. Protein levels for different antibodies used in the TCGA for BAK (Bak-R-E and Bak-R-C) and BCL(X)L (Bcl-xL-R-C and Bcl-xL-R-V) were merged. We did not find a statistical difference in the distribution of protein levels between the respective antibodies (not shown).
PAM50 subtypes and proliferation score analysis
The PAM50 subtype [23] of the TCGA patients was determined using the ‘genefu’ R-Package [24] and mRNA expression levels (mRNA SeqV2). The proliferation score [25] was determined by averaging the normalised expression of 11 genes (RNASeq: BIRC5, CCNB1, CDC20, CDCA1, CEP55, KNTC2, MKI67, PTTG1, RRM2, TYMS, UBE2C) as described by Nielsen et al. [25].
DR_MOMP
DR_MOMP was employed as previously described [8, 9, 11] and run with Matlab R2007b (V.7.5.0.342; The MathWorks, USA). Details of the model are described in the supplementary Methods of Lindner et al. [8]. Briefly, the stress dose required to induce MOMP in a cell was determined by mathematically modelling reversible protein binding of the pro- and anti-apoptotic proteins BAK, BAX, BIM, BCL2, BCL(X)L, MCL1, PUMA, NOXA and VDAC2 based on mass action kinetics [8]. BIM and PUMA are modelled to activate BAK and BAX. BAX and BAK are modelled to subsequently form homo-dimers and higher oligomers. Pores are defined as BAK and BAX hexamers and higher oligomers [8]. MOMP is assumed to occur if at least 10% of total BAK and BAX are associated in pores as determined previously [8]. Levels of BAK, BAX, BCL2, BCL(X)L and MCL1 were used as input to calculate the DR_MOMP stress dose. The TCGA dataset did not include MCL1 protein levels and values were set to zero for the calculation of the DR_MOMP stress dose. In the TNBC BREAST-PREDICT cohort, setting MCL1 levels to 0 nM led to a modest, on average 8%, reduction of the DR_MOMP stress dose. The calculated stress dose required to induce MOMP was normalised in TNBC patients from the TCGA cohort by determining the z score, excluding extreme outliers. The z score in TNBC served as a reference for other breast cancer subtypes.
Statistics
Employed statistical tests are stated with the p value (p) for each analysis. Statistical significance was considered with p < 0.05. Survival analyses were performed using the ‘survival’ package (V.2.38–3) in R (V.3.2.3, The R Foundation). Statistically significant differences between overall survival (OS) curves were determined using log-rank tests (logrank). We computed hazard ratios (HR) and 95% confidence intervals (CI) using unadjusted and multivariate Cox proportional hazards models to estimate the relative risks of death. Statistical significance was determined using Wald tests (Wald). The proportional hazards assumption (PHA) was tested by using the cox.zph function. Violation of the PHA was accounted by integrating the time-transform functions ‘tt(BCL2) = BCL2 * time’ into the Cox models (Table 2). Extreme outliers (< Q1−3(Q3−Q1) and > Q3 + 3(Q3−Q1), with Q1 = 25th, Q2 = 50th and Q3 = 75th percentile) were excluded for calculating protein profiles, mean or median values. Bootstrapping was applied to estimate the CI of the median of a dataset (bootstrap 95%CI). Datasets were resampled with replacements with the length of the resample being 100 times the original length. After the median was determined, this routine was repeated 100 times. The 0.025 and 0.975 quantiles of the computed medians were used as the 95% CI.
Data availability
Data generated or analysed during this study are available in the supplementary tables 1 and 7.
Results
We determined BCL-2 family of protein profiles in fresh-frozen primary tumour samples from histopathologically confirmed and non-metastatic female TNBC patients. Patient information and tumour pathology characteristics are provided in supplementary Table S1. Protein levels of BAK, BAX, BCL2, BCL(X)L and MCL1 were quantified by quantitative Western blotting. To determine absolute levels of BAK, BAX, BCL2, BCL(X)L and MCL1, protein concentrations were normalised to HeLa cells in which BCL-2 protein levels were previously quantified [8]. Pro- and anti-apoptotic BCL-2 family proteins were heterogeneously expressed in individual tumour samples (Fig. 1a). We found a median level of 585 nM (bootstrap 95% CI 95–1143 nM) and 396 nM (bootstrap 95% CI 218–495 nM) for the pro-apoptotic proteins BAK and BAX respectively. We found a median level of 2037 nM (bootstrap 95% CI 750–3542 nM), 0 nM (bootstrap 95% CI 0–37 nM) and 7 nM (bootstrap 95% CI 0–34 nM) for the anti-apoptotic proteins BCL2, BCL(X)L and MCL1, respectively. BAK, BAX and MCL1 levels of the TNBC samples were not differentially expressed compared to colon or rectal cancer patients in which levels were previously determined by the identical method (Wilcoxon rank-sum test) [8]. BCL2 was significantly higher expressed (Wilcoxon rank-sum test p < 0.01) in the TNBC tissue compared to colon or rectal cancer tissue (median = 595 nM; bootstrap 95%CI 195–840 nM; Fig. 1b). In contrast, BCL(X)L levels in the TNBC samples were significantly lower (Wilcoxon rank-sum test p = 0.02) compared to colon and rectal cancer tissue (median = 19 nM; bootstrap 95% CI 5–64 nM). None of the individual proteins were associated with clinical outcome (not shown; Cox models, Wald test p = 0.3–0.9). We performed IHC staining for BCL2 for five TNBC samples, selected to reflect low, mean and high BCL2 protein levels, and five colorectal cancer (CRC) samples, randomly selected from a previous study [27] (Fig. 1c; Supplementary Fig. S1 and Table S4). The averaged Aperio score of the TNBC samples strongly correlated with the BCL2 protein levels quantified by Western blotting (Pearson’s correlation coefficient r = 0.94, p = 0.02; Fig. 1d). The mean averaged Aperio score of the TNBC sections (0.29) was numerically tenfold higher compared to the mean averaged Aperio score of the randomly selected CRC sections (0.27; Fig. 1e).

BCL-2 family protein profiling and risk score calculations in TNBC patients. a BAK, BAX, BCL2, BCL(X)L and MCL1 protein levels as determined by quantitative Western blotting in 19 TNBC patients from the BREAST-PREDICT cohort. b BCL2 protein levels were found to be significantly higher in triple negative breast cancer (TNBC, Irish Cancer Society BREAST-PREDICT cohort) patients compared to colorectal cancer [8] (CRC) and glioblastoma [26] (GBM) patients (p < 0.01; Tukey HSD post hoc ANOVA). c IHC staining of the BCL2 protein in three representative TNBC and CRC sections (all ten sections in Supplementary Fig. S1). d The averaged Aperio score strongly correlated with the BCL2 protein levels quantified by Western blotting (Pearson’s correlation coefficient r = 0.94, p = 0.02). e Aperio scores of TNBC and randomly selected CRC sections. f Protein profiles were used as input for DR_MOMP [8] to determine g the stress dose required to induce apoptosis in the patients’ cancer samples. h The stress doses were not significantly different among different tumour grades and stages
We employed DR_MOMP [8] to calculate the stress dose required to induce mitochondrial outer membrane permeabilisation (MOMP) in each tumour sample as an indicator of apoptosis sensitivity, by using the determined BCL-2 protein profiles as model inputs (Fig. 1f). We found considerable differences in the DR_MOMP stress dose among TNBC tumour samples (Fig. 1g), independent of tumour grade or TN-staging (Fig. 1d). Analysis of clinical follow-up by determining 5-year disease-free survival (DFS) and breast cancer-related overall survival (OS) suggested that the stress dose was not significantly associated with clinical outcome of TNBC patients, as both very high and very low stress doses were observed in patients with unfavourable clinical outcome (Fig. 1h).
The above findings suggested that apoptosis signalling through BCL-2 family proteins was not indicative of clinical outcome in TNBC or may contribute to clinical outcome only in specific molecular subtypes of breast cancer. To validate and further explore these findings in an external cohort, we inferred protein expression data of 845 patients from the TCGA cohort that included 91 TNBC, 590 ER/PR+ and 127 human epidermal growth factor receptor 2 positive (HER2+) samples (Fig. 2a). A total of 74 patients were removed from the analysis (Fig. 2a; see ‘Materials and Methods’).

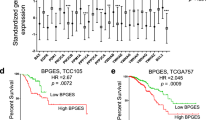
DR_MOMP analysis of the TCGA BRCA cohort. a Workflow and patient criteria for TCGA analysis. TNBC patients of the TCGA cohort were matched with the TNBC BREAST-PREDICT cohort to calculate BCL2 protein profiles among TCGA patients. b BCL2 expression varied between the IHC subtype among TCGA patients. c The MOMP score was not different among the various cancer stages, but varied d among the IHC and e PAM50 molecular subtypes (Tukey HSD post hoc ANOVA). f Patients with a MOMP score > 0 had significantly higher overall survival compared to patients with MOMP score ≤ 0 (logrank-test). g The MOMP score was not a prognostic marker within the various IHC subtypes, h but showed prognostic properties among Luminal B patients (Cox models, Wald p test < 0.01)
BCL2 protein profiles of the TNBC patients of the TCGA cohort were first matched to profiles of the TNBC BREAST-PREDICT cohort by linear regression, as described in ‘Materials and Methods’. The fitted functions were then applied to calculate protein profiles for the remaining non-TNBC TCGA patients. Analysis of BCL-2 profiles did not reveal major differences in BAX, BAK and BCL(X)L levels between TNBC, ER/PR+ and HER2+ breast cancer (Fig. 2b). Protein levels of BCL2 in ER/PR+ breast cancer further exceeded those seen in TNBC (Fig. 2b), confirming previous observations that ER/PR+ breast cancer overexpresses BCL2 [14]. MCL1 protein levels were not available in the TCGA cohort and levels were set to 0 nM for DR_MOMP.
Calculation of the DR_MOMP stress dose revealed a median of 2286 nM (bootstrap 95% CI 2134–2377 nM) across all breast cancers patients. We next performed a data normalisation step by calculating z scores of the DR_MOMP stress dose (MOMP score), with TNBC as reference cohort. Previously, the mean MOMP score (= 0) was identified as an excellent threshold to differentiate colorectal cancer patients with favourable and unfavourable clinical outcome [9]. We did not observe a statistical difference in MOMP score among stage I–IV patients (one way ANOVA, p = 0.11; Fig. 2c), confirming our observations in the smaller BREAST-PREDICT cohort. MOMP scores were significantly lower in TNBC when compared to ER/PR+ (p < 0.0001) and HER+ tumours (p < 0.001; Tukey HSD post hoc ANOVA; Fig. 2d). Analysis of PAM50 molecular subtypes [23] in this cohort revealed a high MOMP score in Luminal A and Luminal B subtypes compared to Basal-like and HER2-enriched patients, reflecting findings observed in ER/PR+ cancers (p < 0.0001, Tukey HSD post hoc ANOVA; Fig. 2e).
OS probability across all breast cancers was significantly higher in patients with a MOMP score ≤ 0 (logrank p < 0.01) and the risk of death was halved (hazard ratio HR 0.48, 95% CI 0.30–0.77; Wald p < 0.01) compared to patients with a MOMP score > 0 (Fig. 2f). Results were similar after adjusting for tumour stage, age at diagnosis and IHC subtype (HR 0.56, 95% CI 0.33–0.93; Wald p = 0.03; Table 1). This finding was in stark contrast to findings in colorectal cancer, where mortality risk was significantly increased in patients with a MOMP score > 0 [9].
We next explored whether the prognostic potential of the MOMP score depended on the different molecular subtypes investigated, as each molecular subtype is expected to have differences in disease biology. However, we did not find a difference in risk assessment based on MOMP scores among the different receptor-based (IHC) subtypes, i.e. ER/PR+, HER2+ and TNBC subtypes (Fig. 2g). Within the PAM50 classification, an increase in the score decreased the risk of death by 1/3 per unit in Luminal B patients (Fig. 2h; HR 0.67, 95% CI 0.50–0.89, Wald p < 0.01). There was no significant association among Luminal A patients (HR 1.04, 95% CI 0.99–1.08, Wald p = 0.10). We also tested for a possible interaction between MOMP score and PAM50 molecular subtypes (Supplementary Table S5). We found that there was an interaction between the MOMP score and whether or not the cancer was classified as Luminal A (interaction p = 0.03) or Luminal B (interaction p < 0.01).
Next we addressed the question whether high MOMP scores were also associated with impairment of mitochondrial apoptosis by interrogating levels of cleaved caspase-7 in the TCGA cohort. Cleaved caspase-7 is activated downstream of MOMP and apoptosome formation [28, 29]. Cleaved caspase-3 levels were not available in the TCGA dataset. The Pearson’s correlation coefficient between BCL2 and cleaved caspase-7 was negative (− 0.10; 95% CI − 0.17—− 0.03, p < 0.01), and mean levels of cleaved caspase-7 were significantly lower in patients with a MOMP score > 1 compared to patients with a MOMP score ≤ 0 (Fig. 3a, p < 0.0001, Tukey HSD post hoc ANOVA). These findings indicated that high MOMP scores were associated with impaired apoptosis execution in breast cancer patients.

High cleaved caspase-7 levels are associated with a decreased risk. a Breast cancer samples with a MOMP score > 0 had significantly lower levels of cleaved caspase-7 compared to samples with a MOMP score ≤ 0 (Tukey HSD post hoc ANOVA). b Patients with cleaved caspase-7 levels > mean (0.11 A.U.) had significantly higher OS compared to patients with cleaved caspase-7 levels ≤ mean (logrank test). c Testing of the prognostic value of cleaved caspase-7 levels in PAM50 subtypes (Cox models, Wald p test). Patients with BCL2 protein levels > mean (0.22 A.U.) had significantly d lower PAM50 proliferation scores and e lower levels of cleaved caspase-7 compared to patients with BCL2 protein levels ≤ mean (Wilcoxon Rank Sum). f There was no difference in the proliferation score within the various PAM50 subtypes, comparing samples with BCL2 protein levels > individual means and ≤ individual means (− 0.50 Basal, − 0.44 Her2, 0.47 Luminal B, 0.64 Luminal A; Wilcoxon Rank Sum)
We next explored whether the levels of cleaved caspase-7 were associated with lower or higher OS in breast cancer. We hypothesised that at least two scenarios would be possible: (a) The ability of cells to activate caspase-dependent apoptosis has an adverse effect on clinical outcome. In this scenario, high cleaved caspase-7 levels, similar to low MOMP scores, should associate with unfavourable outcome. This scenario is supported by recent studies suggesting that caspase-dependent apoptosis may trigger compensatory cell proliferation and relapse in breast cancer [27, 30]. (b) The ability of cells to activate caspase-dependent apoptosis is associated with improved clinical outcome, but other functions of BCL2 proteins (cell proliferation, gene expression, bioenergetics) contribute to disease progression and ‘overshadow’ the anti-apoptotic functions of BCL2 proteins. In this scenario, cleaved caspase-7 levels may be an alternative prognostic biomarker of clinical outcome.
Interestingly, we found that patients with cleaved caspase-7 levels greater than the mean had a 60% reduced risk of death compared to patients with cleaved caspase-7 levels less than or equal to the mean (HR 0.4, 95% CI 0.3–0.7, Wald p = 0.001). This suggested that impairment of caspase-dependent apoptosis was indeed associated with poor clinical outcome. In contrast to the MOMP score, the risk associated with cleaved caspase-7 levels was also similar in all PAM50 molecular subtypes (Fig. 3c). We did not find evidence of an interaction between cleaved caspase-7 levels and PAM50 subtypes (Luminal A interaction p = 0.35; Luminal B interaction p = 0.50).
From a quantitative perspective, the anti-apoptotic BCL2 protein was found to be the most abundant BCL-2 family protein in breast cancer, exceeding the levels of pro-apoptotic BAK and BAX which represented the second and third most abundant BCL-2 family proteins (Figs. 1a and 2b). We therefore also evaluated the prognostic power of individual BCL2 family proteins. Since we found a strong association between BCL2 levels and ER/PR-status, which was previously reported to be time dependent [31], we tested and accounted for the violation of the proportional hazards assumption (PHA) in further analysis. Within the TCGA cohort, we found evidence that BCL2 protein levels (cox.zph p < 0.01) as well as the MOMP scores (cox.zph p = 0.03) violated the PHA (Supplementary Fig. S2), suggesting a time-varying effect of BCL2 levels. Levels of cleaved caspase-7 did not violate the PHA (cox.zph p = 0.91). BCL2 protein levels above the mean (determined in TNBC patients) had a significantly higher OS compared to levels below the mean (logrank p < 0.0001; Supplementary Fig. S3) and we determined a HR = 0.37 (95% CI 0.23–0.61, Wald p < 0.0001; Table 2) accounting for the violation of the PHA. Differences in BCL2 protein levels were not associated with a significant effect on patients’ risk between 5 and 10 years (HR 0.83, 95% CI 0.61–1.13). HRs were similar after we adjusted for PAM50 and IHC subtypes (not shown). Levels of BAK (HR 1.14, 95% CI 0.55–2.36, Wald p = 0.72), BAX (HR 1.03, 95% CI 0.48–2.19, Wald p = 0.94) or BCL(X)L protein (HR 0.61, 95% CI 0.21–1.80, Wald p = 0.37) were not prognostic.
Of note, patients with BCL2 protein levels above the mean also had a significantly lower proliferation score (Fig. 3d; Wilcoxon rank-sum p < 0.001) compared to patients with BCL2 levels below the mean. Patients with high BCL2 protein levels also had lower levels of cleaved caspase 7 (Fig. 3e; Wilcoxon rank-sum p < 0.001). Analysis of PAM50 molecular subtypes indicated that the association of high BCL2 levels with low cell proliferation might be driven by the Luminal B subtype and not by BCL2 levels themselves, since we did not observe a BCL2-dependent variation of the proliferation score within the Basal, Her2 nor Luminal A subtypes (Fig. 3f). The BCL2 protein has potentially complementary roles beyond cell death and proliferation, such as in bioenergetics [32] and cell migration [33, 34]. Indeed, expression levels of a regulator of actin filament dynamics (LIM kinase) [35] were associated with increased risk in a univariate analysis (HR 1.82, 95% CI 1.39–2.38; Wald p < 0.0001), as well as in a multivariate analysis together with BCL2 (HR 1.51, 95% CI 1.07–2.13; Wald p = 0.02; Supplementary Table S6).
Finally, in multivariate Cox-models adjusting for tumour stage, IHC subtype and age at diagnosis, we found that cleaved caspase-7 levels and BCL2 protein levels were associated with an approximately 50% reduced mortality risk (cleaved caspase-7 HR 0.58, 95% CI 0.40–0.84, Wald p < 0.01; BCL2 HR 0.44 95% 0.26–0.73, Wald p < 0.01; Table 2). We next divided patients into groups with cleaved caspase-7 levels greater than the mean (> 0.06 a.U; n = 365; Fig. 4a) and below the mean (n = 511). We further subdivided the latter into patients with BCL2 levels greater (> 0.22 a.U.; n = 317) and below the cohort’s mean (n = 194). We did not find that these BCL2 groups violated the PHA (cox.zph p = 0.08 and cox.zph p = 0.40, respectively). Focusing on long-term survival (5–10 years, n = 186 patients), we found that patients with high BCL2 levels and low cleaved caspase-7 levels had a sevenfold increased risk of death (HR 7.0, 95% CI 1.5–32.2, Wald p = 0.01), and that patients with low BCL2 and low cleaved caspase-7 levels were 11 times more likely to die (HR 11.3, 95% CI 2.2–58.2, Waldp < 0.01), compared to patients with high cleaved caspase-7 levels. Of note, we also found that PAM50 subtypes were scattered across the three groups (Fig. 4b). Sixty percent of Her2-enriched and 62% Basal-like patients were in the high cleaved caspase-7 level group. Sixty-one of Luminal A patients were in the group with low cleaved caspase-7 levels and high BCL2 protein levels.

The combination of BCL2 and cleaved caspase-7 levels is a promising prognostic biomarker. a Patients were divided into those with high (> 0.06 A.U.; solid line, blue) and low cleaved caspase-7 levels. We further subdivided the latter cohort according to high (> 0.22 A.U.; dashed line, purple) and low BCL2 protein levels (dotted line, red). b Distribution of the PAM50 subtypes among the defined patient subgroups. c Kaplan–Meier analysis of patient subgroups. Patients with low cleaved caspase-7 and low BCL2 proteins levels had lower OS compared to patients with high BCL2 or high cleaved caspase-7 levels (logrank-test)
Discussion
The ability of cancer cells to induce the BCL-2 family-controlled mitochondrial apoptosis pathway, when analysed by quantitative systems and computational modelling approaches such as DR_MOMP, was shown to be a reliable indicator of genotoxic therapy responses in colon cancer cells [8] and an excellent biomarker for favourable clinical outcome in colorectal cancer patients [8, 9, 11]. Employing an identical quantitative tissue profiling approach in fresh-frozen tissue material [8, 11], we found that protein levels of the anti-apoptotic protein BCL2 were about sevenfold higher in TNBC tissues when compared to colon or rectal tumours, or when compared to other solid tumours such as glioblastoma (Fig. 1b, e). This was not the case for any of the other pro- or anti-apoptotic BCL-2 family proteins investigated. Systems analysis of BCL-2 protein levels in the in-house BREAST-PREDICT cohort indicated that BCL-2 proteins were not prognostic in a cohort of TNBC patients (Fig. 1), a finding that was validated in the TCGA TNBC cohort (Fig. 2e). Quantitative analysis of BCL-2 protein levels in the TCGA cohort revealed that high BCL2 protein levels were associated with favourable (rather than unfavourable) outcome in ER/PR+ breast cancer and the Luminal B PAM50 subtype. These findings are in agreement with previous studies showing that high levels of BCL2 expression are enriched in ER/PR+ breast cancer and associated with favourable clinical outcome [12, 13], but are counter-intuitive when considering the known roles of BCL2 family proteins in cell death regulation and chemo- and radiotherapy responses. Because most breast cancer patients in the cohorts studied were treated with chemo- and radiotherapy or targeted therapies, it is possible that treatments reduced BCL2 levels particularly in high BCL2 expressing and potentially BCL2-dependent breast cancers. However, studies in the neo-adjuvant setting suggested that alterations in BCL2 levels did not correlate with response to treatment [36, 37] or that BCL2 levels increased in responders [38]. In line with an anti-apoptotic role of BCL2, we observed that tumours with high levels of BCL2 protein had a reduced proficiency to activate the mitochondrial apoptosis pathway, as evidence by reduced caspase-7 activation. However, we also observed that tumours with low levels of cleaved caspase-7 were associated with unfavourable clinical outcome in cancer patients. Collectively, this suggested that the BCL2 protein may exert other activities that contribute to favourable clinical outcome such as regulation of cell proliferation, gene expression or bioenergetics [17, 18, 32]. In this context, we also observed a strong negative correlation between high BCL2 protein levels and cell proliferation scores. Previous studies have shown that BCL2 can delay G(0)-G(1) cell cycle transition [20] and potentially has additional effects in S phase [19], the mechanisms of which are largely unexplored. Interestingly, BCL2 is also localised to the nuclear envelope [39] and has recently been shown to exert extra-mitochondrial effects by regulating gene expression [18]. Additionally, BCL2 may play a joint role with PI3K in the regulation of cell migration and morphology, by inhibiting cell adhesion, spreading and motility [33, 34]. Overexpression of BCL2 in ER/PR+ breast cancer cells has been reported to reduce the risk of metastasis [33]. Indeed, we found that expression of LIM kinase, an essential regulator of actin filament dynamics [35], was associated with increased risk in a multivariate analysis. A holistic model incorporating cell death, proliferation and cell migration might be required to understand the delicate balanced of BCL2’s negative and positive impacts on patients’ clinical outcome.
Alternatively, high BCL2 levels may be merely an indicator of a subtype of breast cancer that has a favourable clinical prognosis and is responsive to ER targeted-therapy [40]. In both scenarios, these biological activities would dominate over BCL2’s anti-apoptotic activity in terms of long-term prognosis. To identify improved prognostic signatures, we combined cleaved caspase-7 and BCL2 protein levels. Patients with impaired apoptosis execution as indicated by low cleaved caspase-7, and high proliferation as indicated by low BCL2 protein levels, had the lowest OS and were 3 times more likely to die. Patients with high BCL2 protein levels did not show a different OS within the first 5 years of follow-up; however, differences in survival became evident after this period. We observed that the effect of BCL2 protein levels varied over time but that levels of cleaved caspase-7 did not. Time dependence of the ER status was previously demonstrated [31, 41], and the 2-year peak in mortality rate of ER-negative cancer patients after diagnosis was used to explain the time dependency [31]. In our study, the patient subgroup with high BCL2 protein levels and low cleaved caspase 7 levels mostly consisted of ER+ (Luminal A and B) cancer. Our study suggests that breast cancers with high levels of cleaved caspase 7 have a higher sensitivity to (neo-) adjuvant therapy; therefore, these patients may have a good prognosis for both short- and long-term survival. Patients with low cleaved caspase 7 but high BCL2 protein levels may show higher survival after surgery, since their cancer is less invasive and slower proliferating [13, 14]. However, the impairment of apoptosis might be disadvantage for long-term survival. This disadvantage could be potentially addressed by targeting caspase activation or MOMP with apoptosis sensitisers including IAP antagonists [42] and BCL-2 antagonists.
BCL2 displays a high binding affinity for activated BAX [43] and most BH3-only proteins [44]. A ‘MODE 1’ prevention of apoptosis (sequestering of direct activators of BAK and BAX) was described to be easily disrupted by BCL2-antagonists [45]. Drugs targeting BCL2 such as Venetoclax/ABT-199 were shown to sensitise ER+ breast cancer cells to tamoxifen in mouse xenograft models [46], presumably by disrupting the binding of BCL2 to the pro-apoptotic BH3-only protein BIM [46, 47]. Indeed, we found a Pearson correlation coefficient = 0.52 (95% CI 0.46–0.57, p < 0.0001) between BIM and BCL2 protein levels among the samples of the TCGA cohort. Both markers were associated with decreased risk in a univariate Cox-model and no statistically significant interaction between BCL2 and BIM protein levels was observed (interaction p = 0.07, Supplementary Table S7). Moreover, in contrast to BAX, BAK is normally held in check predominantly by BCL(X)L and MCL1 [48]. Due to the low levels of BCL(X)L and MCL1 in breast cancer patients, BAK should be readily available to induce apoptosis subsequent to a stress- or therapy-induced activation of BH3-only proteins. In most in vitro and in vivo systems, activation of BAK is sufficient to induce apoptosis in the absence of BAX [49].
Collectively, our study suggests that the protein levels of the anti-apoptotic protein BCL2 (as a potential indicator of cell proliferation and/or molecular subtype) and levels of cleaved caspase 7 (as an indicator for apoptosis impairment) can be combined to deliver a novel prognostic signature across all breast cancers. Low levels of both proteins are indicative for highly unfavourable clinical outcome, independent of patients’ age, tumour stage and PAM50 classification.
Abbreviations
- a.U. :
-
arbitrary unit
- ANOVA :
-
analysis of variance
- BAK, BCL2 :
-
antagonist/killer 1 (BAK1)
- BAX, BCL2 :
-
associated X, apoptosis regulator (BCL2L4)
- BCL2 :
-
B-cell lymphoma 2
- BCL(X)L :
-
B-cell lymphoma-extra large (BCL2L1)
- BIM :
-
Bcl-2 interacting mediator of cell death (BCL2L11)
- BH3 :
-
BCL2 homology 3rd domain
- CI :
-
confidence interval
- DFS :
-
disease-free survival
- ER :
-
oestrogen receptor 1 (ESR1)
- HER2 :
-
Receptor tyrosine-protein kinase erbB-2 (ERBB2)
- HR :
-
hazard ratio
- IAP :
-
inhibitor of apoptosis
- IHC :
-
immunohistochemistry
- MCL1 :
-
myeloid cell leukaemia sequence 1 (BCL2L3)
- MOMP :
-
mitochondrial outer membrane permeabilisation
- nM :
-
nanomolar (10−9 mol/L)
- NOXA :
-
horbol-12-Myristate-13-Acetate-Induced protein 1 (PMAIP1)
- OS :
-
overall survival
- p :
-
probability value
- PAM50 :
-
50-gene signature
- PHA :
-
proportional hazards assumption
- PR :
-
progesterone receptor (PGR)
- PI3K :
-
phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit
- PUMA :
-
p53 upregulated modulator of apoptosis (BBC3)
- RPPA :
-
reverse protein phase array
- TCGA :
-
The Cancer Genome Atlas
- TNBC :
-
triple negative breast cancer
- TNM :
-
tumour/lymph node/metastasis staging system
- Tukey HSD :
-
Tukey Honest significant differences post-hoc test
- VDAC2 :
-
voltage-dependent anion channel 2
- Wald p :
-
Wald test probability value
References
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674
Johnstone RW, Ruefli AA, Lowe SW (2002) Apoptosis: a link between cancer genetics and chemotherapy. Cell 108(2):153–164
Delbridge AR, Grabow S, Strasser A, Vaux DL (2016) Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer 16(2):99–109
Lakhani SA, Masud A, Kuida K, Porter GA Jr, Booth CJ, Mehal WZ et al (2006) Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science 311(5762):847–851
Strasser A, Harris AW, Jacks T, Cory S (1994) DNA damage can induce apoptosis in proliferating lymphoid cells via p53-independent mechanisms inhibitable by Bcl-2. Cell 79(2):329–339
Hector S, Prehn JH (2009) Apoptosis signaling proteins as prognostic biomarkers in colorectal cancer: a review. Biochim Biophys Acta 1795(2):117–129
Juin P, Geneste O, Gautier F, Depil S, Campone M (2013) Decoding and unlocking the BCL-2 dependency of cancer cells. Nat Rev Cancer 13(7):455–465
Lindner AU, Concannon CG, Boukes GJ, Cannon MD, Llambi F, Ryan D, Boland K, Kehoe J, McNamara DA, Murray F, Kay EW, Hector S, Green DR, Huber HJ, Prehn JHM (2013) Systems analysis of BCL2 protein family interactions establishes a model to predict responses to chemotherapy. Cancer Res 73(2):519–528
Lindner AU, Salvucci M, Morgan C, Monsefi N, Resler AJ, Cremona M, Curry S, Toomey S, O'Byrne R, Bacon O, Stühler M, Flanagan L, Wilson R, Johnston PG, Salto-Tellez M, Camilleri-Broët S, McNamara DA, Kay EW, Hennessy BT, Laurent-Puig P, van Schaeybroeck S, Prehn JHM (2016) BCL-2 system analysis identifies high-risk colorectal cancer patients. Gut 66:2141–2148
Lucantoni F, Lindner AU, O'Donovan N, Dussmann H, Prehn JHM (2018) Systems modeling accurately predicts responses to genotoxic agents and their synergism with BCL-2 inhibitors in triple negative breast cancer cells. Cell Death Discov 9(2):42
Flanagan L, Lindner AU, de Chaumont C, Kehoe J, Fay J, Bacon O, Toomey S, Huber HJ, Hennessy BT, Kay EW, McNamara DA, Prehn JHM (2015) BCL2 protein signalling determines acute responses to neoadjuvant chemoradiotherapy in rectal cancer. J Mol Med (Berl) 93(3):315–326
Gasparini G, Barbareschi M, Doglioni C, Palma PD, Mauri FA, Boracchi P, Bevilacqua P, Caffo O, Morelli L, Verderio P (1995) Expression of bcl-2 protein predicts efficacy of adjuvant treatments in operable node-positive breast cancer. Clin Cancer Res 1(2):189–198
Lipponen P, Pietilainen T, Kosma VM, Aaltomaa S, Eskelinen M, Syrjanen K (1995) Apoptosis suppressing protein bcl-2 is expressed in well-differentiated breast carcinomas with favourable prognosis. J Pathol 177(1):49–55
Leek RD, Kaklamanis L, Pezzella F, Gatter KC, Harris AL (1994) bcl-2 in normal human breast and carcinoma, association with oestrogen receptor-positive, epidermal growth factor receptor-negative tumours and in situ cancer. Br J Cancer 69(1):135–139
Knight WA, Livingston RB, Gregory EJ, McGuire WL (1977) Estrogen receptor as an independent prognostic factor for early recurrence in breast cancer. Cancer Res 37(12):4669–4671
Dong L, Wang W, Wang F, Stoner M, Reed JC, Harigai M, Samudio I, Kladde MP, Vyhlidal C, Safe S (1999) Mechanisms of transcriptional activation of bcl-2 gene expression by 17beta-estradiol in breast cancer cells. J Biol Chem 274(45):32099–32107
Sheridan C, Delivani P, Cullen SP, Martin SJ (2008) Bax- or Bak-induced mitochondrial fission can be uncoupled from cytochrome C release. Mol Cell 31(4):570–585
Wu X, Zhang LS, Toombs J, Kuo YC, Piazza JT, Tuladhar R, Barrett Q, Fan CW, Zhang X, Walensky LD, Kool M, Cheng SY, Brekken R, Opferman JT, Green DR, Moldoveanu T, Lum L (2017) Extra-mitochondrial prosurvival BCL-2 proteins regulate gene transcription by inhibiting the SUFU tumour suppressor. Nat Cell Biol 19(10):1226–1236
Belanger S, Cote M, Lane D, L'Esperance S, Rancourt C, Piche A (2005) Bcl-2 decreases cell proliferation and promotes accumulation of cells in S phase without affecting the rate of apoptosis in human ovarian carcinoma cells. Gynecol Oncol 97(3):796–806
Janumyan YM, Sansam CG, Chattopadhyay A, Cheng N, Soucie EL, Penn LZ, Andrews D, Knudson CM, Yang E (2003) Bcl-xL/Bcl-2 coordinately regulates apoptosis, cell cycle arrest and cell cycle entry. EMBO J 22(20):5459–5470
Ward E, Vareslija D, Charmsaz S, Fagan A, Browne AL, Cosgrove N et al (2018) Epigenome-wide SRC-1-mediated gene silencing represses cellular differentiation in advanced breast cancer. Clin Cancer Res https://doi.org/10.1158/1078-0432.CCR-17-2615
Flanagan L, Meyer M, Fay J, Curry S, Bacon O, Duessmann H, John K, Boland KC, McNamara DA, Kay EW, Bantel H, Schulze-Bergkamen H, Prehn JHM (2016) Low levels of caspase-3 predict favourable response to 5FU-based chemotherapy in advanced colorectal cancer: caspase-3 inhibition as a therapeutic approach. Cell Death Dis 7:e2087. https://doi.org/10.1038/cddis.2016.7
Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T et al (2009) Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 27(8):1160–1167
Gendoo DM, Ratanasirigulchai N, Schroder MS, Pare L, Parker JS, Prat A et al (2016) Genefu: an R/Bioconductor package for computation of gene expression-based signatures in breast cancer. Bioinformatics 32(7):1097–1099
Nielsen TO, Parker JS, Leung S, Voduc D, Ebbert M, Vickery T et al (2010) A comparison of PAM50 intrinsic subtyping with immunohistochemistry and clinical prognostic factors in tamoxifen-treated estrogen receptor-positive breast cancer. Clin Cancer Res 16(21):5222–5232
Murphy AC, Weyhenmeyer B, Schmid J, Kilbride SM, Rehm M, Huber HJ, Senft C, Weissenberger J, Seifert V, Dunst M, Mittelbronn M, Kögel D, Prehn JHM, Murphy BM (2013) Activation of executioner caspases is a predictor of progression-free survival in glioblastoma patients: a systems medicine approach. Cell Death Dis 4:e629. https://doi.org/10.1038/cddis.2013.157
Huang Q, Li F, Liu X, Li W, Shi W, Liu FF, O'Sullivan B, He Z, Peng Y, Tan AC, Zhou L, Shen J, Han G, Wang XJ, Thorburn J, Thorburn A, Jimeno A, Raben D, Bedford JS, Li CY (2011) Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med 17(7):860–866
Fernandes-Alnemri T, Takahashi A, Armstrong R, Krebs J, Fritz L, Tomaselli KJ, Wang L, Yu Z, Croce CM, Salveson G (1995) Mch3, a novel human apoptotic cysteine protease highly related to CPP32. Cancer Res 55(24):6045–6052
Lippke JA, Gu Y, Sarnecki C, Caron PR, Su MS (1996) Identification and characterization of CPP32/Mch2 homolog 1, a novel cysteine protease similar to CPP32. J Biol Chem 271(4):1825–1828
Boland K, Flanagan L, Prehn JH (2013) Paracrine control of tissue regeneration and cell proliferation by Caspase-3. Cell Death Dis 4:e725. https://doi.org/10.1038/cddis.2013.250
Dawson SJ, Makretsov N, Blows FM, Driver KE, Provenzano E, Le Quesne J et al (2010) BCL2 in breast cancer: a favourable prognostic marker across molecular subtypes and independent of adjuvant therapy received. Br J Cancer 103(5):668–675
Lucantoni F, Dussmann H, Llorente-Folch I, Prehn JHM (2018) BCL2 and BCL(X)L selective inhibitors decrease mitochondrial ATP production in breast cancer cells and are synthetically lethal when combined with 2-deoxy-D-glucose. Oncotarget 9(40):26046–26063
Ke H, Parron VI, Reece J, Zhang JY, Akiyama SK, French JE (2010) BCL2 inhibits cell adhesion, spreading, and motility by enhancing actin polymerization. Cell Res 20(4):458–469
Wan G, Pehlke C, Pepermans R, Cannon JL, Lidke D, Rajput A (2015) The H1047R point mutation in p110 alpha changes the morphology of human colon HCT116 cancer cells. Cell Death Discov 1:15044
Yamaguchi H, Condeelis J (2007) Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta 1773(5):642–652
Makris A, Powles TJ, Dowsett M, Osborne CK, Trott PA, Fernando IN, Ashley SE, Ormerod MG, Titley JC, Gregory RK, Allred DC (1997) Prediction of response to neoadjuvant chemoendocrine therapy in primary breast carcinomas. Clin Cancer Res 3(4):593–600
Chang J, Powles TJ, Allred DC, Ashley SE, Clark GM, Makris A, Assersohn L, Gregory RK, Osborne CK, Dowsett M (1999) Biologic markers as predictors of clinical outcome from systemic therapy for primary operable breast cancer. J Clin Oncol 17(10):3058–3063
Chang J, Ormerod M, Powles TJ, Allred DC, Ashley SE, Dowsett M (2000) Apoptosis and proliferation as predictors of chemotherapy response in patients with breast carcinoma. Cancer 89(11):2145–2152
Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W, Reed JC (1993) Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res 53(19):4701–4714
Elledge RM, Green S, Howes L, Clark GM, Berardo M, Allred DC, Pugh R, Ciocca D, Ravdin P, O'Sullivan J, Rivkin S, Martino S, Osborne CK (1997) bcl-2, p53, and response to tamoxifen in estrogen receptor-positive metastatic breast cancer: a Southwest Oncology Group study. J Clin Oncol. 15(5):1916–1922
Hilsenbeck SG, Ravdin PM, de Moor CA, Chamness GC, Osborne CK, Clark GM (1998) Time-dependence of hazard ratios for prognostic factors in primary breast cancer. Breast Cancer Res Treat 52(1–3):227–237
Gyrd-Hansen M, Meier P (2010) IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat Rev Cancer 10(8):561–574
Ku B, Liang C, Jung JU, Oh BH (2011) Evidence that inhibition of BAX activation by BCL-2 involves its tight and preferential interaction with the BH3 domain of BAX. Cell Res 21(4):627–641
Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DCS (2005) Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 17(3):393–403
Llambi F, Moldoveanu T, Tait SW, Bouchier-Hayes L, Temirov J, McCormick LL et al (2011) A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell 44(4):517–531
Vaillant F, Merino D, Lee L, Breslin K, Pal B, Ritchie ME, Smyth GK, Christie M, Phillipson LJ, Burns CJ, Mann GB, Visvader JE, Lindeman GJ (2013) Targeting BCL-2 with the BH3 mimetic ABT-199 in estrogen receptor-positive breast cancer. Cancer Cell 24(1):120–129
Merino D, Khaw SL, Glaser SP, Anderson DJ, Belmont LD, Wong C, Yue P, Robati M, Phipson B, Fairlie WD, Lee EF, Campbell KJ, Vandenberg CJ, Cory S, Roberts AW, Ludlam MJC, Huang DCS, Bouillet P (2012) Bcl-2, Bcl-x(L), and Bcl-w are not equivalent targets of ABT-737 and navitoclax (ABT-263) in lymphoid and leukemic cells. Blood 119(24):5807–5816
Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DC (2005) Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev 19(11):1294–1305
Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor G, Thompson CB, Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292(5517):727–730
Acknowledgements
We are grateful to the patients and their families who participated in this study. We thank Ciaran de Chaumont and Lance Hudson for technical assistance, Ramphul Eimear, Dr. Róisín M. Dwyer and Prof. Michael J. Kerin for sharing of samples, and Dr. Triona Ni Chonghaile for critical review of the manuscript. The results published here are in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov which we also gratefully acknowledge.
Funding
Funding support was provided by the Irish Cancer Society Collaborative Cancer Research Centre grant, BREAST-PREDICT (to J.H.M.P. and W.M.G.) and a Science Foundation Ireland Investigator Award to J.H.M.P. (13/IA/1881).
Author information
Authors and Affiliations
Contributions
A.U.L., F.L. and D.V. generated data. A.U.L. and A.R. performed data analysis. D.V., B.M.M., A.D.K.H. and L.S.Y. acquired clinical data and provided material. J.H.M.P. and W.M.G. conceived this study and provided funding. J.H.M.P. and L.S.Y. supervised the study. A.U.L. and J.H.M.P. wrote the manuscript. All authors read, reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Informed consents were collected following ethical approval from Beaumont Hospital Medical Research Ethics Committee.
Conflict of interest
AUL and JHMP filed a patent application at the EPO (Appl.No. EP20120166187 and EP20130728324), USPTO (Appl. No. 14/397697) and WIPO (Appl. No. PCT/EP2013/059051). The other authors declare no further conflict of interest.
Ethics approval and consent to participate
For the BREAST-PREDICT cohort, informed consents were collected following ethical approval from Beaumont Hospital Medical Research Ethics Committee.
Rights and permissions
About this article

Cite this article
Lindner, A.U., Lucantoni, F., Varešlija, D. et al. Low cleaved caspase-7 levels indicate unfavourable outcome across all breast cancers. J Mol Med 96, 1025–1037 (2018). https://doi.org/10.1007/s00109-018-1675-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-018-1675-0